

Summary
The restriction factor SAMHD1 limits HIV-1 replication in non-cycling cells. SIV and HIV-2 overcome this restriction via the accessory protein Vpx, which targets SAMHD1 for degradation through interactions with the host ubiquitin ligase adaptor, DCAF1. However, the factors used by HIV-1 to replicate in macrophages, despite the presence of the restriction factor SAMHD1, are unknown. Using a yeast 2-hybrid screen, we identified Cyclin L2 as a DCAF1-interacting protein required for HIV-1 replication in macrophages. Knockdown of Cyclin L2 results in severe attenuation of HIV-1 replication in macrophages, but not cycling cells, and this effect is lost in the absence of SAMHD1. Cyclin L2 and SAMHD1 form a molecular complex that is partially dependent on the presence of DCAF1 and results in SAMHD1 degradation in a proteasome- and DCAF1-dependent manner. Thus, Cyclin L2-mediated control of SAMHD1 levels in macrophages supports HIV-1 replication.
Introduction
HIV-2 and its counterpart, Simian Immunodeficiency Virus (SIV), encode the accessory protein Vpx which facilitates efficient infection of quiescent non-cycling cells like macrophages, resting T cells, and dendritic cells (Ueno et al., 2003; Srivastava et al., 2008). Vpx also endows HIV-1 with the ability to replicate efficiently in non-dividing cells when it is supplied in trans or packaged into incoming virions, suggesting that Vpx disables a restriction factor in the very early steps of viral replication. Recently, SAMHD1 was identified as the critical restriction factor targeted by Vpx. Degradation of SAMHD1 by Vpx in macrophages, dendritic cells and resting T cells allows for efficient infection by HIV-2/SIV (Laguette et al., 2011; Hrecka et al., 2011a; Baldauf et al., 2012; Laguette et al., 2011). In addition, depletion of SAMHD1 from non-dividing cells, either by Vpx or by genetic knockdown, leads to more effective HIV-1 replication. Furthermore, Vpx binds to SAMHD1, and promotes its degradation in the proteasome (Brandariz-Nunez et al., 2012; Ahn et al., 2012). The degradation process requires Vpx to also bind to DCAF1, the Cul4A ubiquitin ligase adaptor (Wei et al., 2012a; Zhu et al., 2013).
Despite the critical role of SAMHD1 as a restriction factor in non-dividing cells, little is known about how it is regulated. SAMHD1 was shown earlier on as one of the genes mutated in children with Aicardi–Goutières syndrome (Rice et al., 2009; Dale et al., 2010). In this rare genetic disorder, children present with symptoms resembling those of an overwhelming viral infection, the result of an excessive type I interferon response to circulating nucleic acids. SAMHD1 has an N-terminal SAM (sterile alpha motif) domain and a C-terminal histidine aspartic acid (HD) domain. The HD domain acts as a deoxyguanosine triphosphate (dGTP) dependent triphosphohydrolase(St et al., 2012; Goldstone et al., 2011; Zhu et al., 2013). Several groups found that depletion of dNTPs by SAMHD1 reduces the nucleotide pools in non-dividing cells, and prevents efficient HIV replication. Limited levels of dNTPs in non-dividing cells may explain why SAMHD1 restricts HIV replication in macrophages, dendritic cells and resting T cells but not in actively dividing T lymphocytes. In addition, the antiviral activity of SAMHD1 innon-cycling compared to cycling cells may be explained by post-translational modification. SAMHD1 is phosphorylated by Cyclin A2/Cdk1 in dividing but not in non-dividing cells. Phosphorylated SAMHD1 is unable to restrict HIV, but retains dNTPase activity(Cribier et al., 2013; White et al., 2013). Although differentiated macrophages express large amounts of SAMHD1, HIV-1 is able to replicate in these cells. Thus, the restriction imposed by SAMHD1 on HIV-1 in macrophages is incomplete; suggesting that HIV-1 has a mechanism to overcome SAMHD1, or HIV-1 utilizes a cellular factor that regulates SAMHD1 activity.
Since Vpx requires interaction with DCAF1 for efficient macrophage infection by SIV/HIV-2, we postulated that other DCAF1-interacting proteins may play a role in HIV infection of macrophages. Hence we performed a yeast-2-hybrid screen using a T-cell library from Clontech and identified Cyclin L2 as a DCAF1-interacting protein. Cyclin L2 is part of the recently discovered family of cyclin L proteins, consisting of Cyclin L1 and Cyclin L2. It possesses an N-terminal cyclin box and a C-terminal serine arginine (SR) domain, and it has been shown to be involved in cell cycle regulation and pre-mRNA splicing(Yang et al., 2004; de et al., 2004; Li et al., 2007; Loyer et al., 2008; Zhuo et al., 2009).
In this study, we show that depletion of Cyclin L2 attenuates HIV replication in macrophages, but not in dividing cells. We found that Cyclin L2 interacts with and targets SAMHD1 for degradation in a proteasome- and DCAF1-dependent manner. Moreover, we found that during the early phase of HIV infection in macrophages, the level of Cyclin L2 is negatively correlated with that of SAMHD1. We present several lines of evidence to show that Cyclin L2 is an important endogenous regulator of SAMHD1 and a critical HIV dependency factor in macrophages.
Results
Screen of putative DCAF1-interacting proteins identifies Cyclin L2 as an HIV-dependency factor
The HIV accessory proteins Vpr and Vpx are important for HIV replication in macrophages. Both proteins require interaction with the Cul4A ubiquitin ligase adaptor DCAF1 for their ability to induce cell cycle arrest in the case of Vpr, and degradation of the restriction factor SAMHD1 by Vpx (Fregoso et al., 2013; Romani and Cohen, 2012; Wei et al., 2012b; Srivastava et al., 2008). We postulated that other DCAF1-interacting proteins may play a role in HIV replication, especially in quiescent cells like macrophages where the effects of Vpx and Vpr on virus replication are most prominent. To test this idea, we performed a screen for DCAF1-interacting proteins using the yeast 2-hybrid method, employing full length and truncated DCAF1 proteins as baits. Based on the proteins identified in the screen, and those recently identified as putative DCAF1-interacting proteins (Hrecka et al., 2011b; Laguette et al., 2011), we selected five gene products to test their ability to restrict or potentiate HIV replication in macrophages. For this purpose we generated stable THP-1 monocytoid cell lines selected in puromycin using a cocktail of shRNAs for each gene expressed in the lentiviral vector, pLKO.puro. Significant depletion of each of these gene products was obtained (Figure 1A). After differentiation in PMA for 48h, cells were infected with VSV-G pseudotyped HIV-1Luc, HIV-2Luc or HIV-2ΔVpxLuc reporter viruses for 48hrs. We expected that compared to controls, restriction factors will show increased HIV replication upon mRNA knockdown, while dependency factors will show reduced HIV replication, as measured by intracellular luciferase luminescence read-outs in infected cells. The assay was validated using the knockdown of the restriction factor SAMHD1 as a positive control. There was a 4-fold increase in HIV-1 replication with SAMHD1 depletion, as expected (Figure S1A, B). Out of the five genes screened, knockdown CCNL2, which encodes Cyclin L2, resulted in a significant reduction in the replication of HIV-1 (5-fold), HIV-2 (2.7-fold) and HIV-2ΔVpx (4-fold) (Figure 1B–D), indicating that it could be a potential HIV dependency factor.
Figure 1. Screening of putative DCAF1-interacting proteins for their effect on HIV replication identifies Cyclin L2.
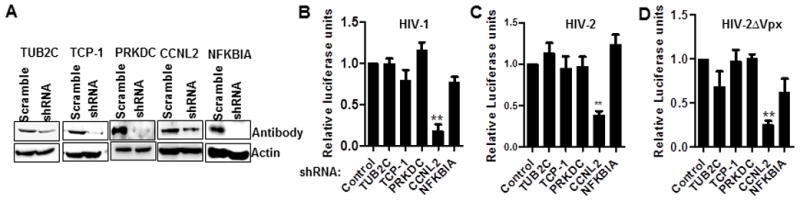
(A) Western blot (WB) showing shRNA knockdown of the indicated genes in PMA differentiated THP-1 cells. (B–D) THP-1 cells stably expressing the indicated or control shRNA were differentiated with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA) for 48h and infected with HIV-1Luc, HIV-2Luc or HIV-2ΔVpxLuc. After 2 days, cells were lysed for luciferase luminescence. Luciferase luminescence for four triplicate experiments were pooled and expressed as fold change of the control shRNA. See also Figure S1. Data indicate means; error bars indicate ±SEM (n≥3). **, p < 0.01 (ANOVA).
Cyclin L2 is a DCAF1 interacting protein
Since Cyclin L2 was identified as a DCAF1-interacting protein in yeast, we confirmed that it is expressed and interacts with DCAF1 in mammalian cells. Figure 2A shows that Cyclin L2 is expressed ubiquitously in various cell types relevant to HIV biology, including monocyte-derived macrophages, THP-1 cells, and T cell lines. To confirm that Cyclin L2 interacts with DCAF1 in mammalian cells, immunofluorescence and immunoprecipitation experiments were performed. Expression of Cyclin L2 in HeLa cells showed that it is localized in the nucleus and partially colocalized with DCAF1 (Figure 2B, Figure S2A). Cells transfected with Cyclin L2 showed intranuclear vacuoles as previously described (Yang et al., 2004). Moreover, with a pull down of either protein, Cyclin L2 and DCAF1 were together in immunoprecipitates, indicating that the two proteins are closely associated (Figure 2C, 2D). More importantly, we could detect interaction between Cyclin L2 and DCAF1 when immunoprecipitations were performed in untransfected THP-1 cells (Figure 2E, Figure S2B). Moreover, DDB1 and Cul4a, other components of the ubiquitin ligase complex, were also detected in the immunoprecipitates. Taken together, these data show that Cyclin L2 is an HIV dependency factor that interacts with DCAF1.
Figure 2. Cyclin L2 is ubiquitously expressed and interacts with DCAF1 in mammalian cells.
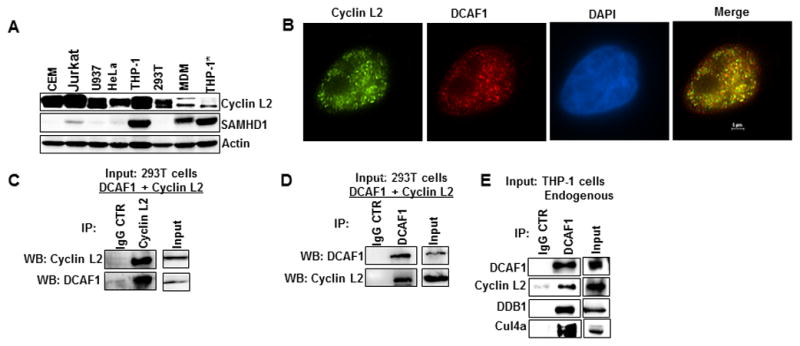
(A) WB showing expression levels of Cyclin L2 in various cell lines and monocyte derived macrophages (MDM). *Differentiated THP-1 (B) Cyclin L2 and DCAF1 show partial colocalization in the nucleus. HeLa cells were transfected with myc-Cyclin L2 and stained for myc and endogenous DCAF1.See also Figure S2. (C, D) 293T cells were transfected with Cyclin L2 and DCAF1 for 48 h, then cells were lysed and immunoprecipitation was performed using polyclonal DCAF1 or Cylin L2 antibody or IgG control. Western blots were probed with DCAF1 or Cyclin L2 antibodies. (E) Differentiated THP-1 cells were lysed, immunoprecipitation performed with DCAF1 antibody and blotted for the indicated proteins. The blots shown represent one of two or three independent experiments.
Depletion of Cyclin L2 inhibits HIV replication in non-dividing cells
To further define the effect of Cyclin L2 on HIV replication, we made different individual shRNAs targeting the CCNL2 mRNA in THP-1 cells. Knockdown of Cyclin L2 was achieved with several of the shRNAs in THP-1 cells differentiated with PMA for 48 hours (Figure 3A).
Figure 3. Cyclin L2 is required for HIV replication in macrophages.
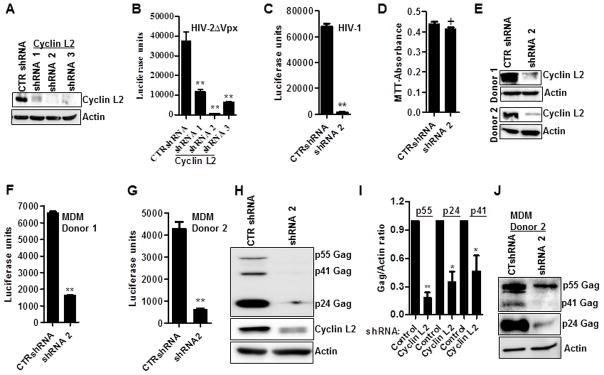
(A, B) Stable THP-1 cell lines expressing different shRNAs to Cyclin L2 or control shRNA (CTR) were differentiated for 48h in PMA and infected with HIV-2ΔVpxLuc for 48h. Western blot (A) shows knockdown of Cyclin L2 prior to infection. Viral replication is shown as luciferase units (B) in infected cell lysates. (C) Differentiated THP-1 cells expressing shRNA2 (from A) or control were infected with HIV-1Luc for 48h and luciferase luminescence measured in cell lysates. (D) Differentiated THP-1 cells expressing indicated shRNAs, were infected with HIV-1Luc for 48h, washed and used for proliferation (MTT) assay. (E–G) Human peripheral blood monocyte derived macrophages (MDM) expressing control or Cyclin L2 shRNAs were infected with HIV-1Luc for 48h and HIV replication measured as luciferase units in infected cells. Western blot of Cyclin L2 knockdown from two different donors. Three parallel experiments were performed for each donor using the different shRNA preparations. HIV replication in control vs Cyclin L2 shRNA expressing cells (F, G) are shown. (H) Differentiated THP-1 cells expressing control or Cyclin L2 shRNA were infected with HIV-1 for 72h and immunoblotted for Cyclin L2, HIV-1 Gag and actin. (I) Quantification (ratio of Gag to Actin band intensities). (J) MDMs infected with HIV-1 from donor 2 were immunoblotted for HIV-1 Gag and Actin. Longer exposure times were required for p55 and p41. Data indicate means; error bars indicate ±SEM (n≥3). **, p < 0.001, *p<0.05, + p>0.05 (ANOVA for multiple comparisons and student t test for pairwise comparisons).See also Figure S1 and S3.
Differentiated THP-1 cells were infected with HIV-2ΔVpxLuc reporter virus. In each of the shRNA knockdowns, we detected a significant reduction in HIV-2ΔVpx replication as measured by luciferase activity (Figure 3B). The shRNA with the greatest effect on HIV-2ΔVpx replication, designated shRNA2, was used to examine the effect of Cyclin L2 knockdown on HIV-1 in subsequent experiments. We found a profound decrease in HIV-1 replication in THP-1 cells when Cyclin L2 was depleted (Figure 3C). The decrease in HIV-1 replication was not due to reduced proliferation or enhanced apoptosis of cells transduced with Cyclin L2 shRNA as evidenced by similar levels of MTT incorporation in control and Cyclin L2 depleted cells (Figure 3D). In addition, HIV entry was not affected by Cyclin L2 as shown by intracellular p24 ELISA assay at 2h post infection (Figure S1C). We then tested the effect of Cyclin L2 in human monocyte-derived macrophages from two different donors. Human monocyte derived macrophages were transduced with control or Cyclin L2 shRNA, infected with HIV-1Luc and luciferase read-outs measured on cell lysates. In both donors, there was more than six-fold reduction in HIV-1 replication upon knockdown of Cyclin L2 (Figure 3E–G). In order to confirm that the effect seen in luciferase activity translated into HIV-1 gene products, we examined intracellular Gag production. Compared to controls, there was a significant decrease in HIV-1 Gag protein with Cyclin L2 depletion (Figure 3H–J). However, we did not detect a change in HIV-1 replication when Cyclin L2 was depleted or overexpressed in 293T cells (Figure. S3A–D), or depleted in HeLa cells (Figure S3E, S3F) suggesting that the effect of Cyclin L2 is restricted to non-dividing cells. Taken together these data confirm Cyclin L2 as a significant HIV dependency factor in macrophages.
Cyclin L2 interacts with SAMHD1
We found Cyclin L2 to be critical for HIV replication, but the effect was restricted to differentiated THP-1 cells and monocyte-derived macrophages, cell types that are terminally differentiated and not actively dividing. The cell types in which Cyclin L2 is active for HIV replication are similar to those in which SAMHD1 acts as a restriction factor. In addition, Cyclin L2 interacts with DCAF1, which is also required for SAMHD1 degradation through Vpx in the context of HIV-2 infection. Hence we wondered if Cyclin L2 and SAMHD1 interact with each other. Upon expression of SAMHD1 in HeLa cells, we found significant nuclear colocalization with Cyclin L2 (Figure 4A, Figure S4). We also performed protein-interaction studies using 293T cells, and found that SAMHD1 and Cyclin L2 co-immunoprecipitated (Figure 4B, C). The co-immunoprecipation between Cyclin L2 and SAMHD1 was present, but reduced in DCAF1-depleted cells (Figure 4D) indicating that the interaction between Cyclin L2 and SAMHD1 depends at least partially on DCAF1. Moreover, we found interaction between endogenous Cyclin L2 and SAMHD1 in differentiated THP-1 cells (Figure 4E), attesting to the physiological significance of these interactions.
Figure 4. Cyclin L2 interacts with SAMHD1.
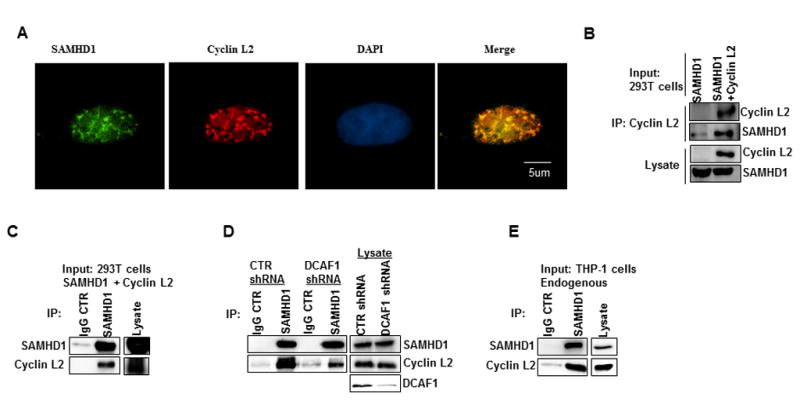
(A) HeLa cells were transfected with SAMHD1 overnight and immunostained for SAMHD1 and endogenous Cyclin L2. See also Figure S4. (B) 293T cells were transfected with control, SAMHD1 or both Cyclin L2 and SAMHD1 expression plasmids. After 48h, Cyclin L2 was immunoprecipitated using myc monoclonal antibody. Western blots were probed with Cyclin L2 and SAMHD1 antibodies. (C) 293T cells were transfected with Cyclin L2 and SAMHD1 and immunoprecipitation was performed using SAMHD1 or isotype IgG control antibody. (D) 293T cells stably expressing control or DCAF1 shRNA were transfected with Cyclin L2 and SAMHD1 plasmid and immunoprecipitation performed with SAMHD1 or IgG antibody. (E) Differentiated THP-1 cells were lysed and immunoprecipitation performed with SAMHD1 antibody or isotype IgG control. The blots shown represent one of two or three independent experiments. CTR, control.
Cyclin L2 degrades SAMHD1 through the proteasome
Since Cyclin L2 and SAMHD1 exhibited colocalization and co-immunoprecipitation, we sought to determine if there was a functional relationship between these two proteins. One possibility that could explain the sensitivity of HIV-1 and HIV-2 to Cyclin L2 depletion is that Cyclin L2 depletion leads to increased SAMHD1 levels. This would suggest that Cyclin L2 is a natural regulator for SAMHD1 in terminally differentiated cells. To test this idea, we co-transfected SAMHD1 and Cyclin L2 in 293T cells using increasing concentrations of a Cyclin L2 expression plasmid. We found that SAMHD1 was degraded by Cyclin L2 overexpression, in a dose-dependent manner (Figure 5A). This degradation was partially rescued upon addition of the proteasome inhibitor MG132 (Figure 5B). In addition, SAMHD1 degradation was potentiated when protein synthesis was blocked with cycloheximide (Figure 5C). Since Vpx degradation of SAMHD1 is dependent on DCAF1, and Cyclin L2 also interacts with DCAF1, we tested whether Cyclin L2-dependent degradation of SAMHD1 was DCAF1-dependent. As shown in Figure 5D, degradation of SAMHD1 by Cyclin L2 was abolished in cell lines where DCAF1 was depleted (Figure 5D, E). In addition knockdown of DNA binding protein 1 (DDB1) abrogated the effect of Cyclin L2 overexpression on SAMHD1 (Figure 5F, G), showing that Cyclin L2-mediated degradation of SAMHD1 requires an intact DCAF1-DDB1 complex. When we overexpressed SAMHD1 in increasing amounts, Cyclin L2 was not degraded (Figure S5A) suggesting that SAMHD1 is brought to the DCAF1 complex by Cyclin L2, and not vice versa. In knockdown experiments in differentiated THP-1 cells, depletion of Cyclin L2 led to increased levels of SAMHD1 (Figure 5H, I). Taken together, these findings show a role for Cyclin L2 in controlling the abundance of SAMHD1, which could explain the effect of Cyclin L2 on HIV replication in macrophages. Given recent evidence that SAMHD1 is phosphorylated by Cyclin A/Cdk1(Cribier et al., 2013; White et al., 2013), we determined whether Cyclin L2 depletion has an effect on SAMHD1 phosphorylation. As shown in Figure S5B, depletion of Cyclin L2 increased the overall levels of SAMHD1 but did not have an effect on SAMHD1 phosphorylation, as examined by Phos-Tag SDS PAGE.
Figure 5. Cyclin L2 degrades SAMHD1 through the proteasome.
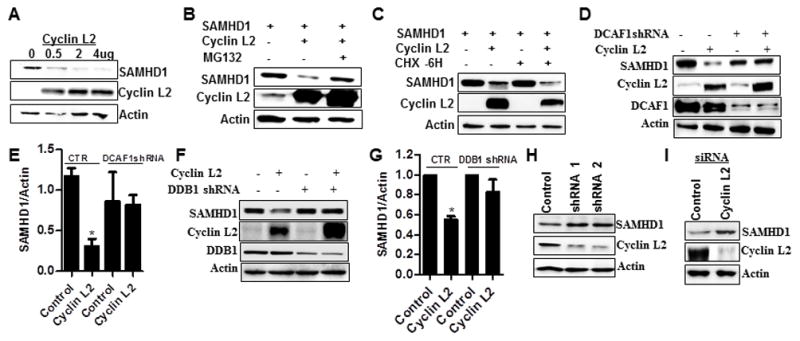
(A) 293T cells were transfected with increasing concentrations of Cyclin L2 and 1 ug of SAMHD1 for 48h and the expression levels of the two proteins determined by Western blot. (B) Co-transfection of Cyclin L2 and SAMHD1 in 293T cells with 10uM of MG132 added after 24 h. WB was performed after an additional 16 hrs. (C) 293T cells were transfected with Cyclin L2 and SAMHD1. After 48h, protein synthesis was blocked with 10uM of cycloheximide (CHX) for 6 h and WB performed on cell lysates. (D) Stable 293T cells expressing control or DCAF1 shRNA were transfected with Cyclin L2 and SAMHD1 and WB performed after 48h. (E) Quantification of SAMHD1/Actin band intensities from experiments in D. (F) Stable 293T cells expressing control or DDB1 shRNA were transfected with Cyclin L2 and SAMHD1 and WB performed after 48h. (G) Quantification of SAMHD1/Actin band intensities from experiments in F. (H, I) THP-1 cells were transfected with Cyclin L2 siRNA or transduced with shRNA, differentiated for 48h and WB performed for their expression levels. See also Figure S5.
Effect of Cyclin L2 depletion on HIV-1 replication is aborted upon SAMHD1 knockdown
Given that SAMHD1 depletes triphosphate nucleotide levels, which restricts HIV reverse transcription (RT), we determined if depletion of Cyclin L2 results in lower levels of HIV-1 RT products. We found a three-fold reduction of HIV-1 late RT products upon Cyclin L2 depletion in differentiated THP-1 cells (Figure 6A), showing that depletion of Cyclin L2 results in increased SAMHD1 and less HIV-1 replication. For direct evidence that HIV-1 replication in Cyclin L2 depleted cells is dependent on SAMHD1 levels, we made stable THP-1 cells with knockdown of both genes. Our reasoning was that if depletion of Cyclin L2 promotes SAMHD1 action, double knockdown will restore HIV-1 replication. When the cells were infected with HIV-1, SAMHD1 knockdown restored HIV-1 replication close to control levels in Cyclin L2 depleted cells (Figure 6B). This was also seen with measurement of HIV-1 p55 in immunoblots (Figure 6C, D) showing that Cyclin L2 controls SAMHD1 levels in vivo, and the effect is borne out in HIV replication. In addition, it shows that HIV-1 replication in macrophages is dependent on the presence of Cyclin L2, when SAMHD1 is depleted by means other than through Cyclin L2. It is noteworthy that when measured by HIV-1 p55 Gag, the upregulation of HIV-1 upon SAMHD1 depletion is not as robust compared to luciferase measurements.
Figure 6. Effect of Cyclin L2 depletion on HIV-1 replication requires intact SAMHD1.
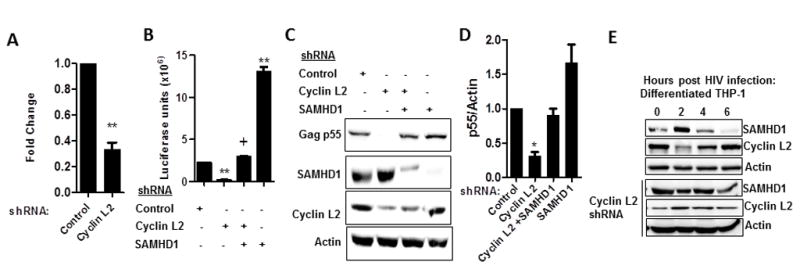
(A) Differentiated THP-1 cells stable expressing control or Cyclin L2 shRNA were infected with HIV for 24h. Whole cell DNA was isolated and PCR performed to measure HIV-1 gag gene. Results are expressed as fold change relative to control. (B) Differentiated THP-1 cells expressing shRNA to the indicated genes were infected with HIV-1Luc for 48h and viral replication determined by luciferase luminescence. (C) Differentiated THP-1 cells expressing the shRNA to the indicated proteins were infected with HIV-1 and WB performed after 48 h. (D) Quantification of HIV-1 Gag p55/actin for three experiments from C. (E) Differentiated THP-1 cells with or without shRNA to Cyclin L2 were infected with HIV-1 and cellular lysates taken at the indicated time points for Western blot. Blots represent one of 3 or more experiments. Data indicate means; error bars indicate ±SEM (n≥3). **, p < 0.001, *p<0.05, + p>0.05 (ANOVA for multiple comparisons and student t test for pairwise comparisons).
Since several lines of evidence indicated that Cyclin L2 controls SAMHD1 levels to enable efficient HIV infection in macrophages, we wondered if there was a temporal relationship between the two proteins during HIV infection. To test this idea, we performed time course immunoblotting for levels of Cyclin L2 and SAMHD1 upon HIV-1 infection of differentiated THP-1 cells. Prior to infection, Cyclin L2 and SAMHD1 are in equilibrium in THP-1 cells (Figure 6E). Upon HIV-1 infection, we detected initial down regulation of Cyclin L2 levels followed by increased levels at 6 hours post-infection, which correlated with decreased SAMHD1 levels. When the same experiment was repeated in Cyclin L2 depleted cells, SAMHD1 levels remained constant. These data further strengthen our hypothesis that Cyclin L2 controls SAMHD1 levels in macrophages to permit HIV infection.
Discussion
In this study, we have identified Cyclin L2 as a critical HIV dependency factor in macrophages the levels of which are negatively correlated with SAMHD1 upon HIV infection. Knockdown of Cyclin L2 in macrophages decreased HIV replication several fold in differentiated THP-1 cell lines and primary macrophages. Cyclin L2 interacts with SAMHD1, resulting in proteasomal degradation of SAMHD1. In cells in which SAMHD1 is depleted, the effect of Cyclin L2 knockdown on HIV is abrogated. Hence the effect of Cyclin L2 on HIV replication is at least in part through its regulation of SAMHD1 levels.
Cyclin L2 is the dominant member of the Cyclin L group of cyclins consisting of several isoforms of Cyclin L1 and L2 (Herrmann et al., 2007; Yang et al., 2004; de et al., 2004). Unlike SAMHD1 which is not expressed in T cell lines or U937 cells (Hrecka et al., 2011b), Cyclin L2 was expressed in all cell types examined, and has been found to be expressed in several tissues (Loyer et al., 2008). In dividing cells, Cyclin L2 is important for pre-mRNA splicing through association with splicing factors (Herrmann et al., 2007; Yang et al., 2004). This suggests that Cyclin L2 may be an essential protein in dividing cells. In line with this idea, whenever we selected dividing cells in puromycin with shRNA to Cyclin L2, we could not achieve knockdown (data not shown). When selected THP-1 cells were differentiated however, knockdown was readily achieved. In terminally differentiated cells, cyclins may play a role other than cell cycle regulation. Several cyclins have been implicated in HIV replication, notably cyclin T1 and most recently Cyclin A/B (Cribier et al., 2013; Welbourn et al., 2013). Here, Cyclin L2 is added to that growing list. Our findings show that cyclins can be co-opted to participate in efficient replication of HIV, enabling replication to occur in macrophages even in the presence of potent restriction factors like SAMHD1. Consistent with this assertion, levels of Cyclin T1 has been shown to be induced during the early phase of HIV-1 infection in monocytes/macropohages, with levels significantly going down after about a week of infection, through proteasome mediated proteolysis. The levels of Cyclin T1 can increase again in response to pathogen associated molecular patterns (PAMPs) raising the possibility that Cyclin T1 may play a role in HIV replication during viral reactivation in latently infected cells(Liou et al., 2006; Liou et al., 2004; Liou et al., 2002). Whether or not Cyclin L2 plays a role in HIV reactivation or viral alternative splicing remains to be determined.
In the context of HIV-2 infection, Vpx acts as a bridge between SAMHD1 and DCAF1 resulting in efficient proteasomal degradation of SAMHD1 (Guo et al., 2013; DeLucia et al., 2013). While SAMHD1 is established as a restriction factor for several retroviruses in non-dividing cells, how it is regulated in the absence of viral infection is only now beginning to be elucidated. Given the importance of SAMHD1 in regulating nucleotide levels, especially in noncycling cells, it must be tightly regulated to prevent excessive nucleotide levels, and vice versa. Here we propose that Cyclin L2 is an important factor that targets SAMHD1 for degradation through DCAF1 in the absence of Vpx.
Recent evidence indicates that SAMHD1 is regulated during different phases of the cell cycle (Rossi, 2014; Kretschmer et al., 2014). SAMHD1 is highly phosphorylated in cycling cells but remains largely unphosphorylated in quiescent cells where it is active as a restriction factor (St et al., 2014; Cribier et al., 2013; St et al., 2012). Our study did not find a change in phosphorylation status for SAMHD1 when Cyclin L2 was depleted. Rather, depletion of Cyclin L2 resulted in overall increases in the levels of SAMHD1 in differentiated THP-1 cells. Since the bulk of SAMHD1 in differentiated macrophages remain unphosphorylated, it is reasonable to suggest that depletion of Cyclin L2 increases the portion of SAMHD1 relevant for HIV restriction. Just like Vpx, Cyclin L2 also interacts with both DCAF1-DDB1 and SAMHD1, and in the process tags SAMHD1 for proteasomal degradation. Since SAMHD1 levels change with different phases of the cell cycle, it is not surprising that in the absence of HIV infection, a DCAF1-interacting protein will mark SAMHD1 for proteasomal degradation as several important players in the cell cycle undergo the same fate. A recent report shows that premature activation of the SLX4 complex by Vpr is critical for the G2/M cell cycle arrest induced by Vpr, a process that also requires the DCAF1-DDB1-Cul4a complex. Given that SLX4 is also involved in the cell cycle, and Cyclin L2 interacts with the DCAF1-DDB1 complex, it will be interesting to find out if Cyclin L2 has a role in the Vpr-mediated cell cycle arrest (Gritenaite et al., 2014; Laguette et al., 2014).
We observed a several-fold decrease in HIV-1 reverse transcription products in Cyclin L2 depleted cells. This was expected given that Cyclin L2 depletion led to increases in SAMHD1 levels. While depletion of SAMHD1 in macrophages led to increased replication of HIV likely due to the availability of nucleotides, knockdown of Cyclin L2 led to marked reduction in HIV-1 replication. The effect of Cyclin L2 could be rescued when SAMHD1 was depleted. This rescue, while significant, was not complete indicating that factors other than Cyclin L2 are involved in determining SAMHD1 abundance, and remain to be determined. Our finding that knockdown of Cyclin L2 was less effective in attenuating HIV-2 replication compared to HIV-1 also supports our hypothesis that Cyclin L2 controls SAMHD1 levels. The presence of Vpx in HIV-2 most likely reduces the levels of SAMHD1, and thus blunts the effects of Cyclin L2 depletion. Hitherto, the factors that allow HIV-1 to replicate in macrophages, despite the abundance of SAMHD1 in this cell type, have not been determined. Here we propose that Cyclin L2 is an important factor in macrophages, which controls the levels of SAMHD1 to promote viral replication. Further work will be needed to delineate other players in this interaction and explore Cyclin L2 and its interacting partners as potential therapeutic targets for HIV in non-dividing cells.
Materials and Methods
Cell lines
For the preparation of macrophages, human monocytes were prepared from HIV-negative donors by density gradient centrifugation (400 g for 30 min) through a Ficoll-Hypaque gradient (GE Healthcare). Monocyte differentiation was performed for 7 days using 50ng/ml of rh-MCSF. THP-1, Jurkat, CEM and U937 cells were maintained in RPMI supplemented with glucose, glutamine, penicillin/streptomycin, and pyruvate. Where required, THP-1 cells were differentiated for 24–48 h in 50 ng/ml of phorbol 12-myristate 13-acetate (PMA). HeLa and 293T cells were maintained in DMEM supplemented with glutamine and FBS and antibiotics
Plasmids and shRNA
Myc-DDK Cyclin L2 clone was obtained from Origene. SAMHD1 was expressed in a pcDNA3.1 vector with a Flag tag at its N-terminus. DCAF1 was expressed in a pcDNA3.1 vector with a Flag-Myc-HA tag at its N-terminus. The HIV-1-Luc reporter virus from a pNL4-3 background, HIV-2Luc and HIV-2Δvpx-Luc are from an HIV-2-Rod background with luciferase replacing the Nef gene in all three molecular clones. All viruses were pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G) for infections. All shRNAs were obtained from the Washington University Genome Center. Sequences of the Cyclin L2 shRNAs used are shRNA #1-5′CTTGCAGCTTTATGCTCGGAA3′, #2-5′GTGGTCTCAATGTATTGACTT3′, #3-5′GCGTCTCCTAAGAGGAGGAAA3′ #4-5′CCCTACAAAGGCTCTGAGATT3′ and #5-5′GCCTACCAGATTCTGGTGATT3′. shRNA #2 was most effective at knocking down Cyclin L2 expression and was used in all experiments except for the initial screen in Figure 1 where a cocktail of all 5 were used, and the experiment shown in Fig 3A, where the designated shRNAs were used.
Transfections
Cells were transfected using the nucleoporation protocol (Lonza) as described previously (Chua and Deretic, 2004). Cycin L2 siRNA knockdown was achieved using siGENOME SMART pool (Thermo Fisher Scientific). All effects of siRNA were compared with non-targeting siRNA pool (Thermo Fischer Scientific).
Virus generation and infections
HIV-1Luc, HIV2ΔVpxLuc pseudotyped with the envelope glycoprotein of the VSV were obtained as cleared supernatants of 293T cells 48 h, after calcium phosphate co-transfection. Viral infections were carried out as previously described (Olivetta and Federico, 2006). Briefly, infection of THP-1, monocyte derived macrophages (MDM), 293T, HeLa cells were performed by addition of 100, 50, or 10 ng p24 of VSV-G pseudotyped HIV-1Luc depending on the experiment. 293T and HeLa cells were infected with the lower concentrations of virus. Higher concentrations of virus was used in differentiated THP-1 cells for assays involving Western blots of intracellular Gag. Cells were transduced overnight, washed and allowed to go for another 24–48 hours in single round infection assays. Viral replication was measured by assessing firefly luciferase activity or HIV-1 Gag in cell lysates. In Figure 1, typical numbers for the normalized reads of luciferase in control shRNA were: 10,000 units for HIV-1 and HIV-2ΔVpx and 50,000 units for HIV-2Luc with a background read of <300units. We used lower concentrations of virus in Figure 1 to increase the chances of observing a biological effect from the various gene knockdowns.
Small hairpin RNA (shRNA) virus-like particles were obtained after co-transfection of 293T with the pLKO.puro vector containing the shRNA sequence, Gag-pol obtained from HIV-1 and VSV-G at a ratio of 10:5:1. Cleared supernatant was used for transduction of THP-1 cells. Stable cells lines were obtained with puromycin selection. For knockdown of Cyclin L2, freshly isolated PBMCs were transduced for 24 h with shRNA VLPs prior to differentiation to macrophages.
Antibodies and chemicals
Cyclin L2 rabbit polyclonal antibodies were obtained from Novus Biologicals and ProSci Inc. SAMHD1 mouse polyclonal, rabbit Cul4a and DDB1 antibodies were from Abcam. Goat polyclonal Actin, goat polyclonal Cyclin L2, mouse monoclonal DCAF1 antibodies were obtained from Santa Cruz Biotechnologies. Rabbit DCAF1 antibody was from Proteintech. Myc and FLAG antibodies were obtained from Clontech and Sigma, respectively. MG132 was from Sigma. Mouse monoclonal Gag antibody was obtained through the NIH AIDS Reagent Program. MTT assay reagents were obtained from Sigma and performed according to manufacturer’s instructions. Briefly, differentiated THP-1 cells in 12-well plates were infected with HIV-1 for 48h and MTT assay reagent was added. Cells were incubated for 4h, lysed in isopropanol and the optical density measured.
Western blots and immunoprecipitations
Cells were washed in PBS and lysed with buffer containing 0.2% NP40 and protease inhibitor cocktail (Roche). 50 μg of protein was loaded and separated on a 12.5% SDS-polyacrylamide gel and transferred to nitrocellulose. The membrane was probed overnight at 4°C in 5% milk in PBS/Tween 20 (0.05%). After washing with PBS/Tween, the blot was probed with appropriate HRP-conjugated secondary antibody for 1 h at room temperature and stained with Femto supersignal. Actin was used as a loading control. Where indicated, Western blots were quantified with the ImageJ software. For immunoprecipitations, transfected 293T cells or differentiated THP-1 cells were lysed with buffer containing 0.2% Nonidet P-40 with protease and phosphatase inhibitors for 1 h, followed by centrifugation to remove cell debris. For Cyclin L2/DCAF1 interaction in 293T cells, we added Bafilomycin A1(Sigma) to stabilize the levels of DCAF1. Co-immunoprecipitations involving overexpression of Cyclin L2 and SAMHD1 required expression of more SAMHD1 to account for its degradation by Cyclin L2). Supernatants were incubated for overnight with the immuoprecipitating antibody at 4°C. The immune complexes were captured with protein G–agarose beads (EMD Biosciences) for 1–2h or overnight at 4°C. Immunoprecipitates were washed three times with PBS in 0.1%NP40, eluted with SDS-PAGE sample buffer for 5 min at 100°C, and subjected to immunoblot analysis with appropriate antibodies. Phost-tag immunoblots were performed according to manufacturer’s instructions (Wako Chemicals). Phos tag gel is able to separate proteins into phosphorylated (higher molecular weight) and unphosphorylated portions without the need for phospho-antibodies.
Immunofluorescence
Cells on coverslips were fixed for 10 min with 1% paraformaldehyde, washed twice with PBS, and permeabilized with 0.1% Triton X-100. Blocking with 1% BSA in PBST was done for 30mins at room temperature. Primary antibodies were incubated overnight after which cells were washed and secondary antibody staining with appropriate Alexa Fluorescent antibodies (Molecular Probes) done for 30mins. Secondary antibody controls were routinely performed and showed no staining pattern without primary antibodies. Images were taken and processed on Nikon Epifluorescense microscope. Single cells were imaged to show vesicular colocalization more clearly.
Quantification of HIV-1 total DNA
Differentiated THP-1 cells were infected with HIV-1 for 24 hrs. DNaseI treatment of virus was done for 1 hour prior to infection. Total DNA was isolated using a kit from Qiagen. Quantitative PCR was performed using gag primers. Quantitation of DNA present was done using a standard curve and subtraction of values obtained from non-infected cells and normalized to control. Actin was used as internal control.
Supplementary Material
Figure S1 (related to figures 1 and 3)
(A) THP-1 cells stably expressing the indicated or control shRNA were differentiated with PMA for 48h and infected with HIV-1Luc. After 2 days, cells were lysed for luciferase luminescence. Average of two independent experiments are shown. (B) Western blot showing SAMHD1 knockdown by shRNA in differentiated THP-1 cells. (C) Differentiated THP-1 cells were infected with HIV-1Luc for 2 h. Cells were washed vigorously three times, lysed and p24 ELISA performed on the cell lysates.
Figure S2 (related to figure 2)
(A) Cyclin L2 and DCAF1 show partial colocalization in the nucleus. HeLa cells were transfected with myc-Cyclin L2 and stained for myc and endogenous DCAF1. Blue staining in the top panel represents DAPI (B) THP-1 cells were lysed and immunoprecipitation performed with Cyclin L2 antibody and Western blots probed for Cyclin L2, DCAF1 and DDB1.
Figure S3 (related to figure 3). Cyclin L2 does not affect HIV replication in HeLa and 293T cells
(A) 293T cells were transfected with control or siRNA to Cyclin L2 for 48h and WB performed. (B) 293T cell were transfected with control or Cyclin L2 siRNA. After 48h, cells were infected with HIV-1luc and viral replication determined by luciferase luminescence in cellular lysates (C) 293T cells were transfected with control or Cyclin L2 plasmid and Western blot performed after 48h. Antibodies to epitope tag (myc) and Cyclin L2 on the same blot are shown. (D) 293T cells transfected with Cyclin L2 or control plasmid for 48h were infected with HIV-1luc and viral replication measured by luciferase in cellular lysates (E) HeLa cells were transfected with control or siRNA to Cyclin L2 for 48h and WB performed (F) HeLa cells were transfected with control or Cyclin L2 siRNA. After 48h cells were infected with HIV-1luc and viral replication determined by luciferase luminescence in cellular lysates. Data indicate means; error bars indicate ±SEM (n≥3).
Figure S4 (related to figure 4)
Cyclin L2 shows significant nuclear colocalization with SAMHD1. HeLa cells were transfected with SAMHD1 overnight and immunostained for SAMHD1 and endogenous Cyclin L2.
Figure S5 (related to Figure 5)
(A) 293T cells were transfected with increasing concentrations of SAMHD1 and 1ug of Cyclin L2 for 48h and the expression levels of the two proteins determined by Western blot. (B) Stable THP-1 cells expressing control or Cyclin L2 shRNA were differentiated for 48h in PMA and WB performed on a Phostag gel for SAMHD1 levels.
Acknowledgments
This work was supported by National Institutes of Health Grant R21 AI093175 to LR. GBK is a Harold Amos Scholar from the Roberts Woods Johnson Foundation.
Footnotes
Authors Contributions
GBK, LR and XC came up with the concept of the paper and designed the experiments. GBK, XC and RR performed the experiments. GBK and LR wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, Gronenborn AM, Skowronski J. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem. 2012;287:12550–12558. doi: 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler OT, Keppler OT. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med. 2012 doi: 10.1038/nm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandariz-Nunez A, Valle-Casuso JC, White TE, Laguette N, Benkirane M, Brojatsch J, Diaz-Griffero F. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology. 2012;9:49. doi: 10.1186/1742-4690-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua J, Deretic V. Mycobacterium tuberculosis reprograms waves of phosphatidylinositol 3-phosphate on phagosomal organelles. J Biol Chem. 2004;279:36982–36992. doi: 10.1074/jbc.M405082200. [DOI] [PubMed] [Google Scholar]
- Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 2013;3:1036–1043. doi: 10.1016/j.celrep.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Dale RC, Gornall H, Singh-Grewal D, Alcausin M, Rice GI, Crow YJ. Familial Aicardi-Goutieres syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am J Med Genet A. 2010;152A:938–942. doi: 10.1002/ajmg.a.33359. [DOI] [PubMed] [Google Scholar]
- deGraaf GK, Hekerman P, Spelten O, Herrmann A, Packman LC, Bussow K, Muller-Newen G, Becker W. Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: phosphorylation by DYRK1A and colocalization with splicing factors. J Biol Chem. 2004;279:4612–4624. doi: 10.1074/jbc.M310794200. [DOI] [PubMed] [Google Scholar]
- DeLucia M, Mehrens J, Wu Y, Ahn J. HIV-2 and SIVmac accessory virulence factor Vpx down-regulates SAMHD1 enzyme catalysis prior to proteasome-dependent degradation. J Biol Chem. 2013;288:19116–19126. doi: 10.1074/jbc.M113.469007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregoso OI, Ahn J, Wang C, Mehrens J, Skowronski J, Emerman M. Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 2013;9:e1003496. doi: 10.1371/journal.ppat.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, Pfander B. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014;28:1604–1619. doi: 10.1101/gad.240515.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Wei W, Wei Z, Liu X, Evans SL, Yang W, Wang H, Guo Y, Zhao K, Zhou JY, Yu XF. Identification of critical regions in human SAMHD1 required for nuclear localization and Vpx-mediated degradation. PLoS One. 2013;8:e66201. doi: 10.1371/journal.pone.0066201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann A, Fleischer K, Czajkowska H, Muller-Newen G, Becker W. Characterization of cyclin L1 as an immobile component of the splicing factor compartment. FASEB J. 2007;21:3142–3152. doi: 10.1096/fj.07-8377com. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011b;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer S, Wolf C, Konig N, Staroske W, Guck J, Hausler M, Luksch H, Nguyen LA, Kim B, Alexopoulou D, Dahl A, Rapp A, Cardoso MC, Shevchenko A, Lee-Kirsch MA. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Bregnard C, Hue P, Basbous J, Yatim A, Larroque M, Kirchhoff F, Constantinou A, Sobhian B, Benkirane M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell. 2014;156:134–145. doi: 10.1016/j.cell.2013.12.011. [DOI] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Wang TS, Li XY, Li N, Huang DZ, Chen Q, Ba Y. Overexpression of cyclin L2 induces apoptosis and cell-cycle arrest in human lung cancer cells. Chin Med J (Engl) 2007;120:905–909. [PubMed] [Google Scholar]
- Liou LY, Haaland RE, Herrmann CH, Rice AP. Cyclin T1 but not cyclin T2a is induced by a post-transcriptional mechanism in PAMP-activated monocyte-derived macrophages. J Leukoc Biol. 2006;79:388–396. doi: 10.1189/jlb.0805429. [DOI] [PubMed] [Google Scholar]
- Liou LY, Herrmann CH, Rice AP. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J Virol. 2002;76:10579–10587. doi: 10.1128/JVI.76.21.10579-10587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou LY, Herrmann CH, Rice AP. Human immunodeficiency virus type 1 infection induces cyclin T1 expression in macrophages. J Virol. 2004;78:8114–8119. doi: 10.1128/JVI.78.15.8114-8119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyer P, Trembley JH, Grenet JA, Busson A, Corlu A, Zhao W, Kocak M, Kidd VJ, Lahti JM. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: influence of cyclin L isoforms on splice site selection. J Biol Chem. 2008;283:7721–7732. doi: 10.1074/jbc.M708188200. [DOI] [PubMed] [Google Scholar]
- Olivetta E, Federico M. HIV-1 Nef protects human-monocyte-derived macrophages from HIV-1-induced apoptosis. Exp Cell Res. 2006;312:890–900. doi: 10.1016/j.yexcr.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Cohen EA. Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol. 2012;2:755–763. doi: 10.1016/j.coviro.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D. SAMHD1: a new gene for CLL. Blood. 2014;123:951–952. doi: 10.1182/blood-2013-12-545384. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008;4:e1000059. doi: 10.1371/journal.ppat.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St GC, de SS, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, Wu L. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology. 2012;9:105. doi: 10.1186/1742-4690-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St GC, de SS, Hach JC, White TE, Diaz-Griffero F, Yount JS, Wu L. Identification of Cellular Proteins Interacting with the Retroviral Restriction Factor SAMHD1. J Virol. 2014;88:5834–5844. doi: 10.1128/JVI.00155-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno F, Shiota H, Miyaura M, Yoshida A, Sakurai A, Tatsuki J, Koyama AH, Akari H, Adachi A, Fujita M. Vpx and Vpr proteins of HIV-2 up-regulate the viral infectivity by a distinct mechanism in lymphocytic cells. Microbes Infect. 2003;5:387–395. doi: 10.1016/s1286-4579(03)00042-x. [DOI] [PubMed] [Google Scholar]
- Wei W, Guo H, Han X, Liu X, Zhou X, Zhang W, Yu XF. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell Microbiol. 2012b;14:1745–1756. doi: 10.1111/j.1462-5822.2012.01835.x. [DOI] [PubMed] [Google Scholar]
- Welbourn S, Dutta SM, Semmes OJ, Strebel K. Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J Virol. 2013;87:11516–11524. doi: 10.1128/JVI.01642-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe. 2013;13:441–451. doi: 10.1016/j.chom.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li N, Wang C, Yu Y, Yuan L, Zhang M, Cao X. Cyclin L2, a novel RNA polymerase II-associated cyclin, is involved in pre-mRNA splicing and induces apoptosis of human hepatocellular carcinoma cells. J Biol Chem. 2004;279:11639–11648. doi: 10.1074/jbc.M312895200. [DOI] [PubMed] [Google Scholar]
- Zhu C, Gao W, Zhao K, Qin X, Zhang Y, Peng X, Zhang L, Dong Y, Zhang W, Li P, Wei W, Gong Y, Yu XF. Structural insight into dGTP-dependent activation of tetrameric SAMHD1 deoxynucleoside triphosphate triphosphohydrolase. Nat Commun. 2013;4:2722. doi: 10.1038/ncomms3722. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Gong J, Yang R, Sheng Y, Zhou L, Kong X, Cao K. Inhibition of proliferation and differentiation and promotion of apoptosis by cyclin L2 in mouse embryonic carcinoma P19 cells. Biochem Biophys Res Commun. 2009;390:451–457. doi: 10.1016/j.bbrc.2009.09.089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to figures 1 and 3)
(A) THP-1 cells stably expressing the indicated or control shRNA were differentiated with PMA for 48h and infected with HIV-1Luc. After 2 days, cells were lysed for luciferase luminescence. Average of two independent experiments are shown. (B) Western blot showing SAMHD1 knockdown by shRNA in differentiated THP-1 cells. (C) Differentiated THP-1 cells were infected with HIV-1Luc for 2 h. Cells were washed vigorously three times, lysed and p24 ELISA performed on the cell lysates.
Figure S2 (related to figure 2)
(A) Cyclin L2 and DCAF1 show partial colocalization in the nucleus. HeLa cells were transfected with myc-Cyclin L2 and stained for myc and endogenous DCAF1. Blue staining in the top panel represents DAPI (B) THP-1 cells were lysed and immunoprecipitation performed with Cyclin L2 antibody and Western blots probed for Cyclin L2, DCAF1 and DDB1.
Figure S3 (related to figure 3). Cyclin L2 does not affect HIV replication in HeLa and 293T cells
(A) 293T cells were transfected with control or siRNA to Cyclin L2 for 48h and WB performed. (B) 293T cell were transfected with control or Cyclin L2 siRNA. After 48h, cells were infected with HIV-1luc and viral replication determined by luciferase luminescence in cellular lysates (C) 293T cells were transfected with control or Cyclin L2 plasmid and Western blot performed after 48h. Antibodies to epitope tag (myc) and Cyclin L2 on the same blot are shown. (D) 293T cells transfected with Cyclin L2 or control plasmid for 48h were infected with HIV-1luc and viral replication measured by luciferase in cellular lysates (E) HeLa cells were transfected with control or siRNA to Cyclin L2 for 48h and WB performed (F) HeLa cells were transfected with control or Cyclin L2 siRNA. After 48h cells were infected with HIV-1luc and viral replication determined by luciferase luminescence in cellular lysates. Data indicate means; error bars indicate ±SEM (n≥3).
Figure S4 (related to figure 4)
Cyclin L2 shows significant nuclear colocalization with SAMHD1. HeLa cells were transfected with SAMHD1 overnight and immunostained for SAMHD1 and endogenous Cyclin L2.
Figure S5 (related to Figure 5)
(A) 293T cells were transfected with increasing concentrations of SAMHD1 and 1ug of Cyclin L2 for 48h and the expression levels of the two proteins determined by Western blot. (B) Stable THP-1 cells expressing control or Cyclin L2 shRNA were differentiated for 48h in PMA and WB performed on a Phostag gel for SAMHD1 levels.