

Summary
Protein design provides an ultimate test of our knowledge about proteins and allows the creation of novel enzymes for biotechnological applications. While progress has been made in designing proteins that mimic native proteins structurally1–3, it is more difficult to design functional proteins4–8. In comparison to recent successes in designing non-metalloproteins4,6,7,9,10, it is even more challenging to rationally design metalloproteins that reproduce both the structure and function of native metalloenzymes5,8,11–20, since protein metal binding sites are much more varied than non-metal containing sites, in terms of different metal ion oxidation states, preferred geometry and metal ion ligand donor sets. Because of their variability, it has been difficult to predict metal binding site properties in silico, as many of the parameters for metal binding sites, such as force fields are ill-defined. Therefore, the successful design of a structural and functional metalloprotein will greatly advance the field of protein design and our understanding of enzymes. Here, we report a successful, rational design of a structural and functional model of a metalloprotein, nitric oxide reductase (NOR), by introducing three histidines and one glutamate, predicted as ligands in the active site of NOR, into the distal pocket of myoglobin. A crystal structure of the designed protein confirms that the minimized computer model contains a heme/non-heme FeB center that is remarkably similar to that in the crystal structure. This designed protein also exhibits NOR activity. This is the first designed protein that models both the structure and function of NOR, offering insight that the active site glutamate is required for both iron binding and activity. These results show that structural and functional metalloproteins can be rationally designed in silico.
NOR is a metalloenzyme in the denitrification pathway of anaerobic bacteria that is responsible for the two electron reduction of NO to N2O21. It is a key enzyme in the nitrogen cycle that is critical for all life. Furthermore, well-studied denitrification enzymes may provide structural and spectroscopic models for mammalian enzymes that produce and utilize NO in signal transduction pathways. Progress in understanding NOR has been hampered by difficulties in obtaining enzyme in high yield and the lack of a three-dimensional structure. Peptide sequence alignments and structural threading have suggested that NORs are structurally homologous to subunit I of heme-copper oxidases (HCOs)22. All six His ligands to the active site of HCO (two to the low spin heme, one to the high spin heme, and three to CuB) are conserved in NOR at the same predicted positions in HCO. The major structural difference is the replacement of a copper binding site (CuB) with a non-heme iron binding site (FeB) in NOR21,23. Additionally, several conserved glutamates (not found in HCOs), have been identified near the catalytic heme/non-heme FeB site of NOR, and are predicted to play a role in iron binding and/or catalysis24,25. Synthetic models of NOR have been reported21,26,27. Despite these progresses, computational design and 3D structural confirmation of structural and functional models remain elusive, and it is often difficult to address long-range non-covalent interactions important for enzymatic functions using these models.
We chose to use sperm whale myoglobin (swMb) as a scaffold protein in which to engineer an NOR active site because it is much easier to prepare and crystallize than both native proteins and synthetic models of NOR. Myoglobin and its mutants are also more amenable to biophysical characterization due to the presence of a single metal center, free from interference by other chromophores. Although swMb contains a heme center, it lacks the FeB center and NO reduction activity of NOR.
To design an FeB center in swMb, we examined its distal pocket for locations to introduce the putative non-heme metal binding site, consisting of three histidines and one glutamate. The histidine residues were introduced first14, as crystal structures of HCOs containing three conserved histidines are readily available (e.g., CcO, cytochrome c oxidase). Based on overlays of the minimized structure of swMb and CcO, the distal His64 in swMb was chosen as one of the histidines, while Leu29 and Phe43 were mutated to histidines, to produce a non-heme metal binding site. The V68E mutation to swMb was chosen based on its proximity and angle to the heme and the three His. The minimized computer model of the resultant triple mutant (swMb L29H, F43H, V68E), named FeBMb, is shown in Fig. 1A. The FeB site (modeled as Zn(II)) is within bonding distance to Nε of all three His residues (2.13 Å to H29, 2.20 Å to H43, and 2.09 Å to H64) and both O atoms of E68 (2.11 Å and 1.96 Å), indicating that the proposed mutations would support iron binding in myoglobin, forming an FeB site. A comparison of the computer model overlaid with the crystal structure of native CcO (see Fig. S1A) suggests structural similarity between the two centers.
Figure 1.
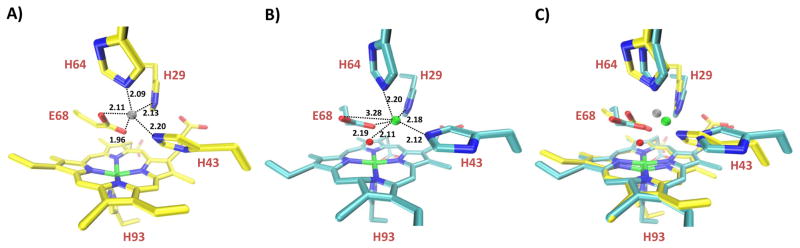
Crystal structure of rationally designed FeBMb overlays closely with minimized computer model. A) Minimized computer model of FeBMb with Zn(II) in the FeB site. B) Crystal structure of Fe(II)-FeBMb collected at Fe-edge absorption (1.7309 Å) at the Brookhaven National Synchrotron Light Source X12C beamline (Upton, NY) with 1.72 Å resolution. The Fe•••Fe distance is 4.82 Å, while the Fe-O-Fe angle is 115° (OE1 atom of E68). C) Overlay of FeBMb model (yellow) with Fe(II)-FeBMb crystal structure (cyan). In general, Fe(II) of the FeB site is represented by a green sphere; Zn(II) (grey sphere) was used to model the FeB site. A water molecule in the heme pocket is represented by a red sphere.
To test designed FeBMb for iron binding, Fe2+ was titrated into deoxy (reduced heme iron) FeBMb that contains heme, but not any metal ions in the FeB site. The Soret band of deoxy FeBMb shifted slightly from 433 nm to 434 nm with Fe2+ addition. Concurrently, the peak at 557 nm was split into two shallow bands at 550 nm and 572 nm (Fig. 2Aa). This observation provided evidence that FeBMb is capable of incorporating iron into its designed metal binding site, an important step toward modeling the FeB site of NOR. In contrast, titration of Fe2+ into either wtMb (Fig. 2Ab) or a Mb derivative with the three His but no Glu (called FeBMb(-Glu), Fig. 2Ac) resulted in no changes to the UV-vis spectra, indicating that both the histidines and particularly the glutamate, are required for Fe2+ binding.
Figure 2.

Designed FeBMb has NOR activity in the presence of Fe2+ and NO. Control proteins show no evidence of NO activity under identical conditions. A) UV-vis spectra of 10 μM of deoxy FeBMb (a), deoxy wtMb (b), and deoxy FeBMb(-Glu) (c) in the absence (black spectra) or presence (red spectra) of 2 eq Fe2+. B) UV-vis spectra of the NO reaction of 10 μM of deoxy FeBMb (a), deoxy wtMb (b), and deoxy FeBMb(-Glu) (c) in the absence (black spectra) or presence (red spectra) of 2 eq Fe2+ after 20 min of incubation with ~17 eq NO. The deoxy protein was formed by reaction of met protein with excess dithionite in a glove box. Gel filtration purification was used to remove excess dithionite to preclude its reaction with NO.
Further evidence of Fe2+ binding to the designed FeB site comes from a high resolution crystal structure (1.72 Å) of Fe(II)-FeBMb (Fig. 1B, S1, S5, Table S1). The non-heme iron in the FeB site is five coordinate, with bonds to all three histidines (2.18 Å to H29, 2.12 Å to H43, and 2.20 Å to H64), one O atom of E68 (2.19 Å) and one water molecule (2.11 Å). A weaker interaction between the non-heme iron and the other O atom of E68 at 3.28 Å is also present. The crystal structure presented here is consistent with the proposal that a glutamate in the active site of NOR helps stabilize iron binding to the FeB site. In addition to structural roles, the glutamate, histidines and water in the active site may also provide the two protons required for NO reduction.
The overlay of the crystal structure of Fe(II)-FeBMb with the computer model (Fig. 1C) is striking. Although His43 does not overlay as well since it is located in a flexible loop, the other two His and Glu68 overlay well with one another. Zn(II) in the model and Fe(II) in the FeB site do not overlay exactly, but are in a similar position, 1.08 Å away from each other. Furthermore, the model did not predict the recruitment of a water molecule into the heme pocket, possibly because Zn(II) was used to model Fe(II), in the absence of Fe(II) parameters. Future development of Fe(II) parameters (not in a heme) will likely help resolve this issue. The heme in the computer model is puckered instead of planar, causing the iron to be pulled ~ 1 Å out of the heme plane. If out-of-plane distortion of the model is considered, the FeB to heme iron distance is ~ 4.8 Å for both the model and crystal structure. Therefore, the distinct similarities between the crystal structure and computer model suggest that metalloprotein active sites can be rationally designed.
Since the FeB iron is only ~ 4.8 Å away from the heme iron, it is possible for spin-coupling to occur between the two irons, as is shown in native NOR23,28. To find out if the designed FeBMb mimics this aspect of native NOR, we titrated different equivalents of Fe2+ into deoxy FeBMb and then added blue copper Azurin as a redox partner. In the absence of non-heme Fe2+ and Azurin, deoxy FeBMb displayed no EPR signal, as Fe(II) heme has an EPR-inactive integer electron spin in the perpendicular mode of EPR. Upon addition of Cu(II)-Azurin, a high spin Fe(III) heme EPR signal centered at g~6 was observed (Fig. S2), suggesting that Azurin can serve as a redox partner of FeBMb and oxidize the Fe(II) heme to Fe(III) heme. Interestingly, the intensity of the high spin heme signal decreased with Fe2+ binding to the FeB site of FeBMb, which suggests that Fe2+, when oxidized by Azurin, is spin-coupled to the Fe(III) heme, similar to that observed in native NOR. This conclusion is also supported by electrochemical redox potential determination, as the presence of iron in the FeB site resulted in a dramatic increase of the heme reduction potential (from −158 ± 4 mV to −46 ± 2 mV vs. NHE; Fig. S3).
To test its NO activity, deoxy FeBMb was reacted with excess NO (~17 eq) in the presence of Fe2+ and monitored by UV-vis (Fig. 2B). These conditions approximate one turnover as only two electrons are available for reaction, one from reduced heme iron and one from ferrous FeB. Furthermore, the reduction of NO to N2O is highly thermodynamically favorable (E0′ = + 1.2 V)29, such that many common water soluble reductants can reduce NO non-enzymatically. Studying NO activity under single turnover conditions precludes non-enzymatic NO reduction and allows the reaction to be easily followed by spectroscopy. The appearance of visible peaks at 546 nm and 582 nm ~ 1 s after NO addition indicate fast NO binding. However, the NO-bound protein is not stable and a species with peaks at 408 nm, 550 nm and 577 nm forms immediately after NO binding and is stable with no other changes in the spectrum over the course of 20 min (Fig. 2Ba, red spectrum). The Soret band at 408 nm is characteristic of Fe(III) heme and the visible peaks are characteristic of an NO-bound heme, similar to the NO-bound oxidized Fe(III) heme or met form of FeBMb (Fig. S4, black spectrum). Therefore, it appears that NO addition to Fe(II)-FeBMb results in a single species, namely the met-NO form. In contrast, when the FeB site of FeBMb is empty, NO addition results in NO binding with no additional changes in the UV-vis spectrum over 20 min (Fig. 2Ba, black spectrum). Therefore, these observations provide evidence that FeBMb with an intact FeB site can react with NO, whereas FeBMb with a vacant FeB site binds, but does not react with NO.
As further controls, deoxy wtMb lacking an FeB site, and an FeBMb derivative lacking the glutamate (FeBMb(-Glu)) were reacted with NO in the presence or absence of Fe2+. After 20 min incubation with NO, the UV-vis spectra of both proteins in the presence of Fe2+ remains unchanged (Fig. 2Bb and Bc), suggesting the lack of NO activity for both proteins. These results are not surprising since Fe2+ binding studies (Fig. 2Ab and 2Ac) indicate no evidence of iron binding for either proteins. Therefore, the histidines and glutamate are important not only for iron binding, but also for NOR activity.
The product of NO reduction was identified by GC/MS under single turnover conditions by monitoring NO formation in the headspace of the solution. Because of the high solubility of N2O (~ 25 mM in water at room temperature), this method cannot be used to quantify the rates of the NO reduction under 1–2 turnover conditions (see Supplemental Information, Fig. S8). Typical MS of NO (MW 30) and N2O (MW 44) are shown in Fig. S6. When Fe(II)-FeBMb was incubated with NO, N2O production was observed by the appearance of a second peak in the GC with a longer retention time than NO (Fig. 3A). Although the amount of N2O formed by Fe(II)-FeBMb is low, it is consistently more than that produced by Fe2+ alone (without protein, Fig. 3B) over multiple samples. After no additional changes were observed over a period of 1–2 hr, 2 eq of dithionite was added at 6 hr to reduce the heme and FeB sites again, approximating a second turnover. Although considerable N2O was produced in both samples (dithionite can non-enzymatically reduce NO to N2O), Fe(II)-FeBMb produced ~ 2 times more N2O than the control sample without protein, as observed in the chromatogram of only the 44 MW component, representing the main ion peak of N2O (Fig. S7, bottom blue traces). As an additional control, wtMb in the presence of Fe2+ was reacted with NO with no N2O formation observed (Fig. 3C), indicating that non-specifically bound iron, if present, does not contribute to NOR activity. The yield of N2O production by Fe(II)-FeBMb was estimated to be ~ 30%. These results, together with UV-vis data (Fig. 2), indicate that designed FeBMb is a functional model of NOR.
Figure 3.

Product of NO reaction by FeBMb is N2O. Time dependent GC/MS measurements of N2O formation by Fe(II)-FeBMb (A). Control reactions of NO with Fe2+ only (B) and wtMb (C). NO (~17 eq) was reacted with Fe(II)-FeBMb (0.6 mM protein, 1.5 mM or 2.5 eq Fe2+), Fe2+ (1.5 mM, no protein), or with wtMb (1 mM) and Fe2+ (2 mM or 2 eq). At 6 hr, 2 eq dithionite (1.2 mM) was added and allowed to react to simulate a second turnover. NO2 (MW 46) was not detected. GC peaks have been normalized. N2O yield (30%) was determined from a comparison of the ratio of the NO:N2O peaks of the 30 MW:44 MW single ion chromatograms 2 hr after dithionite addition (after which additional N2O formation was not observed), to that of known dithionite concentrations (2 hr after addition). Background N2O formation (i.e., Fe2+ without protein) was subtracted from that of Fe(II)-FeBMb to estimate the yield.
We have rationally and successfully designed a protein model that both structurally and functionally mimics native NOR. We have demonstrated that both the glutamate and histidine ligands, which are conserved in NOR, are essential for iron binding and NOR activity. Being much smaller, easier to express in high yields, and free of other chromophores, this designed NOR protein will serve as an excellent model for mechanistic studies of NOR.
METHODS
FeBMb computer modeling
A computer model of the designed protein FeBMb was constructed with the L29H, F43H, and V68E mutations to the crystal structure of swMb (PDB: 1JP6) using the modeling extension in VMD (Visual Molecular Dynamics)31. Rendering was also performed in VMD. H43 was rotated about Cα to ensure interaction with the heme pocket as opposed to the bulk solvent. To assess the ability of the designed FeB site to bind an iron ion, molecular dynamics simulations were performed. To mimic an iron ion, the water molecule found in crystal structure of the distal pocket of swMb, was changed to a Zn(II) ion. The resulting computer model was then minimized with NAMD (Molecular Dynamics Simulator)32 using 5000 minimization steps at 0 K, then 10,000 molecular dynamics steps (1 fs/step) via an NVT ensemble at 310 K.
Construction, expression and purification of FeBMb
The triple mutant L29H/F43H/H64/V68E of sperm whale myoglobin (called FeBMb) was constructed using a protocol described previously14,33 and confirmed by DNA sequencing at the Biotechnology Center of the University of Illinois. The DNA was transformed into BL-21(DE3) cells, expressed overnight, and harvested by centrifugation. The bacterial pellet from the equivalent of a 1 L growth was resuspended in 100 mL of 10 mM Tris (2-amino-2-hydroxymethyl-propane-1,3-diol) pH 8 containing 1 mM EDTA, 0.5 mM DTT (dithiothreitol), 35 units/mL DNaseI, 4 units/mL RNaseI, 200 mg lysozyme and a few crystals of PMSF (phenylmethylsulphonyl fluoride). The resultant solution was stirred at 4 °C overnight, diluted to 200 mL with the resuspension buffer, and centrifuged at 10,000xg for 45 min. The resulting grey/white pellet was resuspended in 50 mL of 7 M Guanidine HCl (GuHCl) and shaken at 4 °C for ~ 1 hr. A solution of 25 mg KCN/7.5 mg hemin in 5 mL H2O was prepared and diluted by rapid mixing into 450 mL of cold H2O. Heme incorporation was achieved by dropwise addition of the protein/GuHCl to the hemin-CN solution over 30 min with rapid stirring at 4 °C. The resulting cloudy red solution was stirred for ~ 1 hr after the final addition and centrifuged for 30 min at 8000xg to remove precipitate. The supernatant (500 mL) was dialyzed against 8 L of H2O and 4 L of 100 mM (ionic strength) potassium phosphate (Kpi) at pH 7 to remove excess GuHCl. The protein was refolded with three additional buffer changes at least 6 hr between changes. The protein was then centrifuged for 30 min at 8000xg and concentrated with a stirred cell concentrator (8000 MWCO membrane; Millipore) to 10 mL. The concentrated protein was then loaded onto a gel filtration column (Sephacryl S-100 High Resolution; GE Healthcare) equilibrated with 100 mM Kpi pH 7 and eluted with the same buffer. The fractions with high (≥4.5) R/Z (A406nm/A280nm) were pooled, concentrated, and dialyzed twice against 100 mM Kpi pH 7 containing 30 mM EDTA (ethylenediaminetetraacetic acid), then twice with 100 mM Kpi pH 7 treated with a chelating resin (Chelex 100, iminodiacetic acid, Sigma) containing 100 mM NaCl, and finally three times with 100 mM Kpi pH 7 treated with chelating resin to remove adventitious metal. The purity and identity of purified FeBMb was confirmed by SDS-PAGE and ESI-MS (cal’d MW=17375 Da; exp’l MW= 17376+/−1 Da). Yield: ~ 13 mg protein/L of BL-21(DE3) bacterial growth.
UV-Vis characterization of FeBMb under one turnover conditions
UV-vis spectra were collected on either a Cary 3E (Varian) spectrometer connected to a circulating water bath equipped with a digital temperature controller (Polyscience), or a Cary 5000 (Varian) spectrometer equipped with a Quantum Northwest peltier temperature controller and stirring module. Kinetic UV-vis data were also collected to probe the reaction of NO with FeBMb in the absence or presence of Fe2+ using a Hewlett-Packard 8453 spectrometer with the kinetics module. A protein solution was degassed and transferred to a glove box where it was reduced, purified by size exclusion column, and reconstituted with 2 molar equivalents of Fe2+ (FeCl2, Alfa Aesar) when required.
EPR characterization of spin-coupling of FeBMb in the presence of Fe2+
To prepare EPR samples, FeBMb was first reduced by dithionite in a glove box to form deoxy FeBMb. Excess dithionite was removed with a size exclusion column. Different molar equivalents of Fe2+ were then titrated (i.e., 0, 0.5, 1.0, 1.5, 2.0 eq) into the deoxy FeBMb solution. After stirring 20 min, 3 molar equivalents of oxidized wt Cu(II)-Azurin was added to each sample to oxidize FeBMb as well as the Fe(II) (Fig. S2). After 5 min, the sample was then transferred into an EPR tube and flash frozen in liquid N2. EPR samples contained 0.5 mM FeBMb, 1.5 mM Azurin and 6.8% glycerol in 50 mM Bis-Tris (bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane) pH 7.0. Control experiments of wtswMb only, FeBMb(-Glu) only and Fe2+ only were prepared under identical conditions. Spectra were recorded at 4 K on a Bruker ESP 300 equipped with an Oxford liquid helium cryostat and an ITC4 temperature controller.
Spectroelectrochemical determination of reduction potential
Protein reduction potentials were measured using an optically transparent thin layer electrode (OTTLE) as previously described30,34, with the following changes. A 200 μM protein solution, ~0.6 μL, containing 2 molar equivalents of Fe2+ when required, and 100 mM NaCl in 50 mM Bis Tris pH 7 buffer or 100 mM Kpi pH 7 buffer without added NaCl were used. Each protein solution also contained 80 μM each of phenazine methosulfate (PMS, Sigma) and anthraquinone-2-sulfonate (AQS, Sigma) as electron transfer mediators. For the spectroelectrochemistry study of deoxy FeBMb in the presence of Fe2+ (starting from the reduced deoxy metal bound form and transitioning to the oxidized met form), the protein was reduced in the glove box first with dithionite and purified of excess dithionite before mediator (AQS only) and Fe2+ addition, and subsequent transfer to the degassed OTTLE cell. The headspace of the OTTLE cell was purged with N2 during the experiment to prevent O2 contamination.
The potential of the working electrode was applied in the negative direction for deoxy FeBMb and in the positive direction for deoxy FeBMb + Fe2+. The UV-vis spectrum of the protein at each potential was recorded with a Cary 3E or Cary 5000 spectrophotometer until no further spectral changes were observed.
Data analysis
The spectra from 300–800 nm were analyzed by global analysis using singular value decomposition (SVD) and nonlinear regression modeling software (SpecFit/32, Spectrum Software Associates, Inc). The reduction potential of each protein solution was obtained by fitting the data to the best fit model, an A ↔ B model, in SpecFit/32 and checked by generating a Nernst plot at a single wavelength30. The Ag/AgCl (3 M KCl) reduction potentials were converted to NHE potentials by adding 0.209 V. Since the reduction potentials obtained from the Nernst plots and from SpecFit/32 modeling were generally within ± 16 mV of each other, only the potentials derived from SpecFit/32 were reported (Fig. S3).
Purification of NO and NO saturated solution preparation
NO gas (Matheson Trigas, 99%) was purged of N2O3, NO2, and higher NxOy impurities by passing it through a bubbler containing 1 M NaOH and a second bubbler of water before it entered a reaction vessel filled with ultrapure water35. The reaction solution was first purged with helium to remove O2 to make a ~1.1 mM saturated NO solution, as measured by spectroscopic titrations of a saturated solution of NO to oxy wtMb to form met wtMb. NO was added via a gas-tight syringe to a protein and/or metal solution.
X-ray crystallographic studies
Fe2+ incorporated FeBMb was prepared as above. FeBMb crystals were grown anaerobically in a glove box at room temperature. Red crystals were observed after 2–3 days. Diffractable crystals were obtained from a 1.5 mM protein solution in 20 mM Kpi pH 7 mixed 1:1 with well buffer (0.2 M sodium acetate trihydrate, 0.1 M sodium cacodylate trihydrate pH 6.5 and 30 % w/v PEG 8000). The well of the crystal tray was filled with 300 μL of the same solution. Fe(II)-FeBMb crystals were soaked in a cryoprotectant solution and flash frozen in liquid N2 in a glove box before data collection.
Data collection
Diffraction data of Fe(II)-FeBMb were collected using Fe-edge absorption (1.7309 Å) as wavelengths of data collection at the Brookhaven National Synchrotron Light Source X12C beamline. All data were integrated using the program HKL200036.
Crystal structure determination
The crystal structure was solved by the molecular replacement method using MOLREP in the CCP4 Package37. Refinement was performed using X-plor38 and SHELX’9739. The position of Fe2+ in the FeB site, and the E68 and H43 residues were rebuilt by the program O40.
NO activity studies by GC/MS
NO reduction was monitored by GC/MS using an HP-Plot Q column (Aligent), installed in a Hewlett Packard 5890 gas chromatography system equipped with an online mass spectrometer (Micromass 70-VSE). As dithionite can reduce NO non-enzymatically, the NO activity of FeBMb was probed under one turnover conditions whereby the two electrons required for reduction, one from the reduced Fe(II) heme and one from the ferrous FeB site of Fe(II)-FeBMb, were rate limiting. Where applicable, an extra 2 molar equivalents of dithionite was added to the protein/metal solution to induce an additional turnover. Glucose, glucose oxidase and catalase were added to scavenge residual O2 as described by Giuffre41. Under anaerobic conditions, a 2.67 mL reaction sample consisting of 0.6 mM protein, 10 mM glucose, glucose oxidase (100 μg/mL) and catalase (50 μg/mL) in 20 mM Bis Tris (treated with chelating agent) pH 7 was prepared in a Schlenk flask fitted with a septum. The head-space of the flask was then exchanged to helium. Purified NO gas (1 atm) was injected into the head space of the reaction flask via a gas-tight syringe and the reaction mixture was stirred at room temperature (the molar ratio of NO:protein = ~ 17:1). Typically, 250 μL of head space gas was analyzed via GC/MS at different time intervals to observe NO and N2O levels. MS of NO (30 MW) and N2O (44 MW) are shown in Fig. S6. NO2 (46 MW) formation was not observed.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Mark J. Nilges for his help with EPR analysis, Dr. Steven L. Mullen and Furong Sun for aiding in GC/MS data collection, Dr. E. Lee for help with computational modeling, Nicholas M. Marshall for providing Azurin protein, Jonathan R. Askim for help in FeBMb expression and purification, and Dr. Takahiro Hayashi and Prof. Pierre Moënne-Loccoz for suggestions regarding N2O detection in solution. This work is supported by the US National Institutes of Health (GM062211).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions N.Y. and Y.-W.L performed most of the experimentation and authored most of the manuscript. B.S.R. helped with the initial design of mutants and experiments. X. Z. and L. L. assisted in experimentation. K.D.M. performed computational modeling. Y.-G.G. guided crystallization and refined the crystal structure. H.R. collected crystal diffraction data. Y.L. designed, guided the project and edited the manuscript.
Crystallographic data for FeBMb have been deposited in the Protein Data Bank with the following PDB code: 3K9Z.
References
- 1.Regan L, DeGrado WF. Characterization of a helical protein designed from first principles. Science. 1988;241:976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 2.Hecht MH, Richardson JS, Richardson DC, Ogden RC. De novo design, expression, and characterization of Felix: a four-helix bundle protein of native-like sequence. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 4.Bolon DN, Mayo SL. Enzyme-like proteins by computational design. Proc Natl Acad Sci U S A. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc Natl Acad Sci U S A. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shifman JM, Mayo SL. Exploring the origins of binding specificity through the computational redesign of calmodulin. Proc Natl Acad Sci U S A. 2003;100:13274–13279. doi: 10.1073/pnas.2234277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang SG, Saven JG. Computational protein design: structure, function and combinatorial diversity. Curr Opin Chem Biol. 2007;11:329–334. doi: 10.1016/j.cbpa.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Robertson DE, et al. Design and synthesis of multi-heme proteins. Nature. 1994;368:425–432. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 12.Hay M, Richards JH, Lu Y. Construction and characterization of an azurin analog for the purple copper site in cytochrome c oxidase. Proc Natl Acad Sci U S A. 1996;93:461–464. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeung BK, Wang X, Sigman JA, Petillo PA, Lu Y. Construction and characterization of a manganese-binding site in cytochrome c peroxidase: towards a novel manganese peroxidase. Chem Biol. 1997;4:215–221. doi: 10.1016/s1074-5521(97)90291-x. [DOI] [PubMed] [Google Scholar]
- 14.Sigman JA, Kwok BC, Lu Y. From myoglobin to heme-copper oxidase: design and engineering of a CuB center into sperm whale myoglobin. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 15.Case MA, Mclendon GL. Metal-assembled modular proteins: Toward functional protein design. Acc Chem Res. 2004;37:754–762. doi: 10.1021/ar960245+. [DOI] [PubMed] [Google Scholar]
- 16.Cochran FV, et al. Computational de novo design and characterization of a four-Helix bundle protein that selectively binds a nonbiological cofactor. J Am Chem Soc. 2005;127:1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe Y, Hayashi T. Functionalization of myoglobin. Prog Inorg Chem. 2005;54:449–493. [Google Scholar]
- 18.Ghosh D, Pecoraro VL. Probing metal-protein interactions using a de novo design approach. Curr Opin Chem Biol. 2005;9:97–103. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Petros AK, Reddi AR, Kennedy ML, Hyslop AG, Gibney BR. Femtomolar Zn(II) affinity in a peptide-based ligand designed to model thiolate-rich metalloprotein active sites. Inorg Chem. 2006;45:9941–9958. doi: 10.1021/ic052190q. [DOI] [PubMed] [Google Scholar]
- 20.Koder RL, et al. Design and engineering of an O2 transport protein. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wasser IM, de Vries S, Moeenne-Loccoz P, Schroeder I, Karlin KD. Nitric oxide in biological denitrification: Fe/Cu metalloenzyme and metal complex NOx redox chemistry. Chem Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 22.van der Oost J, et al. The heme-copper oxidase family consists of three distinct types of terminal oxidases and is related to nitric oxide reductase. FEMS Microbiol Lett. 1994;121:1–10. doi: 10.1111/j.1574-6968.1994.tb07067.x. [DOI] [PubMed] [Google Scholar]
- 23.Girsch P, de Vries S. Purification and initial kinetic and spectroscopic characterization of NO reductase from Paracoccus denitrificans. Biochim Biophys Acta. 1997;1318:202–216. doi: 10.1016/s0005-2728(96)00138-7. [DOI] [PubMed] [Google Scholar]
- 24.Hendriks J, Gohlke U, Saraste M. From NO to OO: nitric oxide and dioxygen in bacterial respiration. J Bioenerg Biomembr. 1998;30:15–24. doi: 10.1023/a:1020547225398. [DOI] [PubMed] [Google Scholar]
- 25.Watmough NJ, et al. Nitric oxide in bacteria: synthesis and consumption. Biochim Biophys Acta-Bioenerg. 1999;1411:456–474. doi: 10.1016/s0005-2728(99)00032-8. [DOI] [PubMed] [Google Scholar]
- 26.Wasser IM, Huang H-w, Moeenne-Loccoz P, Karlin KD. Heme/non-heme diiron(II) complexes and O2, CO, and NO adducts as reduced and substrate-bound models for the active site of bacterial nitric oxide reductase. J Am Chem Soc. 2005;127:3310–3320. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- 27.Collman JP, et al. A functional nitric oxide reductase model. Proc Natl Acad Sci U S A. 2008;105:15660–15665. doi: 10.1073/pnas.0808606105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moenne-Loccoz P, et al. Nitric oxide reductase from paracoccus denitrificans contains an oxo-Bridged heme/non-heme diiron center. J Am Chem Soc. 2000;122:9344–9345. [Google Scholar]
- 29.Zumft WG. Nitric oxide reductases of prokaryotes with emphasis on the respiratory, heme-copper oxidase type. J Inorg Biochem. 2005;99:194–215. doi: 10.1016/j.jinorgbio.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 30.Zhao X, Yeung N, Wang Z, Guo Z, Lu Y. Effects of metal ions in the CuB center on the redox properties of heme in heme-copper oxidases: spectroelectrochemical studies of an engineered heme-copper center in myoglobin. Biochemistry. 2005;44:1210–1214. doi: 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- 31.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 32.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Yeung N, Russell BS, Garner DK, Lu Y. Catalytic reduction of NO to N2O by a designed heme copper center in myoglobin: implications for the role of metal ions. J Am Chem Soc. 2006;128:6766–6767. doi: 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taboy CH, Bonaventura C, Crumbliss AL. Anaerobic oxidations of myoglobin and hemoglobin by spectroelectrochemistry. Methods Enzymol. 2002;353:187–209. doi: 10.1016/s0076-6879(02)53048-2. [DOI] [PubMed] [Google Scholar]
- 35.Bonner FT. Nitric oxide gas. Methods Enzymol. 1996;268:50–57. doi: 10.1016/s0076-6879(96)68008-2. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 38.Brunger AT, et al. Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 40.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr, Sect A: Found Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 41.Giuffre A, et al. The heme-copper oxidases of Thermus thermophilus catalyze the reduction of nitric oxide: evolutionary implications. Proc Natl Acad Sci U S A. 1999;96:14718–14723. doi: 10.1073/pnas.96.26.14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.