

Abstract
DNAzymes, sequences of DNA with catalytic activity, have been demonstrated as a potential platform for sensing a wide range of metal ions. Despite significant promise, cellular sensing using DNAzymes has been difficult, mainly due to the ‘always-on’ nature of first generation DNAzyme sensors. To overcome this limitation, we demonstrate herein the design and synthesis of a photoactivatable or photocaged DNAzyme, and its application in sensing Zn(II) in living cells. In this design the adenosine ribonucleotide at the scissile position of the 8–17 DNAzyme is replaced by 2′-O-nitrobenzyl adenosine, rendering the DNAzyme inactive and thus allowing its delivery into cells intact, protected from non-specific degradation within cells. Irradiation at 365 nm restores DNAzyme activity, allowing for temporal control over the DNAzyme’s sensing activity for metal ions. The same strategy has also been applied towards the GR-5 DNAzyme for detection of Pb(II), demonstrating its broad generalizability.
Keywords: fluorescent probes, DNAzymes, biosensors, photolabile protecting groups, cellular sensing
Metal ions have been involved in many critical functions in biology, providing structural stability and catalytic activity to proteins, and alone as signaling molecules.[1] The wide variety of functions carried out in whole or in part by metal ions has led to significant interest in developing sensors to probe the location and distribution of these metal ions in cells.[2] Toward this goal, a number of fluorescent sensors, most notably those based on small organic molecules or proteins, have been developed, allowing for detection of changes in cellular metal ion concentration.[2b, 3] While these reports have demonstrated that cellular metal sensors can enrich our knowledge on the biological functions of metal ions, the current scope of metal ion sensors has been limited to relatively few metal ions, most extensively Ca2+, Zn2+, and Cu2+/+. Further advances in understanding the role of biological metal ions will require the development of new sensors for many more metal ions. However, most methods rely on rational design, and success in designing one metal sensor may not be readily translated into success for another metal sensor, because the difference between metal ions can be very subtle and designing sensors with high selectivity and little or no interference is very difficult. A complementary approach to rational design is combinatorial selection, which does not rely on prior knowledge of metal-binding, and in which sensor selectivity and affinity can be improved by adjusting the stringency of selection conditions.[4]
DNAzymes are a class of functional DNA that offers great promise in improving the process of metal ion sensor development. Discovered in 1994, DNAzymes are sequences of DNA that have catalytic activity, such as RNA hydrolysis, porphyrin metallation or Diels-Alder activity.[4c, 5] Frequently these DNAzymes make use of a metal ion cofactor in order to carry out their functions. Recognizing this important connection, we and other labs have taken advantage of this property to develop corresponding metal ion sensors.[4a, 4b, 6] Unlike other types of sensors, DNAzymes can be obtained through a combinatorial selection process called in vitro selection, from a large DNA library of up to 1015 different sequences.[5b] This selection process does not require immobilization of the metal ion on a solid support and instead relies on DNAzyme cleavage as a measure of metal binding and activity. The selection process allows DNAzymes with specific binding affinity, selectivity, and sensitivity to be obtained.[4c, 4d, 5–6, 6g–h] Since the DNAzyme can be readily modified by different signaling agents, such as fluorophores and gold nanoparticles, we can transform metal-dependent catalytic activity into sensor readouts.[7] In this way we and other labs have developed DNAzyme-based metal ion sensors for a wide range of metal ions, including Zn2+, Pb2+, Cu2+, UO22+, Mg2+ and Hg2+.[4b, 4c, 5–6, 6f–h]
Even though the use of DNAzymes for metal ion sensing has been established for some time, the majority of previously published work has been limited to sensing metal ions in environmental samples such as water and soil, with very few demonstrating detection inside cells.[4b, 6a] In 2013, we have reported that a gold nanoparticle-DNAzyme conjugate is capable of being taken up by cells, enabling the detection of endosomal uranyl.[8] Recently, a dendritic polymer has also been used to deliver DNAzymes for detection of cellular lead ions.[9] While these results are encouraging, a significant unaddressed issue is that the DNAzyme can be active in the presence of its metal cofactor during the cellular delivery and uptake process. Depending on the presence of metal cofactors inside and outside of the cells, the DNAzymes may not be able to reach their cellular destination before they are cleaved. Furthermore, most DNAzymes require an internal RNA base at the cleavage site of the substrate strand. Although chimeric DNA/RNA substrates are relatively stable compared to all-RNA substrates, the RNA site makes the sensor vulnerable to endogenous nuclease activity. Both metal-catalyzed cleavage and nuclease-induced degradation result in loss of dynamic range, negatively affecting the signal-to-background ratio and sensor performance. It is thus necessary to develop a method that allows both the controlled activation of the DNAzyme as well as a method for reversibly protecting the RNA cleavage site from enzymatic degradation.
To overcome this major limitation, we present the design and synthesis of a DNAzyme whose activity is controlled by a photolabile group (called photocaged DNAzyme), and its application for imaging metal ions in cells. While the addition of photolabile or photoswitchable groups has been used to control the activity of DNAzymes previously,[10] no previous report has been able to control both the activity of the DNAzyme and the stability and cleavage of the substrate strand. As a result, despite photolabile group addition having been widely used as a chemical biological tool in the development of photoactivatable proteins,[11] small molecules,[2d, 11c, 11d, 12] and oligonucleotides,[11c, 11d, 13] no such strategy has yet been reported to enable the use of DNAzymes for sensing metal ions in living cells. In addition to showing the intracellular activation of a DNAzyme metal ion sensor, we also demonstrate that this strategy is applicable towards all members of the broader class of RNA-cleaving DNAzymes, making this work a significant step towards achieving the use of DNAzymes as a generalizable platform for cellular metal ion detection and imaging.
The sensor design and photocaging strategy is shown in Figure 1a, using the 8–17 DNAzyme as an example. The DNAzyme contains an enzyme strand and a substrate strand, which are all DNA except for a single adenosine ribonucleotide (rA) in the substrate strand, at the cleavage site. The substrate strand is also functionalized with a 5′-fluorophore (F) such as fluorescein, and a 3′-quencher (Q) such as Black Hole Quencher-1 (BHQ-1), with another quencher (Dabcyl) on the 5′-of the enzyme strand. At ambient conditions, the enzyme and substrate strands can hybridize, as the pair has a melting temperature of 57.5°C. This places the quenchers in close proximity to the fluorophore, resulting in low background fluorescence signal prior to sensing.[6g] In the presence of its metal ion cofactor, the substrate strand is cleaved at the scissile bond, resulting in two fragments with reduced melting temperature (14.7°C) to the enzyme strand. This allows the fluorophore to be separated from the quenchers, giving a dramatic increase in fluorescent signal.
Figure 1.

(a) Design of catalytic beacon DNAzyme sensor, and illustration of caging strategy for DNAzyme protection and activation. F = Fluorophore (fluorescein), Q = Quencher (Dabcyl, BHQ-1), PG = protecting group (o-nitrobenzyl) (b) Fluorescence response of caged (black) and unmodified (red) 8–17 DNAzyme in response to 50 μM Zn2+. (c) Fluorescence response of caged DNAzyme uncaged with increasing doses of 365 nm light, in response to 50 μM Zn2+. (d) HeLa cells transfected with caged active or caged inactive 8–17 DNAzyme (0.5 μM) for 11 hr, then irradiated for 30 m at 365 nm, followed by addition of 50 μM Zn citrate. Scale bar = 20 μm. (e) Normalized fluorescence intensity of cells in part (d).
To prevent the substrate strand from being cleaved, a 2′-O-nitrobenzyl adenosine (called caged or photocaged adenosine) is used in place of the normal adenosine at the cleavage site. Since the 2′-OH of the adenosine plays a critical role in the DNAzyme activity, adding the 2′-O-nitrobenzyl protection group (PG) to this location can render the DNAzyme inactive. In this way, the DNAzymes can be allowed to enter into cells and distribute in different compartments without being cleaved prematurely. In order to restore the active DNAzyme for sensing purposes, we can make use of the photolabile characteristic of the 2′-O-nitrobenzyl group. Under 365 nm light, the PG will be removed from the caged adenosine, uncovering the native adenosine with the 2′-OH and thus restoring activity of the DNAzyme. As with the unmodified DNAzyme, the reactivated (uncaged) DNAzyme will then cleave the substrate strand leading to a fluorescent signal. Because the DNAzyme is highly specific to the metal ion used, this photoactivation strategy allows detection of metal ions in cells. Since deprotection is performed with light, it should be orthogonal to cellular delivery and cellular function, and thus allow temporal control over the uncaging and activation of the DNAzyme sensor.
The performance of the photocaged DNAzyme was first assessed in a buffer under physiological conditions. The substrate strand containing either caged adenosine or native adenosine was annealed to the enzyme strand. The DNAzyme reaction was initiated with the addition of Zn2+. In the absence of 365 nm light, the fluorescent signal increased rapidly only in the case of the unmodified substrate containing the native adenosine (Figure 1b), similar to those observed previously. In contrast, when the substrate strand containing the caged adenosine was used, no increase in fluorescent signal was observed, indicating complete inhibition of the DNAzyme activity.
To test whether the protective effect of the 2′-O-nitrobenzyl PG could be reversed with light activation, we used both a portable hand lamp (Spectroline, 365 nm) and a Blak-Ray B100 365 nm lamp to irradiate samples of the caged 8–17 DNAzyme for different amount of time. While no fluorescent signal increase was observed in the absence of light, the fluorescent signal showed an increase with time after addition of metal ions (Figure 1c). Longer exposure to 365 nm light led to greater increase in fluorescent signal. (Figure 1b). These results strongly suggest that the DNAzyme activity can be restored after light activation: the longer the exposure to light, the more active DNAzyme was uncovered and thus more fluorescent signal increase could be observed. The same conclusion can be drawn from the results of polyacrylamide gel electrophoresis (PAGE, Figure S4 in SI)
For biological applications, the stability of the 2′-protecting group is paramount. To test the stability of caged DNAzymes, substrates containing either caged or native adenosine were annealed to the DNAzyme strand and incubated in either buffer containing 50 μM Zn2+ or 80% human serum for extended periods of time. PAGE analysis revealed that the substrate containing the native adenosine was cleaved in <1 hour under both conditions, but little cleavage was observed even at times up to 7 days in the presence of 50 μM Zn2+, or 2 days in the presence of human serum. (Figures S5, S6 in SI)
Having demonstrated that the caged DNAzymes are stable, we then proceeded to use the caged DNAzymes for sensing metal ions within cultured HeLa cells, using the Zn2+--responsive 8–17 DNAzyme as our model system. As zinc is present in both cells and growth media, the cell-delivery process itself poses a major challenge, because the presence of endogenous Zn2+ can promote DNAzyme-based cleavage of the substrate strand before the DNAzyme can be delivered to the interior of the cells, if no protection strategy is used. The 8–17 DNAzyme containing the caged adenosine was delivered to HeLa cells via Lipofectamine 2000 following a modification of manufacturer’s protocols. Briefly, enzyme and caged substrate strands were heated to 80°C and allowed to anneal. Lipofectamine 2000 (5 μL) and annealed DNAzymes (1 nmol) were incubated separately in Opti-MEM media (Invitrogen) for 5 minutes, then combined and allowed to incubate for an additional 25 minutes. The prepared DNAzyme-Lipofectamine mixture was added to HeLa cells and allowed to incubate for several hours. Confocal microscopy images of the DNAzyme (Figure 1d) showed that the fluorescent DNAzyme was delivered inside the cells, in a diffuse staining pattern mainly localized in the nucleus (determined by colocalization with Hoechst stain). This distribution pattern is in agreement with previous reports demonstrating nuclear accumulation of DNA delivered via cationic liposomes (Lipofectamine PLUS).[14] To localize the DNAzyme probe into other organelles, alternative delivery methods can be used, such as the use of gold nanoparticles for lysosomal distribution.[8]
Upon irradiation with a 365 nm lamp, followed by Zn2+-citrate addition, an increase in fluorescence intensity was observed with time (Figure 1e). To confirm that the observed increase in fluorescence was caused by DNAzyme activity and not nonspecific cleavage by other cellular components, we used an enzyme sequence in which two critical bases in the catalytic loop have been substituted (Supplemental Table S1).[15] Using this inactive DNAzyme, no fluorescence signal increase was observed in buffer (Figure S7), confirming that this inactive 8–17 enzyme was unable to cleave the native adenosine-containing substrate strand, even in the presence of Zn2+. Furthermore, the inactive DNAzyme showed no significant increase in fluorescence over 45 minutes (Figure 1d, e). Together, these results strongly indicate that the caged DNAzyme can be used to detect and image metal ions in living cells.
The 8–17 DNAzyme described in this manuscript has previously been shown to display a linear response to [Zn2+] in the range of 0–500 μM in buffer.[6f] Similarly, the GR-5 DNAzyme described in this manuscript has been reported to show a linear range to [Pb2+] in the range of 0–1 μM.[16] Like many other metal ion sensors reported in the literature,[3d] the current intensity-based sensor design does not allow accurate determination of metal ion concentration within cells, making it difficult to show linearity of the sensor response to intracellular metal ions. To overcome this limitation, we are currently investigating the design of new ratiometric sensors that may allow for better quantification within cells.
After demonstrating the use of 8–17 DNAzyme for cellular sensing and imaging of Zn2+, we investigated whether such a method could be applied generally to other DNAzymes for detection of their respective target metal ions as well. Since the first discovery of DNAzymes in 1994 using in vitro selection, many DNAzymes have been obtained using similar selection methods. As a result, the majority of currently identified DNAzymes share a similar secondary structure consisting of two double stranded DNA binding arms flanking the cleavage site. More interestingly, the sequence identity of the two binding arms are not conserved, as long as they can form Watson-Crick base pairs with the chosen substrate. The metal ion selectivity of DNAzymes comes from the sequence identity of the loop in the enzyme strand. As a result, the exact substrate sequence that can be recognized by a DNAzyme can be arbitrarily chosen. This feature also allows multiple DNAzymes to recognize the same substrate sequence. An attractive advantage of our photocaging strategy is that we can use the same caged substrate strand to achieve sensing of different metal ions by using different enzyme strands. To demonstrate this advantage, we synthesized a DNAzyme sequence that can hybridize to the same caged substrate strand as the 8–17 DNAzyme, but contains the catalytic loop of the GR-5 DNAzyme, (the first DNAzyme, obtained in 1994), which has significant activity in the presence of Pb2+ but not with any other metal ions.[16] As shown in Figure 2, this caged GR-5 DNAzyme showed little cleavage activity in the absence of light activation, but increasing dose-dependent increase of DNAzyme activity upon light activation.
Figure 2.
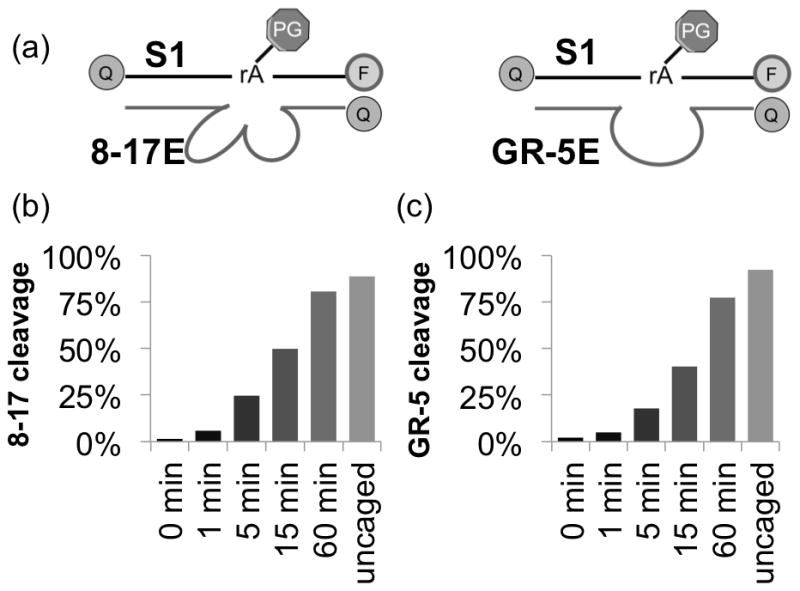
Generalizability of caging strategy. (a) Schematic showing hybridization of different DNAzymes to same substrate strand S1. Caged and uncaged (b) 8–17 and (c) GR-5 activity in presence of Zn2+ (8–17, 500 μM) or Pb2+ (GR-5, 2 μM).
In conclusion, we have demonstrated a general and effective strategy for protecting the substrate of a DNAzyme sensor, enabling its delivery into cells without being cleaved during the process, and allowing it to be used as a cellular metal ion sensor upon photoactivation. This strategy provides enhanced stability (up to multiple days in serum) and allows temporal control over DNAzyme activity. As the only modification to the original DNAzyme is on the substrate strand, we can replace the enzyme strand without needing to re-optimize for each new substrate sequence, greatly improving the generalizability of this protection strategy. Furthermore, the enhanced stability of the caged DNAzyme does not require the use of a specific nanomaterial vehicle as a delivery agent, further demonstrating the wider accessibility of this protection approach. This work will greatly expand the applicability of DNAzymes as versatile biosensors and will greatly improve the field of metal ion sensing.
Supplementary Material
Acknowledgments
This work is supported by the US National Institute of Health (ES016865 to YL) and by the Office of Science (BER), the U.S. Department of Energy (DE-FG02-08ER64568). K. H. was supported by the NIH Molecular Biophysics Training Grant (T32GM008276) and by the Lester E. and Kathleen A. Coleman fellowship at the University of Illinois at Urbana-Champaign. P.W. was supported by NSF Grant 0965918 IGERT: Training the Next Generation of Researchers in Cellular and Molecular Mechanics and BioNanotechnology.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Kevin Hwang, Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA.
Peiwen Wu, Department of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA.
Taejin Kim, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA.
Lei Lei, Department of Bioengineering and Institute of Engineering in Medicine, University of California, San Diego, La Jolla, California, 92093, USA.
Shiliang Tian, Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA.
Prof. Yingxiao Wang, Department of Bioengineering and Institute of Engineering in Medicine, University of California, San Diego, La Jolla, California, 92093, USA
Prof. Yi Lu, Email: yi-lu@illinois.edu, Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA. Department of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, 61801, USA
References
- 1.a) Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; 1994. [Google Scholar]; b) Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T. J Biol Inorg Chem. 2011;16:1123–1134. doi: 10.1007/s00775-011-0797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Clapham DE. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]; d) Outten CE, O’Halloran TV. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]; e) Wegner SV, Sun F, Hernandez N, He C. Chem Commun. 2011;47:2571–2573. doi: 10.1039/c0cc04292g. [DOI] [PubMed] [Google Scholar]
- 2.a) Jiang P, Guo Z. Coord Chem Rev. 2004;248:205–229. [Google Scholar]; b) Kikuchi K. Adv Biochem Eng/Biotechnol. 2010;119:63–78. doi: 10.1007/10_2008_42. [DOI] [PubMed] [Google Scholar]; c) Raimunda D, Khare T, Giometti C, Vogt S, Arguello JM, Finney L. Metallomics. 2012;4:921–927. doi: 10.1039/c2mt20095c. [DOI] [PubMed] [Google Scholar]; d) Mbatia HW, Burdette SC. Biochemistry. 2012;51:7212–7224. doi: 10.1021/bi3001769. [DOI] [PubMed] [Google Scholar]; e) Deibler K, Basu P. Eur J Inorg Chem. 2013;2013:1086–1096. doi: 10.1002/ejic.201200997. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ. Chem Rev. 2009;109:4780–4827. doi: 10.1021/cr900223a. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Kikuchi K, Komatsu K, Nagano T. Curr Opin Chem Biol. 2004;8:182–191. doi: 10.1016/j.cbpa.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 3.a) Domaille DW, Que EL, Chang CJ. Nat Chem Biol. 2008;4:168–175. doi: 10.1038/nchembio.69. [DOI] [PubMed] [Google Scholar]; b) Que EL, Domaille DW, Chang CJ. Chem Rev. 2008;108:1517–1549. doi: 10.1021/cr078203u. [DOI] [PubMed] [Google Scholar]; c) Tomat E, Lippard SJ. Curr Opin Chem Biol. 2010;14:225–230. doi: 10.1016/j.cbpa.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pluth MD, Tomat E, Lippard SJ. Annu Rev Biochem. 2011;80:333–355. doi: 10.1146/annurev-biochem-061009-091643. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dean KM, Qin Y, Palmer AE. Biochim Biophys Acta. 2012;1823:1406–1415. doi: 10.1016/j.bbamcr.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liu J, Karpus J, Wegner SV, Chen PR, He C. J Am Chem Soc. 2013;135:3144–3149. doi: 10.1021/ja3106779. [DOI] [PubMed] [Google Scholar]; g) Sareen D, Kaur P, Singh K. Coord Chem Rev. 2014;265:125–154. [Google Scholar]; h) Fahrni CJ. Curr Opin Chem Biol. 2013;17:656–662. doi: 10.1016/j.cbpa.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Komatsu K, Kikuchi K, Kojima H, Urano Y, Nagano T. J Am Chem Soc. 2005;127:10197–10204. doi: 10.1021/ja050301e. [DOI] [PubMed] [Google Scholar]
- 4.a) Liu J, Cao Z, Lu Y. Chem Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiang Y, Lu Y. Inorg Chem. 2013;53:1925–1942. doi: 10.1021/ic4019103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Joyce GF. Annu Rev Biochem. 2004;73:791–836. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]; d) Nelson K, Bruesehoff P, Lu Y. J Mol Evol. 2005;61:216–225. doi: 10.1007/s00239-004-0374-3. [DOI] [PubMed] [Google Scholar]
- 5.a) Breaker RR, Joyce GF. Chem Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]; b) Sai Lau P, Li Y. Curr Org Chem. 2011;15:557–575. [Google Scholar]; c) Li Y, Breaker RR. Curr Opin Struct Biol. 1999;9:315–323. doi: 10.1016/S0959-440X(99)80042-6. [DOI] [PubMed] [Google Scholar]; d) Li Y, Sen D. Nat Struct Biol. 1996;3:743–747. doi: 10.1038/nsb0996-743. [DOI] [PubMed] [Google Scholar]
- 6.a) Zhang XB, Kong RM, Lu Y. Annu Rev Anal Chem. 2011;4:105–128. doi: 10.1146/annurev.anchem.111808.073617. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li T, Dong S, Wang E. Anal Chem. 2009;81:2144–2149. doi: 10.1021/ac900188y. [DOI] [PubMed] [Google Scholar]; c) Wei H, Li B, Li J, Dong S, Wang E. Nanotechnology. 2008;19:095501. doi: 10.1088/0957-4484/19/9/095501. [DOI] [PubMed] [Google Scholar]; d) Liu J, Lu Y. Methods Mol Biol. 2006;335:275–288. doi: 10.1385/1-59745-069-3:275. [DOI] [PubMed] [Google Scholar]; e) Zhang XB, Wang Z, Xing H, Xiang Y, Lu Y. Anal Chem. 2010;82:5005–5011. doi: 10.1021/ac1009047. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Li J, Zheng W, Kwon AH, Lu Y. Nucleic Acids Res. 2000;28:481–488. doi: 10.1093/nar/28.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Liu J, Lu Y. Anal Chem. 2003;75:6666–6672. doi: 10.1021/ac034924r. [DOI] [PubMed] [Google Scholar]; h) Hollenstein M, Hipolito C, Lam C, Dietrich D, Perrin DM. Angew Chem Int Ed. 2008;47:4346–4350. doi: 10.1002/anie.200800960. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:4418–4422. [Google Scholar]
- 7.a) Liu J, Lu Y. J Am Chem Soc. 2004;126:12298–12305. doi: 10.1021/ja046628h. [DOI] [PubMed] [Google Scholar]; b) Liu J, Lu Y. J Am Chem Soc. 2005;127:12677–12683. doi: 10.1021/ja053567u. [DOI] [PubMed] [Google Scholar]; c) Xiang Y, Lu Y. Nat Chem. 2011;3:697–703. doi: 10.1038/nchem.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu P, Hwang K, Lan T, Lu Y. J Am Chem Soc. 2013;135:5254–5257. doi: 10.1021/ja400150v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Huang H, Xu N, Yin Q. J Mater Chem B. 2014;2:4935–4942. doi: 10.1039/c4tb00680a. [DOI] [PubMed] [Google Scholar]
- 10.a) Chaulk SG, MacMillan AM. Nucleic Acids Res. 1998;26:3173–3178. doi: 10.1093/nar/26.13.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Y, Sen D. J Mol Biol. 2004;341:887–892. doi: 10.1016/j.jmb.2004.06.060. [DOI] [PubMed] [Google Scholar]; c) Lusic H, Young DD, Lively MO, Deiters A. Org Lett. 2007;9:1903–1906. doi: 10.1021/ol070455u. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Richards JL, Seward GK, Wang YH, Dmochowski IJ. Chembiochem. 2010;11:320–324. doi: 10.1002/cbic.200900702. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ting R, Lermer L, Perrin DM. J Am Chem Soc. 2004;126:12720–12721. doi: 10.1021/ja046964y. [DOI] [PubMed] [Google Scholar]; f) Young DD, Lively MO, Deiters A. J Am Chem Soc. 2010;132:6183–6193. doi: 10.1021/ja100710j. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Keiper S, Vyle JS. Angew Chem Int Ed. 2006;45:3306–3309. doi: 10.1002/anie.200600164. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:3384–3387. [Google Scholar]
- 11.a) Wang S, Moffitt JR, Dempsey GT, Xie XS, Zhuang X. Proc Natl Acad Sci U S A. 2014;111:8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Nat Rev Mol Cell Biol. 2005;6:885–891. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]; c) Pelliccioli AP, Wirz J. Photochem Photobiol Sci. 2002;1:441–458. doi: 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]; d) Shao Q, Xing B. Chem Soc Rev. 2010;39:2835–2846. doi: 10.1039/b915574k. [DOI] [PubMed] [Google Scholar]; e) Baker AS, Deiters A. ACS Chem Biol. 2014;9:1398–1407. doi: 10.1021/cb500176x. [DOI] [PubMed] [Google Scholar]; f) Arbely E, Torres-Kolbus J, Deiters A, Chin JW. J Am Chem Soc. 2012;134:11912–11915. doi: 10.1021/ja3046958. [DOI] [PubMed] [Google Scholar]; g) Mizukami S, Hosoda M, Satake T, Okada S, Hori Y, Furuta T, Kikuchi K. J Am Chem Soc. 2010;132:9524–9525. doi: 10.1021/ja102167m. [DOI] [PubMed] [Google Scholar]
- 12.a) Yang Y, Shao Q, Deng R, Wang C, Teng X, Cheng K, Cheng Z, Huang L, Liu Z, Liu X, Xing B. Angew Chem Int Ed. 2012;51:3125–3129. doi: 10.1002/anie.201107919. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2012;124:3179–3183. [Google Scholar]
- 13.a) Graifer D, Karpova G. RSC Adv. 2013;3:2858–2872. [Google Scholar]; b) Monroe WT, McQuain MM, Chang MS, Alexander JS, Haselton FR. J Biol Chem. 1999;274:20895–20900. doi: 10.1074/jbc.274.30.20895. [DOI] [PubMed] [Google Scholar]; c) Yamaguchi S, Chen Y, Nakajima S, Furuta T, Nagamune T. Chem Commun. 2010;46:2244–2246. doi: 10.1039/b922502a. [DOI] [PubMed] [Google Scholar]; d) Walsh S, Gardner L, Deiters A, Williams GJ. Chembiochem. 2014;15:1346–1351. doi: 10.1002/cbic.201400024. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Connelly CM, Deiters A. Methods Mol Biol. 2014;1165:99–114. doi: 10.1007/978-1-4939-0856-1_9. [DOI] [PubMed] [Google Scholar]; f) Yamazoe S, Liu Q, McQuade LE, Deiters A, Chen JK. Angew Chem Int Ed. 2014:10114–10118. doi: 10.1002/anie.201405355. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2014:10278–10282. [Google Scholar]
- 14.Akita H, Ito R, Khalil IA, Futaki S, Harashima H. Mol Ther. 2004;9:443–451. doi: 10.1016/j.ymthe.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 15.Schlosser K, Li Y. Chembiochem. 2010;11:866–879. doi: 10.1002/cbic.200900786. [DOI] [PubMed] [Google Scholar]
- 16.Lan T, Furuya K, Lu Y. Chem Commun. 2010;46:3896–3898. doi: 10.1039/b926910j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.