

Abstract
The cellular responses to two new temozolomide (TMZ) analogues, DP68 and DP86, acting against glioblastoma multiforme (GBM) cell lines and primary culture models are reported. Dose-response analysis of cultured GBM cells revealed that DP68 is more potent than DP86 and TMZ and that DP68 was effective even in cell lines resistant to TMZ. Based on a serial neurosphere assay, DP68 inhibits repopulation of these cultures at low concentrations. The efficacy of these compounds was independent of MGMT and MMR functions. DP68-induced interstrand DNA crosslinks were demonstrated with H2O2-treated cells. Furthermore, DP68 induced a distinct cell cycle arrest with accumulation of cells in S phase that is not observed for TMZ. Consistent with this biological response, DP68 induces a strong DNA damage response, including phosphorylation of ATM, Chk1 and Chk2 kinases, KAP1, and histone variant H2AX. Suppression of FANCD2 expression or ATR expression/kinase activity enhanced anti-glioblastoma effects of DP68. Initial pharmacokinetic analysis revealed rapid elimination of these drugs from serum. Collectively, these data demonstrate that DP68 is a novel and potent anti-glioblastoma compound that circumvents TMZ resistance, likely as a result of its independence from MGMT and mismatch repair and its capacity to crosslink strands of DNA.
Keywords: Temozolomide, MGMT, DNA damage response, resistance, glioblastoma, FANCD2, ATR, ATM
Introduction
The imidazotetrazine prodrug temozolomide (TMZ, Figure S1) concurrent and adjuvant to radiotherapy is now the first line treatment for glioblastoma multiforme (GBM) in North America and Europe; however, intrinsic and acquired resistance ultimately limit the efficacy of therapy (1–3). At neutral pH, TMZ is moderately unstable (t1/2 = 1.24 h, pH 7.4) (4) and is hydrolyzed in a ring-opening reaction to the open chain triazene MTIC (Figure S2A; t1/2 = 8 min, pH 7.4) (5) that fragments to the reactive electrophile, methyldiazonium (t1/2 = 0.39 s, pH 7.4) (6–8). The methyldiazonium ion reacts with nucleophilic groups on DNA, resulting in DNA methylation. Approximately 70% of the methyl groups are located on N7-guanine (N7-G), 10% on N3-adenine (N3-A) and 5% at O6-guanine (O6-G) sites (3, 9). Products of N-methylation are readily repaired by the base-excision repair pathway and are not major contributors to cytotoxicity (10). In contrast, O6-methylguanine (O6-MeG) lesions are reversed by O6-methylguanine methyltransferase (MGMT), and failure to remove these lesions can lead to cytotoxicity and accumulated G→A transition mutations (Figure S2B) (11). The MGMT gene is silenced by promoter methylation in approximately 35% of GBMs (12). In these tumors, persistent O6-MeG lesions form wobble base-pairs with thymidine during replication. These O6-MeG:T pairings trigger futile cycles of mismatch repair (MMR), stalled replication forks and lethal DNA double-strand breaks. Disruption of MMR through mutation or suppressed expression results in a TMZ-tolerant phenotype, while high-level expression of MGMT protein is a major mechanism of inherent TMZ resistance (13).
Attempts have been made to engineer new TMZ derivatives with modified spectra of activity. One strategy is to modify DNA at O6-G in a manner that is not recognized or repaired by MGMT (14, 15). A disulfide linked imidazotetrazine dimer showed a similar response profile as the established agent busulfan in the NCI60 screening panel (16). The design of the imidazotetrazine compounds investigated herein sought a switch of chemical mechanism with the aims of avoiding known mechanisms of TMZ resistance and generating therapeutic benefit from the major reaction site, N7-G (70% for TMZ), rather than the minor (5%) O6-G site (17). In this study, we focus on two of the most promising compounds developed: the mono-functional DP86 and the bi-functional DP68 imidazotetrazines (Figure S1) (17, 18). These compounds are precursors of aziridinium ions, which are reactive intermediates of proven clinical utility related to those generated by nitrogen mustard drugs (Figure S2C–D). The bi-functional, p-methyl substituted compound, DP68, is the most potent compound, and has the least dependence on MGMT function and MMR status. This analogue was selected for detailed investigation; the mono-functional analogue, DP86, was evaluated for comparison. In matrix COMPARE analysis of NCI60 data, the new compounds showed no significant correlation with mitozolomide (0.46 ≥ P ≥ 0.35) so these are distinct new members of the imidazotetrazine class. The putative DNA lesion of the bifunctional agent is a five-atom crosslink, related in structure to those formed by the nitrogen mustard prodrugs; however, no drugs of this class showed strong correlations 0.59 ≥ P ≥ 0.29. Notably, there was also, no similarity to the nitrosoureas, which are also diazonium ion precursors, 0.45 ≥ P ≥ 0.05 or cisplatin, another N7-G reactive agent 0.42 ≥ P ≥ 0.27.(18). This manuscript describes the biological effects of these novel DNA alkylating agents in GBM models and their relative activities in the context of MGMT-overexpression and other mechanisms of TMZ resistance.
Materials and Methods
Cells and Reagents
The glioma cell lines U87MG, T98G, and U251 were purchased from the ATCC in 2011 and 2001 respectively. U118MG was a kind gift from Dr. Larry Recht (Stanford University, 2003). U251 (ATCC), T98G (ATCC), U118MG (IDEXX RADIL) and U87MG (IDEXX RADIL) were authenticated by short tandem repeat analysis in 2013. These cell lines are representative of the diverse GBM genotypes (Table 1). Adherent lines were maintained as monolayer cultures in DMEM supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. U87MG and U118MG were converted to neurosphere cultures, U87NS and U118NS, and maintained in serum-free media consisting of DMEM/F12 1:1 (Gibco), B27 supplement (Gibco), 15 mM HEPES (Gibco) supplemented with 20 ng/mL EGF (Invitrogen) and 20 ng/mL bFGF (Invitrogen). A TMZ-resistant culture, U87NSTMZ, was established from U87NS cells by incrementally increasing, twice-weekly treatments of TMZ to a final concentration of 325 µM. TMZ-resistant cells were maintained with a single weekly TMZ dose. GBM12TMZ (#3080) from the Mayo GBM xenograft panel and U251TMZ have been described (19, 20). Short-term explant cultures, GBM6 and GBM12, were established from Mayo GBM xenograft lines by mechanical disaggregation followed by culture in neurobasal serum-free media (StemPro NSCSFM; Invitrogen Cat#A1050901).
Table 1.
Activity of TMZ, DP68, and DP86 in glioma cultures
| Cell Line | MGMT | EGFR amplified |
P53 | PTEN activity |
IC50 (µM) | TMZ/DP68 | ||
|---|---|---|---|---|---|---|---|---|
| TMZ | DP86 | DP68 | ||||||
| Established glioma cell line CyQuant assay | ||||||||
| T98G | +++ | − | mutant | − | 340 ± 21 | 110 ± 9 | 11.3 ± 2.2 | 30 |
| U118MG | + | − | mutant | − | 135 ± 41 | 42 ± 7 | 10 ± 3.0 | 14 |
| U251 | − | − | mutant | − | 60 ± 21 | 68 ± 7 | 5.2 ± 0.6 | 12 |
| U251TMZ | − | − | ND | − | 306 ± 21 | 70 ± 13 | 3.6 ± 0.5 | 85 |
| U87MG | − | − | WT | − | 43 ± 23 | 67 ± 20 | 14 ± 5 | 3 |
| Established glioma line neurosphere assay | ||||||||
| U118NS | + | − | mutant | − | 71 ± 11 | 42 ± 9 | 2.7 ± 0.2 | 26 |
| U87NS | − | − | WT | − | 7.5 ± 1.0 | 61 ± 2 | 5.9 ± 0.5 | 1.3 |
| U87NSTMZ | − | − | ND | ND | 153 ± 8 | 33 ± 3 | 4.1 ± 0.2 | 37 |
| Patient-derived xenograft neurosphere assay | ||||||||
| GBM6 | +++ | EGFRvIII | mutant | WT | 187 ± 38 | 43 ± 24 | 1.3 ± 0.6 | 144 |
| GBM12 | − | mutant | mutant | WT | 3.2 ± 0.3 | 19 ± 3 | 0.8 ± 0.1 | 4 |
| GBM12TMZ | +++ | ND | ND | ND | 500 ± 132 | 25 ± 2 | 1.4 ± 0.1 | 357 |
ND: not determined
DP68 and DP86 were synthesized as described previously (17), TMZ was from Schering-Plough Corp and Sigma-Aldrich. All 3 agents were prepared as 25 mM stocks in dimethyl sulfoxide (DMSO). O6-benzylguanine (O6-BG) was obtained from Sigma-Aldrich, the ATR inhibitor VE-821 from ChemieTek and the ATM inhibitor KU-60019 from Selleck Chemicals. Antibodies against ATM (ab10939), p-ATM S1981 (ab81292) and p-KAP1 (ab70369) were purchased from Abcam; Chk1 (#04-207), Chk2 (#05-649) and ATR (#PC538) from Millipore; p-Chk1 Ser345 (#MA5-15145) and MGMT (#MS-470-P) from Thermo Scientific; p-Chk2 Thr68 (#2661), KAP1 (#4123), p-H2A.X Ser139 (#2577), and MLH1 (#3515) from Cell Signaling; and FANCD2 (#2986-1) from Epitomics. β-actin antibody was purchased from Sigma-Aldrich. Secondary anti-rabbit and anti-mouse IgG were purchased from Cell Signaling Technologies and Pierce, respectively.
Western Blotting
Cells were lysed in RIPA lysis buffer (R0278, Sigma-Aldrich) supplemented with a protease inhibitor cocktail (Roche). Total proteins were isolated from flash frozen flank xenografts or short-term explant cultures, separated by SDS-PAGE, and electro-transferred onto PVDF membranes. Membranes were blocked in Tris-buffered saline (TBS) containing 5% milk and 0.1% Tween-20 at room temperature. All primary antibodies were incubated overnight at 4 °C followed by room temperature incubation with a secondary antibody conjugated with horseradish peroxidase for 1 h. Detection was performed with Super Signal Chemiluminescent reagent according to the manufacturer’s protocol (Pierce).
CyQUANT cell proliferation assay
A cell proliferation assay was performed using the CyQUANT Cell Proliferation kit (Invitrogen) according to manufacturer’s recommendations. Cells were seeded (1,000 cells/well) in triplicate in 96-well plates, exposed to the DP68, DP86, or TMZ, and incubated for 5 days. In some experiments, O6-BG (10 µM) was added 1 h before DP68, DP86, or TMZ treatment. On day 5, medium was removed, cells were washed with PBS, and plates were stored at −80 °C. The plates were thawed and lysed in CyQUANT GR dye-containing lysis buffer. After 4 min incubation at room temperature, the fluorescence intensity of the DNA-binding dye was measured using a TECAN plate reader with excitation at 480 nm and emission at 520 nm.
Flow cytometry analysis
Cell cycle distribution was analyzed by flow cytometry. Cells were harvested, washed with PBS, and fixed with ice-cold 70% ethanol/30% PBS. Cells were re-suspended in PBS containing propidium iodide (40 µg/ml), RNase A (100 µg/ml) and Triton X-100 (0.05%), and incubated at 37 °C for 30 min. DNA content was determined using a FACScan flow cytometer system (Becton Dickinson), and results were analyzed with Modfit software (Verity Software).
Neurosphere assays
Chemosensitivity (sphere IC50) of glioblastoma cells to TMZ, DP86, and DP68 was analyzed by seeding U87NS and U118NS cells in triplicate in 96-well plates (1,000 cells/100 µL/well)‥ After 24 hrs, vehicle, TMZ, DP86, or DP68 was added to the wells and primary neurospheres were quantified after 7 days. Primary xenograft cultures (GBM6, GBM12, and GBM12TMZ) were plated (500 cells/well), treated, and neurospheres were quantified at day 14. For limiting dilution experiments in Figure S3, GBM12 cells were plated at 1, 10, 30, 100, 300 or 1000 cells/well, drug treated after 24hrs, and counted at day 14. For the recovery and secondary sphere assay (21), cells were plated at clonal density (3,000 cells/mL; 2 mL total) in 6-well plates and treated with TMZ, DP86, DP68, or vehicle. Primary spheres were counted on day 7 and cells were fed with neurosphere medium (2 mL). Spheres for the recovery phase were counted on day 14, dissociated using a basic pH dissociation method (22), and a dilution of the culture re-plated. Secondary spheres were counted on day 21. Spheres with 10 cells or more were counted.
siRNA and shRNAs
Transient knockdown of ATM, ATR, and FANCD2 was achieved using siRNAs from Thermo Scientific/Dharmacon: ATM- AAG CAC CAG TCC AGT ATT GGC; ATR- C CTC CGT GAT GTT GCT TGA; FANCD2- GGUCAGAGCUGUAUUAUUC; control luciferase siRNA- CTT ACG CUG AGU ACU UCG A. siRNAs were mixed with 5–8 million U251 cells and electroporated with two 280 V pulses of 10 ms. The cells were plated, incubated overnight and again electroporated. After a 24 h recovery, cells were plated for subsequent studies.
For MLH1 knockdown experiments, lentiviral (TRC) shRNAs against eGFP and MLH1 (TRCN0000040053, TRCN0000040056) as well as empty pLK0.1 vector construct were purchased from UMASS RNAi Core (Open Biosystems), and lentiviruses were produced using HEK293T. T98G cells were infected by lentivirus using 10 µg/mL polybrene, followed by selection using 2µg/mL puromycin for two weeks to generate stable lines.
Analysis of DNA cross-linking in single cells using alkaline comet assay
U251 cells in exponential growth were exposed to DP68, DP86, TMZ, or melphalan (positive control for cross linking) for 24 h and subsequently treated with 100 µM hydrogen peroxide for 20 min. As described previously (23), cells then were embedded in 0.5% low melting point agarose, spread on agarose coated glass slides, and lysed in ice cold lysis buffer before being transferred to electrophoresis buffer (pH >13). After 30 min incubation to allow DNA unwinding and expression of alkali labile sites, cells were subjected to electrophoresis at 0.6 V/cm for 25 min. Neutralization buffer was added drop-wise to the slides followed by rinsing in distilled water, fixation in 100% ice-cold ethanol and drying overnight. Slides were stained with SYBR™Gold solution (Molecular Probes Inc.) and the comets visualized using an epi-fluorescent microscope (Nikon Eclipse E800). Tail moments were measured on 50 randomly selected comets using Comet Assay III software (Perceptive Instruments, UK). Percentage DNA cross-linking was calculated from (TMC-TMT) / TMC where TMC and TMT represent the tail moment of control and drug treated cells respectively (23).
Pharmacokinetic analysis of imidazotetrazine compounds
DP68 and DP86 were formulated in 0.9% NaCl, 10% (w/v) HP-β-cyclodextrin, and C57B2/6 mice were dosed with a single 25 µmol/kg intraperitoneal injection. Groups of 3 mice were euthanized at times up to 6 h after injection and plasma harvested for analysis. Drug levels were assessed by the LTQ quadrupole ion trap mass spectrometry method (24).
Results
Efficacy of DP68 and DP86 in human glioma cultures is independent of MGMT and MMR expression
The efficacies of DP68 and DP86 were evaluated in MGMT non-expressing and MGMT expressing glioma cell lines (Figure 1A, Table 1). In contrast to TMZ, DP68 was efficacious in both MGMT-negative (U251, IC50= 5.2 µM; U87MG, IC50 =14 µM) and MGMT-positive (T98G, IC50 = 11.3 µM; U118MG, IC50=10 µM) cell lines (Table 1). The bi-functional agent DP68 was significantly more potent than TMZ in all cell lines with IC50 ratios (TMZ:DP68) ranging from 3 to 85 (Table 1). The mono-functional agent DP86 was approximately 10-fold less potent than DP68 in all lines studied, but similar to DP68, the activity of DP86 was independent of MGMT expression. U87NS and U118NS cultures that had been converted to long-term neurosphere growth as well as neurospheres derived from the Mayo xenograft panel were similarly sensitive to DP68 and DP86 (Figure 1B–C and Figure S3A). In models of acquired TMZ resistance, the efficacy of DP68 was essentially unchanged compared to the parental lines with an IC50 for DP68 in U251TMZ of 3.6 µM and in U87NSTMZ of 4.1 µM (Table 1, Figure S3B). In a third TMZ-resistant model developed from primary line GBM12 that has high-level MGMT overexpression (GBM12TMZ), DP68 was highly effective (Figure 1CD; IC50 = 1.4 µM). As before, DP86 was approximately 10-fold less potent with IC50s ranging from 19–43 µM.
Figure 1. DP68 and DP86 exhibit strong anti-glioma activity independent of MGMT.
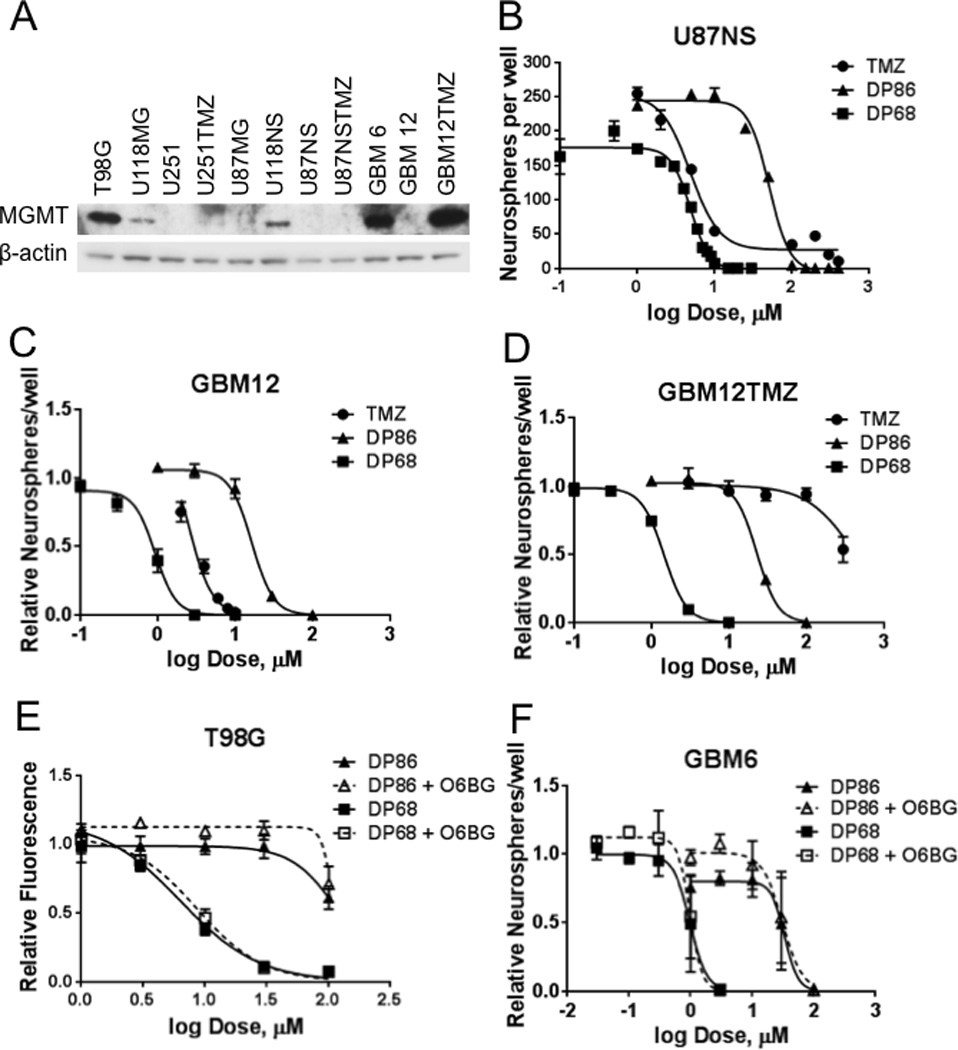
(A) Western blot showing MGMT expression in glioma lines; (B-F) The effects of TMZ, DP68, and DP86 were assessed in neurosphere formation assays for (B) U87NS, (C) GBM12, (D) GBM12TMZ, (E-F) (E) T98G and (F) GBM6 co-treated with 0 or 10 µM O6-BG and DP86 or DP68 were evaluated. The mean ± SEM of three independent experiments are shown.
Because plating density can affect the efficiency of neurosphere formation (25), limiting dilution assays were carried out with GBM12 cells plated at various densities and treated with TMZ, DP68 or DMSO (control). TMZ and DP68 have similar potencies for neurosphere formation regardless of cell density with DP68 demonstrating greater inhibition of neurosphere formation (Figure S3C)
To validate the impact of MGMT activity on DP68 and DP86 activity, T98G and GBM6 cells were co-treated with the MGMT suicide inhibitor O6-BG. Co-treatment with O6-BG increased sensitivity to TMZ in the MGMT-expressing T98G and GBM6 cells (Figure S3D-E), while O6-BG had no impact on the efficacy of DP68 and DP86 in these cells (Figure 1E–F).
Cytotoxicity of TMZ also depends on the integrity of the MMR system, which is activated in response to O6-MeG lesions that mismatch with thymine. To assess the importance of this pathway, we infected T98G cells with shRNAs targeting MLH1 (Figure S4A). Knockdown of MLH1 had no impact on activity of DP86 or DP68 while decreasing the sensitivity to TMZ (empty vector IC50=982 µM, sheGFP IC50=1077 µM, shMLH1 #1 IC50= 2005 µM, shMLH1 #2 IC50= 1748 µM) (Figure S4B). Collectively, these data confirm that activity of the new imidazotetrazine compounds is independent of cellular MGMT and MMR.
DP68 and DP86 inhibit recovery and secondary sphere formation
The effects of imidazotetrazines on long-term cell growth were evaluated in a secondary neurosphere formation assay with MGMT non-expressing glioma line U87NS and MGMT expressing line U118NS. Treatment of U87NS and U118NS with TMZ (100 µM) showed similar reductions in the number of neurospheres at day 7 (‘Treatment’ – Figure 2A) with 95% and 96% reductions, respectively. By day 14, U118NS cultures recovered, showing similar number of neurospheres as the DMSO treated culture (‘Recovery’ – Figure 2A), while there were few spheres present for U87NS cells. The neurospheres were dispersed and replated. By day 21, U87NS and U118NS cultures formed secondary spheres with marked recovery from TMZ treatment (‘Secondary’ – Figure 2A). In contrast, DP68 treatment greatly reduced neurosphere formation after a single treatment and prevented the subsequent recovery of spheres (Figure 2B); secondary sphere formation was completely repressed by 10 to 30 µM DP68. Similar results were seen with DP86 at approximately 10-fold higher concentrations than DP68 (Figure 2C). Thus, in comparison to TMZ, DP68 and DP86 provide more durable inhibitory effects on primary and secondary neurosphere formation.
Figure 2. Single treatment of GBM neurospheres with DP68 or DP86 leads to a reduction of neurospheres and inhibition of secondary sphere formation.
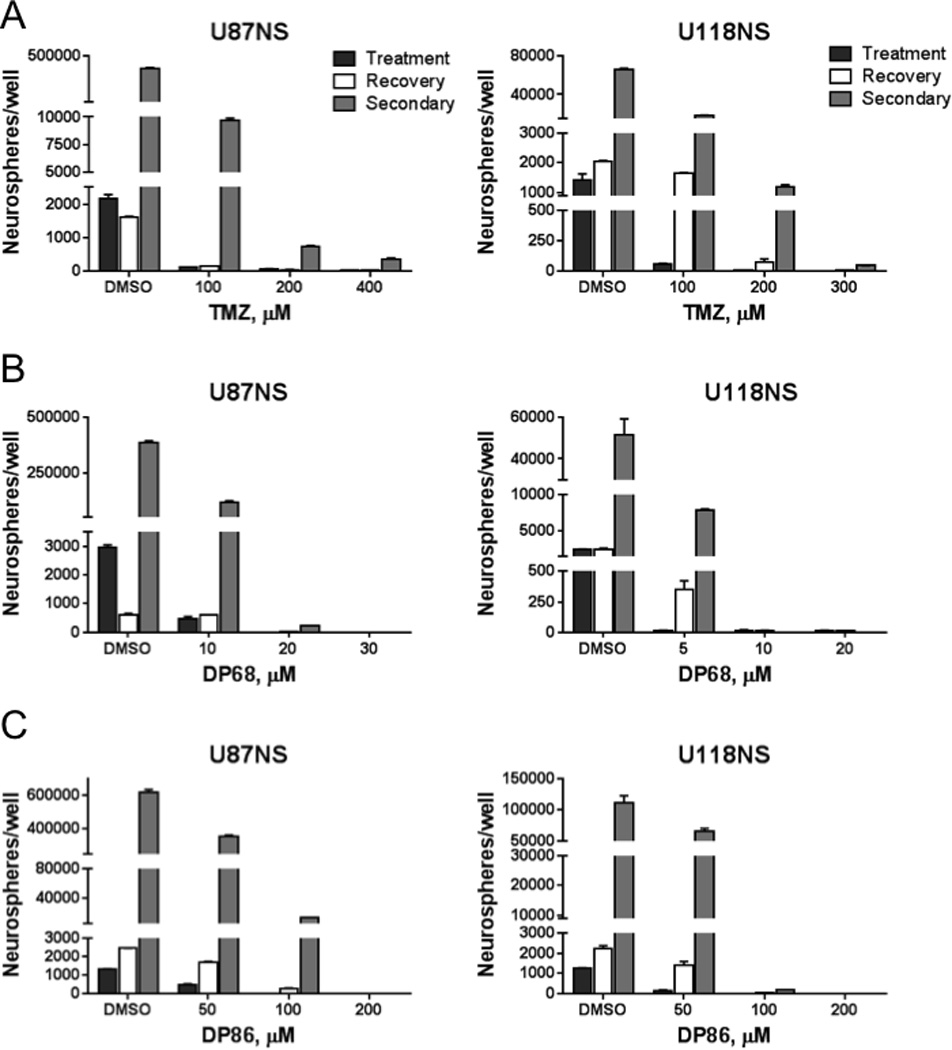
U87NS or U118NS neurospheres were treated with (A) TMZ, (B) DP68, or (C) DP86. Bars show the number of neurospheres following treatment on day 7 (black), recovery at day 14 (white), and secondary spheres at day 21 (grey). Representative results from at least three independent experiments are shown.
DP68 Induces DNA Crosslinks in Glioma Cells
Damage to nuclear DNA can be investigated by single cell electrophoresis (comet assay). This assay detects single and double strand breaks and alkali labile sites and has been adapted to measure DNA inter-strand crosslinking. Pre-treatment with a DNA cross-linking agent will retard the migration of DNA fragments generated by H2O2 treatment (26). The cross-linking agent melphalan (positive control) and DP68 showed significant and comparable, concentration-dependent cross-linking of nuclear DNA in U251 cells (Figure 3A), while DP86 and TMZ showed no evidence of cross-linking (Figure 3A). These data show that DP68 is equipotent with melphalan at DNA cross linking; in contrast, the mono-functional agents do not form cross links.
Figure 3. Cellular responses to DP68.
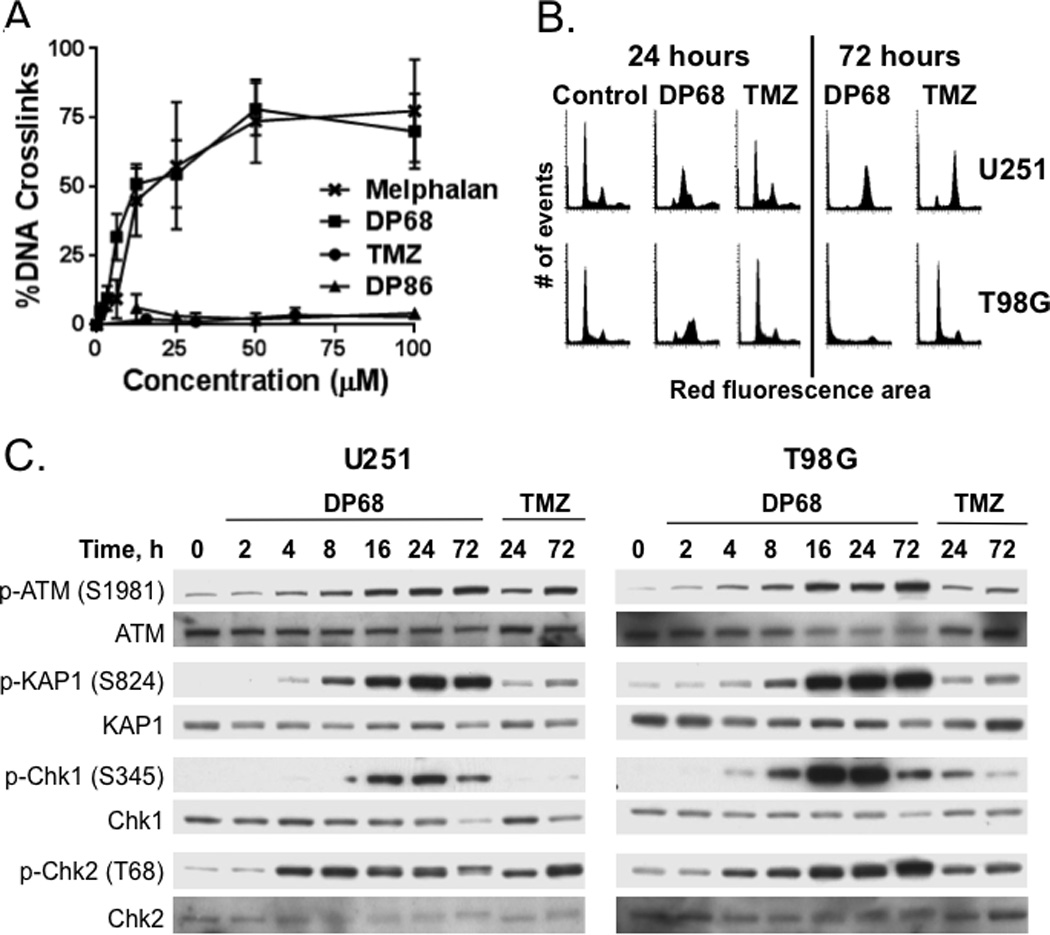
(A) U251 cells were treated with melphalan, TMZ, DP68 and DP86 for 24 h and then H2O2 for 20 min prior to analysis in a Comet assay. Relative amount of DNA cross-linking was calculated from the comet moments (±SD). (B) U251 and T98G cells were treated with vehicle, 10 µM DP68, or 30 µM TMZ for 24 or 72 h, fixed, stained with propidium iodide, and analyzed by flow cytometry. (C) U251 and T98G cells were treated with 0 or 30 µM DP68 or 100 µM TMZ and processed for western blotting at various time points following drug addition. Representative results from three independent experiments are shown.
S-phase accumulation and G2/M arrest by DP68
The effects of DP68 and TMZ on cell cycle distributions were compared in U251 (MGMT non-expressing) and T98G (MGMT expressing) cells. In U251 cells, treatment with 30 µM TMZ had relatively minimal effects on cell cycle until 72 h after treatment, when there was an accumulation of cells in G2/M. In contrast, DP68 induced a marked S-phase accumulation within 16–24 h, and by 72 h the majority of cells accumulated in G2/M (Figure 3B). In contrast, TMZ treatment has no significant effects on cell cycle distribution in TMZ-resistant T98G cells, while DP68 treatment results in a similar cell cycle distribution as in U251 with marked accumulation in S and G2/M 24 h after treatment and significant accumulation in sub-G1 by 72 h after treatment. Thus, DP68 has a cell cycle arrest profile distinct from TMZ, with an early accumulation of cells in S-phase followed by G2/M arrest.
Activation of DNA damage response by low concentrations of DP68
Consistent with the flow-cytometry data, DP68 activated DNA damage signaling pathways within hours of treatment. As seen in Figure 3C, phosphorylation of ATM (S1981) and the canonical ATM substrate Chk2 (T68) were detected 4 to 8 h after treatment with 30 µM DP68, while phosphorylation of the canonical ATR substrate Chk1 was later with reproducible phosphorylation induction at 8 to 16 h. Both ATM and ATR can phosphorylate KAP1 (S824) in response to DNA damage (27), and consistent with activation of these pathways, phosphorylation of KAP1 was evident within 8 h of DP68 treatment and maintained for at least 72 h. In contrast, TMZ-induced damage signaling was evident only in U251 cells and only at a late 72 h time-point. DP68-induced phosphorylation of this DNA damage response network (p-ATM, p-Chk2, p-Chk1, and p-KAP1) and histone variant H2AX (S139; γ-H2AX) at concentrations as low as 3 µM in MGMT-negative U251 and MGMT-positive T98G cells (Figure S5). Collectively, these data are consistent with robust activation of ATM- and ATR-dependent DNA damage signaling at concentrations associated with significant chemosensitivity.
Impact of ATM, ATR and FANCD2 suppression on imidazotetrazine sensitivity
The Fanconi’s Anemia pathway is important for repairing DNA inter-strand crosslinks, and DP68 induces DNA inter-strand crosslinks, activating the ATM and/or ATR damage responses. To assess functional importance, these pathways were evaluated using siRNA and small molecule inhibitors. U251 cells were electroporated with siRNAs directed against ATM, ATR and a core Fanconi Anemia pathway gene, FANCD2 (Figure 4A). Knockdown of ATM had no impact on sensitivity to TMZ, DP68, or DP86 (Figure 4B), while ATR knockdown significantly enhanced the response to TMZ, DP86 and DP68. FANCD2 knockdown resulted in a significantly enhanced activity only with DP68 treatment (relative fluorescence of 0.16 vs. 0.51, p≤0.001), but had no significant effect on TMZ (0.37 vs. 0.44, respectively; p=0.26) or DP86 (0.87 vs. 0.96, respectively; p=0.11) treated cells. The impacts of ATR and ATM signaling on cytotoxicity were confirmed using small-molecule inhibitors of ATM (KU-60019) and ATR (VE-821) kinase activity in U251 (Figure 4C) and T98G (Figure 4D) cells. In both lines, co-treatment with VE-821 markedly enhanced the efficacy of DP68, and to a lesser extent DP86 in T98G cells. In contrast, KU-60019 was less effective than VE-821 when combined with DP68 in T98G cells (p=0.001) and in U251 cells (p=0.01). Similarly, co-treatment with the ATR inhibitor VE-861, but not KU-60019, enhanced the efficacy of DP68 and DP86 in a neurosphere assay with GBM12 or GBM12TMZ (Figures 4E and 4F). These data are consistent with the induction of DNA cross-links by DP68 and suggest that ATR and FANCD2 aid recovery following treatment with DP68.
Figure 4. Recovery from DP68 induced DNA damage is ATR and FANCD2 dependent.
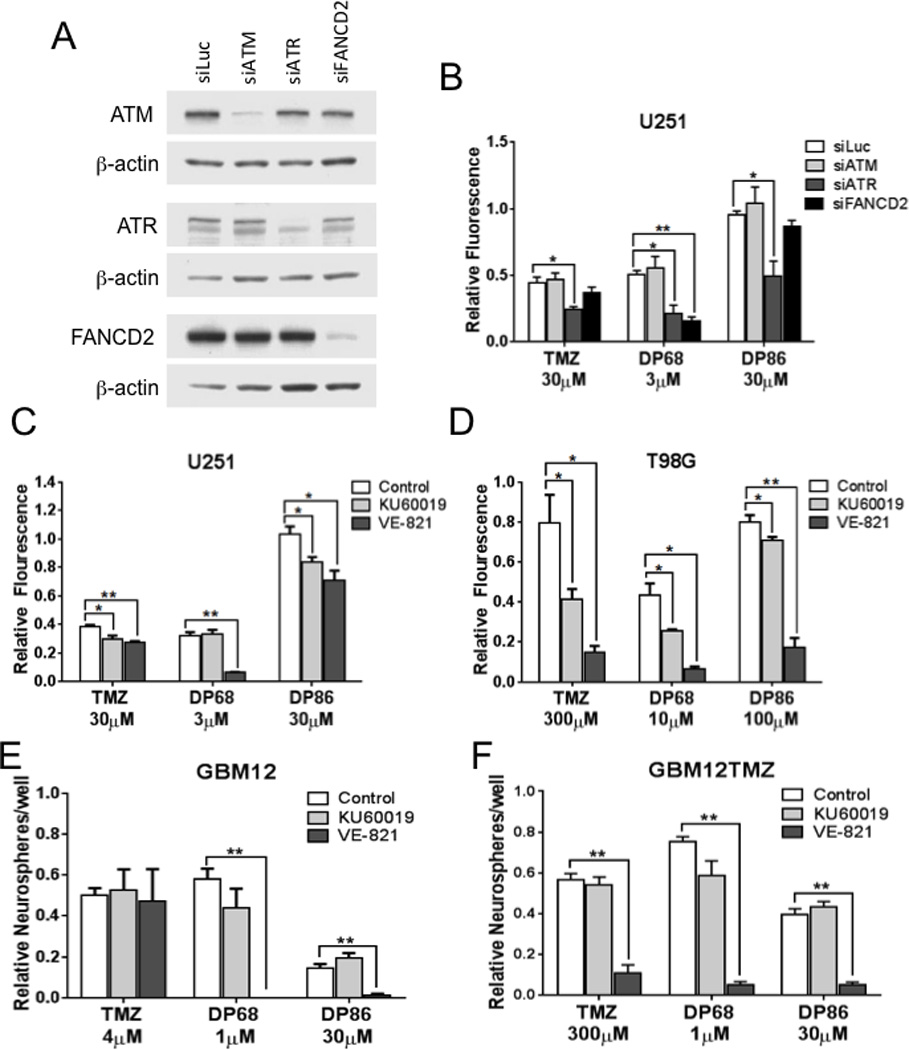
(A) Western blot confirming knockdowns in U251 cells double electroporated with control (fLuc), ATM, ATR, or FANCD2 siRNA. (B) Transfected cells were treated with 0 or 30 µM TMZ, 3 µM DP68 or 30 µM DP86 and assessed for cell growth using a CyQuant assay. (C) U251 and (D) T98G cells were treated with an ATR-inhibitor (1 µM VE-821), an ATM-inhibitor (1 µM KU60019), or vehicle with or without TMZ, DP68, or DP86, and cell survival was analyzed in a CyQuant assay. (E, F) A neurosphere assay was performed in parental GBM12 (E) and TMZ-resistant GBM12TMZ (F) explant cultures treated with 0 or 1 µM VE-821 or 1 µM KU60019 in combination with TMZ, DP68, or DP86. Data points and error bars presented are the mean relative neurosphere number ± SEM from three independent experiments. * (p<0.05), ** (p<0.001)
Initial in vivo characterization of imidazotetrazine compounds
The pharmacokinetic properties of DP68 and DP86 were evaluated in mice for potential dosing strategies. Following a single intraperitoneal injection, DP86 peaked rapidly with a Tmax of 10 min, Cmax of 3230 nM and t1/2 = 14 min (Figure 5A). DP68 had similar pharmacokinetic properties except that absorption was much more limited with a Cmax of 206 nM (Figure 5B). No adverse clinical effects were noted in the mice for up to 6 h after drug injection. However, because of the very short half-life for both drugs and the relatively limited absorption for DP68, further in vivo characterization of these compounds was not pursued.
Figure 5. Pharmacokinetic evaluation of DP86 and DP68.
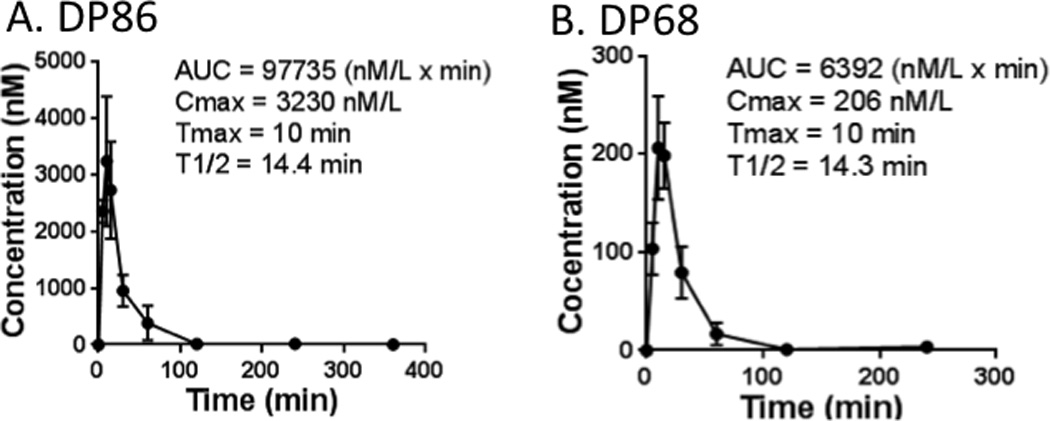
Following a single IP injection of (A) DP86 or (B) DP68, plasma drug levels were determined at times up to 6 hours post-injection. Results shown are mean ± SD at each time-point (n=3 mice per time-point).
We investigated whether the fast elimination could be due to enzymatic degradation using published techniques (24). The half-life of DP68 and DP86 was determined in mouse liver homogenate (DP68 t1/2 = 1.73 ± 0.05 h (n=3); DP86 t1/2 = 1.62 ± 0.18 h (n=3)) and the half-life was roughly the same as in aqueous buffer at pH 7.4 (DP68 t1/2 = 1.75 ± 0.04 h (n=3); DP86 t1/2 = 1.74 ± 0.08 h (n=3)). Therefore, it is likely that DP68 and DP86 are not susceptible to enzymatic degradation.
Discussion
TMZ is a key component of therapy for GBM, but the ultimate benefit is limited by emergence of resistance. Expression of the MGMT DNA repair protein accounts for profound TMZ resistance in the majority of chemotherapy naïve GBM patients, and inactivation or down-regulation of MMR can lead to acquired tolerance of TMZ-induced lesions. Therefore, developing therapeutic agents based on the TMZ structure that circumvent mechanisms of TMZ resistance may provide significant therapeutic gains. In this study, the anti-glioma activity was evaluated for two novel imidazotetrazine analogs: the bivalent DP68 and monovalent DP86. Both compounds exhibited activity in established glioma lines maintained in both serum-containing medium and as neurospheres in defined medium. In short-term explant cultures from primary GBM xenograft lines, both compounds were equipotent in TMZ-sensitive and -resistant GBM models. The bivalent DP68 induced a rapid and profound S-phase accumulation, and was associated with early activation of DNA damage signaling. These data provide a proof-of-concept that novel TMZ analogs can circumvent TMZ resistance in GBM models.
TMZ chemotherapy provides significant benefit to a subset of patients, although progressive tumor growth associated with emergence of TMZ resistance is almost universal. Stem-like cells express drug efflux transporters, have higher DNA repair capacities and may be responsible for re-populating tumors with therapy resistant clones (28–30). They grow as neurospheres in serum-free media, and in this study, the effects of the imidazotetrazine analogs on neurosphere growth were evaluated in two different models. The neurosphere recovery assay provides a 3-dimensional tumor model that measures acute responses to drug therapy and the clonogenic potential of cells following treatment. Both U87NS (TMZ sensitive) and U118NS lines (TMZ resistant), cells were initially responsive to TMZ treatment, but out-growth of secondary neurospheres was only inhibited at TMZ concentrations greater than 100 µM (Figure 2A), which is at the limit of clinically achievable levels of TMZ. A similar pattern of regrowth was observed with DP68, but at a log lower concentration. The impact of drug therapy also was evaluated in neurosphere cultures derived directly from patient derived xenograft lines. In both GBM12 (TMZ sensitive) and GBM6 (TMZ resistant) models, DP68 effectively suppressed neurosphere formation at a log-lower dose of drug than TMZ. Although other criteria for defining tumor stemness were not tested specifically in this study, we have demonstrated tri-lineage differentiation, self-renewal and tumorigenicity in animals for multiple Mayo GBM xenograft lines (31). Thus, these data suggest that DP68 effectively kills stem-like cells and reduce the emergence of TMZ resistance mechanisms.
Three models of acquired TMZ resistance were evaluated in this study. The dose responses for DP68 and DP86 were similar for GBM12 and GBM12TMZ, while the latter line was markedly resistant to TMZ. Resistance in GBM12TMZ has been linked to high-level MGMT expression (32). Co-administration of the MGMT suicide inhibitor O6-BG significantly sensitized MGMT overexpressing cells lines to TMZ (GBM6 and T98G), but had no impact on response to DP68 or DP86. The U87NSTMZ and U251TMZ lines have distinct mechanisms of TMZ resistance unrelated to MGMT. Previous studies have linked mutational inactivation of MSH2 or MSH6 in the MMR pathway to tolerance of cytotoxic O6-MeG lesions, resulting in a TMZ resistance in GBM patients (3, 15). Although we have not tested, we speculate that resistance in these models is related to disruption of MMR or another TMZ-tolerance mechanism. Regardless of mechanism, the data demonstrate equal efficacy of DP68 and DP86 in parental tumor lines (U251, U87NS) and derivative models of acquired TMZ resistance (U251TMZ, U87NSTMZ). Thus, DP68 and DP86 are highly effective in models with diverse mechanisms of TMZ resistance.
The aqueous chemistry of DP68 and DP86 is subtly different from that of TMZ (Figure S2). All three compounds undergo pH-dependent hydrolytic ring-opening reactions to generate reactive diazonium ions. For TMZ these are the final reactive intermediates that covalently modify N7-G, O6-G, and N3-A. In contrast, the diazonium ions from DP68 and DP86 undergo an efficient, intra-molecular trapping reaction to form aziridinium ions, and these intermediates react with DNA, predominantly at N7-G sites (17). The new agents were designed to generate anticancer activity from N7-G adducts but should adducts occur at O6-G, these would be resistant to repair by MGMT (18). Consistent with either mechanism, sensitivity to both compounds was unaffected by MGMT expression in the TMZ-resistant T98G, GBM6, or GBM12TMZ models, and a similar MGMT-independence was observed in A2780 ovarian carcinoma cells (18). The early arrest in S-phase with DP68 treatment is consistent with DNA adducts that cannot be bypassed by the replication machinery, stalling replication forks. Consistent with these observations, DP68 triggered robust DNA damage signaling to the Chk1 and Chk2 checkpoint kinases and the chromatin remodeling protein KAP1 within 4 to 8 h of treatment, and ultimately phosphorylation of H2AX (33). These damage-inducible modifications are typically mediated by ATM in response to DNA double-strand breaks and by ATR in response to replication-induced DNA damage. Consistent with signaling effects triggered predominantly by replication-induced damage, disruption of ATR signaling enhanced the potency of DP68 and DP86, while ATM inhibition had less significant inconsistent effects (Figure 4). Collectively, these data suggest that DP68 and DP86 induce DNA lesions that disrupt DNA replication.
Despite likely similar nucleotide targets, DP68 and DP86 trigger significantly different patterns of DNA damage processing compared to TMZ. DP68 is a bivalent molecule that could generate two reactive aziridinium ions. Given the propensity of aziridinium ions to react with N7-G, we speculated that DP68 would insert five-atom, 3-azapentylene N7-G–N7-G intra- and/or inter-strand crosslinks. The marked suppression of H2O2-induced comet tail moment following treatment with DP68, but not DP86, is consistent with formation of DNA inter-strand crosslinks. Moreover, the Fanconi’s anemia DNA repair pathway specifically repairs cross-link damage (34), and disruption of this repair function, via siRNA suppression of FANCD2, sensitized U251 cells to DP68 but not DP86 or TMZ. These data indicate formation of inter-strand cross-links by DP68, and the 10-fold greater potency compared to DP86 likely reflects this DNA cross-linking activity.
The present study provides proof-of-concept for novel imidazotetrazine analogs that induce DNA adducts insensitive to TMZ-resistance mechanisms. Specifically, bi-functional DP68 with DNA cross-linking activity provided significant gains in potency and was highly effective against cells with the most common mechanism of TMZ resistance: MGMT overexpression. In preliminary pharmacokinetic studies, the half-life for either DP68 or DP86 (14 min) was significantly shorter than temozolomide (55 min). Based on these data, medicinal chemistry approaches are being used to optimize the drug-like properties for improved biodistribution. Essentially all GBM patients treated with TMZ develop refractory disease and ultimately die from progressive tumor. Thus, developing novel chemotherapies specifically effective against, or indeed averting the evolution of TMZ-resistant tumors is a critical unmet clinical need (35). The data presented suggest that mono- and bi-functional imidazotetrazines may be a compound class for treating TMZ-resistant tumors. Future studies with optimized second-generation molecules will focus on defining the in vivo efficacy in orthotopic GBM xenograft models and defining the toxicity profile and therapeutic window for this promising class of agents.
Supplementary Material
Acknowledgments
The authors thank Dr. Larry Karnitz for expert technical advice for electroporation and siRNA knockdown experiments, the UMASS RNAi Core for shRNAs, and the Mayo Flow Cytometry/Optical Morphology Shared Resource for expert assistance in analysis of cell cycle. We thank Arvin Iracheta-Vellve for help with initial IC50 experiments.
JNS has research grants from Merck, Basilea, Genentech and Sanofi-Aventis unrelated to this study.
Grant Support This research was supported by NIH Grants (RO1 NS021716 A.H. Ross; RO1 CA176830 and Mayo Brain Tumor SPORE CA108961 J.N. Sarkaria), Cancer Research UK (C14492/A4884 R.T. Wheelhouse), Academy of Finland (132647 J. Rautio) and Finnish Cultural Foundation (M. Gynther).
Footnotes
Disclosure of Potential Conflicts of Interest The others have no conflict of interests.
References
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:2960–2965. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012;5:102–114. doi: 10.2174/1874467211205010102. [DOI] [PubMed] [Google Scholar]
- 4.Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987;47:5846–5852. [PubMed] [Google Scholar]
- 5.Shealy YF, Krauth CA. Imidazoles. II. 5(or 4)-(Monosubstituted triazeno)imidazole-4(or 5)-carboxamides. J Med Chem. 1966;9:35–38. doi: 10.1021/jm00319a008. [DOI] [PubMed] [Google Scholar]
- 6.Wheelhouse RT, Stevens MFG. Decomposition of the antitumour drug Temozolomide in deuterated phosphate buffer: methyl group transfer is accompanied by deuterium exchange. J Chem Soc Chem Commun. 1993:1177. [Google Scholar]
- 7.McGarrity JF, Smyth T. Hydrolysis of diazomethane-kinetics and mechanism. J Am Chem Soc. 1980;102:7303–7308. [Google Scholar]
- 8.Saleem A, Brown GD, Brady F, Aboagye EO, Osman S, Luthra SK, et al. Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 2003;63:2409–2415. [PubMed] [Google Scholar]
- 9.Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry. 1994;33:9045–9051. doi: 10.1021/bi00197a003. [DOI] [PubMed] [Google Scholar]
- 10.Svilar D, Dyavaiah M, Brown AR, Tang JB, Li J, McDonald PR, et al. Alkylation Sensitivity Screens Reveal a Conserved Cross-species Functionome. Mol Cancer Res. 2012;10:1580–1596. doi: 10.1158/1541-7786.MCR-12-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pegg AE. Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 12.Park CK, Kim JE, Kim JY, Song SW, Kim JW, Choi SH, et al. The Changes in MGMT Promoter Methylation Status in Initial and Recurrent Glioblastomas. Transl Oncol. 2012;5:393–397. doi: 10.1593/tlo.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals (Basel) 2013;6:1475–1506. doi: 10.3390/ph6121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pletsas D, Wheelhouse RT, Pletsa V, Nicolaou A, Jenkins TC, Bibby MC, et al. Polar, functionalized guanine-O6 derivatives resistant to repair by O6-alkylguanine-DNA alkyltransferase: implications for the design of DNA-modifying drugs. Eur J Med Chem. 2006;41:330–339. doi: 10.1016/j.ejmech.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Stevens MF, Hummersone M, Madhusudan S, Laughton CA, Bradshaw TD. Certain imidazotetrazines escape O6-methylguanine-DNA methyltransferase and mismatch repair. Oncology. 2011;80:195–207. doi: 10.1159/000327837. [DOI] [PubMed] [Google Scholar]
- 16.Arrowsmith J, Jennings SA, Langnel DAF, Wheelhouse RT, Stevens MFG. Antitumour imidazotetrazines. Part 39. Synthesis of bis(imidazotetrazine)s with saturated spacer groups. J Chem Soc Perkin Trans. 2000;1:4432–4438. [Google Scholar]
- 17.Garelnabi EAE, Pletsas D, Li L, Kiakos K, Karodia N, Hartley JA, et al. Strategy for imidazotetrazine prodrugs with anticancer activity independent of MGMT and MMR. ACS Med Chem Lett. 2012;3:965–968. doi: 10.1021/ml300132t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pletsas D, Garelnabi EA, Li L, Phillips RM, Wheelhouse RT. Synthesis and Quantitative Structure-Activity Relationship of Imidazotetrazine Prodrugs with Activity Independent of O6-Methylguanine-DNA-methyltransferase, DNA Mismatch Repair, and p53. J Med Chem. 2013;56:7120–7132. doi: 10.1021/jm401121k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-oncol. 2005;7:164–176. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, et al. Inhibition of Histone Deacetylation Potentiates the Evolution of Acquired Temozolomide Resistance Linked to MGMT Upregulation in Glioblastoma Xenografts. Clin Cancer Res. 2012;18:4070–4079. doi: 10.1158/1078-0432.CCR-12-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert CA, Daou MC, Moser RP, Ross AH. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010;70:6870–6879. doi: 10.1158/0008-5472.CAN-10-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sen A, Kallos MS, Behie LA. New tissue dissociation protocol for scaled-up production of neural stem cells in suspension bioreactors. Tissue Eng. 2004;10:904–913. doi: 10.1089/1076327041348554. [DOI] [PubMed] [Google Scholar]
- 23.Phillips RM, Ward TH. Influence of extracellular pH on the cytotoxicity and DNA damage of a series of indolequinone compounds. Anticancer research. 2001;21:1795–1801. [PubMed] [Google Scholar]
- 24.Gynther M, Laine K, Ropponen J, Leppanen J, Mannila A, Nevalainen T, et al. Large neutral amino acid transporter enables brain drug delivery via prodrugs. J Med Chem. 2008;51:932–936. doi: 10.1021/jm701175d. [DOI] [PubMed] [Google Scholar]
- 25.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins AR, Oscoz AA, Brunborg G, Gaivao I, Giovannelli L, Kruszewski M, et al. The comet assay: topical issues. Mutagenesis. 2008;23:143–151. doi: 10.1093/mutage/gem051. [DOI] [PubMed] [Google Scholar]
- 27.White DE, Negorev D, Peng H, Ivanov AV, Maul GG, Rauscher FJ., 3rd KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer research. 2006;66:11594–11599. doi: 10.1158/0008-5472.CAN-06-4138. [DOI] [PubMed] [Google Scholar]
- 28.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreso A, O'Brien CA, van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higgins DM, Wang R, Milligan B, Schroeder M, Carlson B, Pokorny J, et al. Brain tumor stem cell multipotency correlates with nanog expression and extent of passaging in human glioblastoma xenografts. Oncotarget. 2013;4:792–801. doi: 10.18632/oncotarget.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitange GJ, Carlson BL, Mladek AC, Decker PA, Schroeder MA, Wu W, et al. Evaluation of MGMT promoter methylation status and correlation with temozolomide response in orthotopic glioblastoma xenograft model. J Neurooncol. 2009;92:23–31. doi: 10.1007/s11060-008-9737-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2:107–129. doi: 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen CC, Taniguchi T, D'Andrea A. The Fanconi anemia (FA) pathway confers glioma resistance to DNA alkylating agents. J Mol Med (Berl) 2007;85:497–509. doi: 10.1007/s00109-006-0153-2. [DOI] [PubMed] [Google Scholar]
- 35.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.