

Abstract
The PYRIN domain (PYD) is a protein-protein interaction domain, which belongs to the death domain fold (DDF) superfamily. It is best known for its signaling function in innate immune responses and particularly in the assembly of inflammasomes, which are large protein complexes that allow the induced proximity-mediated activation of caspase-1 and subsequently the release of pro-inflammatory cytokines. The molecular mechanism of inflammasome assembly was only recently elucidated and specifically requires PYD oligomerization. Here we discuss the recent advances in our understanding of PYD signaling and its regulation by PYD-only proteins.
Keywords: PYRIN domain, inflammasome, inflammation, Nod-like receptor, NLR, AIM2-like receptor, ALR, PYRIN domain-only protein, POP, PYDC
Introduction
The PYD and other DDF domains are crucial for assembling oligomeric multi-protein signaling platforms in response to pathogens or danger signals, which facilitate activation of caspases, proteolysis of their substrates and subsequently a biological response. Caspases are a family of cysteine-dependent aspartate-directed endoproteases that not only play an important role in immune responses but also in the maintenance of homeostasis, by controlling apoptotic, necrotic and pyroptotic cell death as well as pro-inflammatory cytokine processing and release [1, 2]. They are produced as inactive zymogens (pro-caspases) and each family member recognizes a unique and specific sequence. Caspases can be separated into apoptotic and inflammatory caspases including human caspases-2, -3, -6, -7, -8, -9, -10 or caspases-1, -4, -5, 12 respectively. The apoptotic subgroup can further be divided into initiator and executioner caspases. While initiator caspases are activated by dimerization, executioner caspases are activated by cleavage from initiator caspases. Inflammatory caspases are activated similar as initiator caspases and require oligomerization, which is facilitated by caspase recruitment to oligomeric activation platforms that allow caspase activation through an induced proximity mechanism [3–6]. Protein-protein interaction domains of the DDF superfamily are encoded in proteins involved in innate immunity, inflammation and apoptosis and usually mediate homotypic interactions within their respective four subfamilies: the PYRIN domain (PYD), the caspase recruitment domain (CARD), the death effector domain (DED) and the death domain (DD) [7–9]. All initiator caspases contain either a CARD or a DED, which allow them to be recruited into high molecular weight signaling platforms responsible for caspase activation. Caspase-8 and -10 are activated within the DISC complex downstream of TNF family death receptors (DRs), caspase-2 is activated within the PIDDosome and caspase-1 is activated within the inflammasome downstream of pattern recognition receptors (PRRs) belonging to the Nod-like receptor (NLR) or AIM2-like receptor (ALR) family. In general, upon activation of receptors containing a DDF, adaptor proteins, which usually encode 2 different DDFs, are recruited. While one of their DDF binds through homotypic interactions to the receptor, the other one will interact homotypically with the CARD or DED of pro-caspases. However, some complexes don’t need an adaptor since the DDF of the activated receptor can directly recruit caspases. Specifically, upon binding of cytochrome c to APAF1, its CARD domain interacts with the CARD of pro-caspase-9, which gets activated in the apoptosome [10, 11]. Further, the DD and DED containing adaptor protein FADD is recruited to activated DRs through homotypic DD interactions and binds homotypically to the DED-containing caspase-8 and -10, where they are activated within the DISC complex. Also, the DD and CARD-containing adaptor RAIDD is linked to the genotoxic stress sensor PIDD through homotypic DD interactions and binds homotypically to the CARD-containing pro-caspase-2, which is activated within the PIDDosome [12, 13]. Finally, the inflammasome is assembled via the PYD and CARD-containing adaptor protein ASC, which links the CARD-containing pro-caspase-1 to upstream PYD-containing infection and tissue stress sensing NLRs and ALRs [14]. Besides caspase activation, complexes assembled by DDF interactions are also involved in the transcriptional regulation of inflammatory and apoptotic genes, through interferon regulatory factors (IRFs), NF-κB and MAPK signaling pathways. Intriguingly, the CARD of pro-caspase-4, -5, and -11 has been postulated as a microbial pattern recognition domain by directly binding lipopolysaccharide (LPS) from Gram-negative bacteria [15], thus expanding the DDF function from merely mediating homotypic protein interactions to also directly sensing the signal inducing DDF complex formation – in this case activation of inflammatory caspases as part of the LPS-sensing non-canonical inflammasome [16, 17].
A common structural feature of DDFs is their organization into a six α-helical bundle, but variations in length and direction of helices exist. Furthermore, sequence similarities are limited among DDFs, even within the same subfamily, and specificity of protein interaction is largely mediated by surface features - primarily by charged and hydrophobic pockets [7]. Furthermore, several examples of posttranslational modifications of DDFs have been reported, including disulfide bonds, phosphorylation ubiquitin binding and ubiquitination [18–26].
The PYD was the fourth member to be included in the death domain fold (DDF) superfamily of protein-protein interaction domains [8, 27–32]. Although, it has been linked to apoptosis, NF-κB and type I interferon signaling, it is best known for its essential role in inflammasome assembly.
The inflammasome
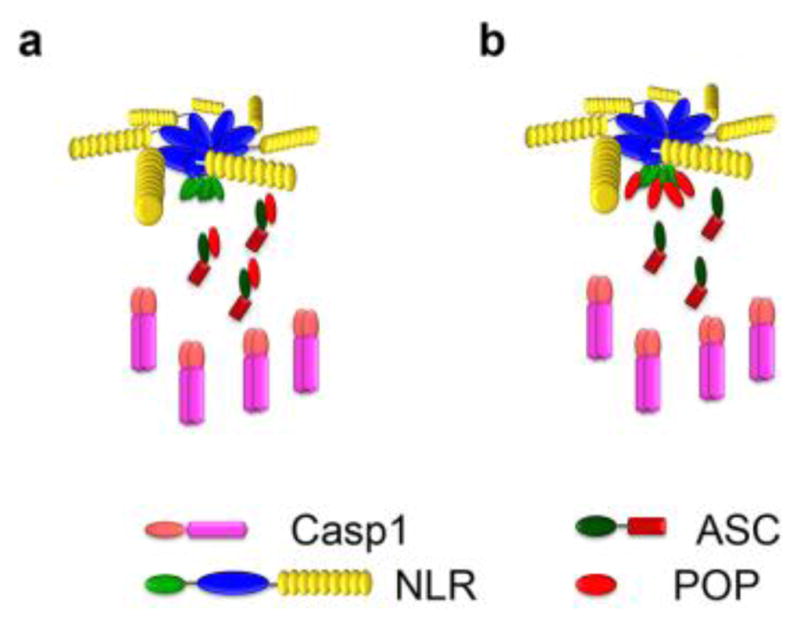
The innate immune system is the first line of defense against infection and tissue damage. This system depends on the specific production and release of soluble mediators, including cytokines, chemokines and interferons to activate and recruit immune cells. Germline-encoded PRRs, which sense extracellular and intracellular pathogen-associated molecular patterns (PAMPs) and self-derived damage-associated molecular patterns (DAMPs) or danger signals, are key components of immune responses culminating in pathogen clearance and wound healing. A core system for monitoring the cytosol is the inflammasome, which is a macromolecular protein complex promoting the proteolytic maturation and release of the key pro-inflammatory cytokines interleukin (IL)-1β and IL-18 and the induction of pyroptotic cell death [14]. This process depends on the activation of pro-caspase-1, which together with human caspases-4, -5 and -12 and mouse caspases-11 and -12 represent the inflammatory caspases [33]. While the mechanism leading to activation of caspases-4, -5 (or -11) and -12 are less well understood, mouse caspase-11 is activated in response to cytosolic delivery of LPS or Gram-negative bacteria that escape the phagosome through the non-canonical inflammasome pathway [17, 34–36]. A similar mechanism is utilized by human caspases-4 and -5 [14, 37, 38]. Any upstream macromolecular protein complex involved in their activation is elusive, but pro-caspase-4, -5, and -11 directly bind LPS through their CARDs, which promotes oligomerization and activation, thus no adaptors and upstream sensors may be necessary [15]. However, activation of caspase-1 occurs through induced proximity-mediated dimerization by the macromolecular inflammasomes [14]. Within inflammasomes, sensory PRRs, in particular members of the cytosolic ALR and NLR families, bridge to the activation of pro-caspase-1 via the adaptor protein ASC, whereby the PRRs recruit ASC through PYD-PYD interaction and ASC then links to pro-caspase-1 through CARD-CARD interaction. Thus, the PYD has a central role in assembly of this ternary complex.
The PYRIN domain
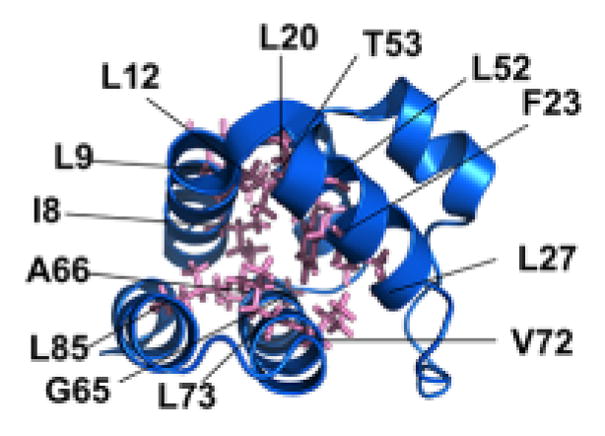
The PYD was originally discovered in the protein Pyrin (Marenostrin, MEFV) [39, 40] and was initially referred to as DAPIN, PAAD and has been shown by structure predictions and homology modeling to acquire the 6 antiparallel α-helical arrangement packed into a Greek key topology characteristic for the DDF [8, 27, 29, 30, 32]. The PYD is an approximately 90 amino acid residue motif and contrary to other DDFs, it is exclusively present at the amino terminal end of proteins. It is encoded in 23 human genes and at least 31 mouse genes (Figure 1). The encoded proteins largely function as inflammasome-activating PRRs, adaptor proteins or negative regulators (Table 1). PYDs are formed around a highly conserved hydrophobic core, which stabilizes the overall structure to which residues from α1, α2, α4, α5 and α6 as well as the α1–2 linker contribute (Figure 2). The surface displays a highly bipolar electrostatic surface patch distribution, which provides the basis for specific PYD-PYD interactions and consequently, protein-protein interaction. Thus, PYD-PYD interactions are primarily mediated by charged and hydrophobic residues. All PYDs from NLRs, ASC, Pyrin and POP1 contain conserved charged surface patches with two acidic patches formed by residues in the α2 and α3 helices and basic patches formed by residues in the α1 and α4 helices. This is evident from the available NMR and crystal structures for the PYDs of NLRP1, NLRP3, NLRP4, NLRP7, NLRP10, NLRP12, NLRP14, AIM2, and POP1 (Figure 3) [18, 41–49]. However, a key observation is that PYDs display a shorter α3 helix than DDs, DEDs and CARDs [9]. Consequently, the α2–α3 loop region in the PYD is extended compared to other DDFs, but the sequence and length of this loop is highly variable among PYDs, likely enabling distinct binding specificities (Figure 4). In some cases, such as NLRP1, the region corresponding to the α3 helix even presents as an unstructured flexible loop [42]. The α3 helix is the most diverse region among PYD structures and is considered to play a crucial role in the specificity of PYD-PYD interactions. Below, we summarize some of the features of individual PYD, for which the NMR or crystal structure has been determined.
Figure 1.
Domain architecture of human and mouse PYD-containing (a) NLRPs, (b) ALRs, and (c) regulatory proteins. The number of the amino acid residues of the longest isoform is indicated.
Table 1.
Human PYD proteins are listed and their function is indicated in relation to the activation of inflammasomes, NF-κB or interferon.
| Gene Name | Alias | Inflammasome | NF-kB | IFN |
|---|---|---|---|---|
| NLRP1 | NAC; CARD7; CIDED; NALP1; SLEV1; DEFCAP; PP1044; VAMAS1; CLR17.1; DEFCAP-L/S | Activate | No | Inhibit |
| NLRP2 | NBS1; PAN1; NALP2; PYPAF2; CLR19.9 | Activate | Inhibit | |
| NLRP3 | FCU; MWS; FCAS; Cias1; Mmig1; NALP3; Pypaf1; AII/AVP; AGTAVPRL | Activate | No | |
| NLRP4 | CT58; PAN2; RNH2; NALP4; PYPAF4; CLR19.5 | Inhibit | Inhibit | |
| NLRP5 | MATER; Op1; NALP5; PAN11; PYPAF8; CLR19.8 | No | No | |
| NLRP6 | AVR; NAVR; PAN3; NALP6; PYPAF5; CLR11.4; NAVR/AVR | Activate | Inhibit | Activate |
| NLRP7 | HYDM; PAN7; NALP7; NOD12; PYPAF3; CLR19.4 | Activate | No | No |
| NLRP8 | PAN4; NALP8; NOD16; CLR19.2 | |||
| NLRP9 | NOD6; NALP9; PAN12; CLR19.1 | No | ||
| NLRP10 | NOD8; PAN5; PYNOD; NALP10; CLR11.1 | Inhibit | Activate | |
| NLRP11 | NOD17; PAN10; NALP11; PYPAF6; CLR19.6 | |||
| NLRP12 | RNO; PAN6; RNO2; FCAS2; NALP12; PYPAF7; CLR19.3 | Activate | Inhibit | Activate |
| NLRP13 | NOD14; PAN13; NALP13; CLR19.7 | Inhibit | No | |
| NLRP14 | NOD5; PAN8; GC-LRR; NALP14; CLR11.2 | Activate | ||
| ALRs | ||||
| AIM2 | PYHIN4 | Yes | Activate | Inhibit |
| IFI16 | PYHIN2; IFNGIP1 | Yes | Activate | Activate |
| MNDA | PYHIN3 | No | Activate | |
| IFIX | PYHIN1 | No | Activate | |
| Other | ||||
| Pyrin | Marenostrin, MEFV | Activate/Inhibit | ||
| adaptors | ||||
| ASC | PYCARD, ASC, CARD5, TMS1 | |||
| regulators | ||||
| POP1 | PYDC1, ASC2, ASCI, PYC1 | Activate | Inhibit | |
| POP2 | PYDC2 | Inhibit | Inhibit | |
| POP3 | PYDC3 | Inhibit | No | Activate |
| POP4 | NLRP2P | No | Inhibit |
Figure 2.

The residues in the ASC-PYD comprising the hydrophobic core, which are necessary for the overall PYD fold, are indicated. A similar hydrophobic core is present in all PYDs.
Figure 3.
PYDs with known NMR or crystal structure are shown as Ribbon diagram and Electrostatic surface representation. The left panel of each PYD (including ribbon and surface) are shown in the same orientation. The right panels of the each PYD is rotated by 180° along the vertical axis. α helices in the Ribbon diagram are shaded in orange (α1), green (α2), yellow (α3), purple (α4), cyan (α5), and magenta (α6). The electrostatic charge surfaces are displayed on a scale of -4 kT/e (red) to 4 kT/e (blue). Notable are the lack of α3 in the NLRP1-PYD, the strongly negatively charged surface in NLRP7, the fused α5–α6 helix in the NLRP14-PYD and the extended α6 helix in the AIM2-PYD. NLRP1-PYD (PDB: 1PN5) [42], NLRP3-PYD (PDB: 3QF2) [18], NLRP4-PYD (PDB: 4EWI) [46], NLRP7-PYD (PDB: 2KM6) [45], NLRP10-PYD (PDB: 2M5V), NLRP12-PYD (PDB: 2L6A) [47], NLRP14-PYD (PDB: 4N1J) [49], AIM2-PYD (PDB: 3VD8) [50], ASC-PYD (PDB: 1UCP) [43], POP1-PYD (PDB: 2HM2) [44, 98, 99].
Figure 4.

CLUSTAL-W alignment of (a) human and (b) mouse PYDs, showing acidic residues (red), basic residues (blue) and hydrophobic residues (grey). α-helices determined by NMR or crystal structure are boxed in black and the Hin-200 specific motifs are boxed in yellow and marked by a yellow line on top, as identified earlier [50].
ASC-PYD
The ASC-PYD conforms to the common PYD electrostatic surface patch distribution with side chains of residues in and adjacent to the α2 and α3 helices providing the positively charged surface and side chains of residues in and adjacent to the α1 and α4 helices providing the negatively charged surface area [43]. Since ASC functions as the inflammasome adaptor, the most likely scenario is that the acidic surface patch of the ASC-PYD packs against the basic surface patch of the PYD in NLRs and ALRs and vice versa, following a type I interaction mode.
NLRP1-PYD
The most striking difference of the NLRP1-PYD compared to other PYDs and even other DDFs is the lack of a structured α3 helix, which instead contains a flexible and unstructured loop [42]. Lys26 within the α2 helix is highly conserved in PYDs and it has been proposed to function in stabilizing the conformation of the α3 helix and the connecting loop through its side chains and therefore to contribute indirectly to PYD-PYD interactions [50]. Interestingly, NLRP1 and NLRP6 show non-conserved residues at this position, which may potentially contribute to the non-structured α3 helix in NLRP1. The NLRP6-PYD structure is not yet available to confirm this hypothesis. One scenario could be that this unstructured region might adopt an α helical conformation upon complex formation or alternatively, through an intra-protein interaction [42]. NLRP1 interacts with ASC through PYD-PYD association to assemble an inflammasome [14]. As discussed above, the α3 helix is predicted to be necessary for binding to the acidic surface of ASC and an inducible-structured α3 helix could provide a regulatory step for this interaction. Alternatively, the acidic surface of the NLRP1-PYD provided by the α1 and α4 helices may interface with the basic patch formed by the α2 and α3 helices in the ASC-PYD. However, several reports also showed that ASC is not necessary for the NLRP1 inflammasome in human and mice, in particular since mouse NLRP1s lack the PYD, but may enhance its activity, and therefore the relevance of such an interaction is not clear [51–53].
NLRP3-PYD
The crystal structure of the NLRP3-PYD revealed an overall similar structure as observed for other PYDs, including the hydrophobic core, but also a second hydrophobic surface exposed patch formed by residues in the α3–α4 loop and the α4 helix, which has been proposed to be conserved among ASC-PYD-interacting PYDs [18]. This second hydrophobic patch is required for formation and stabilization of the α3 helix, which anchors it to the α2 helix and is also found in the PYD of NLRP7 [45]. However, the orientation of the α2 helix and the α2–α3 loop displays opposite orientation compared to the NLRP1-PYD and the NLRP7-PYD. The charged regions are located on the entire peripheral surface of the NLRP3-PYD and may allow interaction with several ASC-PYD molecules, similar to the PIDDosome and DISC [18]. The propensity of PYDs to dimerize has been well established using biochemical approaches, although most structural studies revealed a predominantly monomeric PYD structure. However, the crystal structure of the NLRP3-PYD revealed a symmetric dimer, whereby residues in the α2 and α3 helices interacted with the same residues on the other molecule by electrostatic association [18]. The biological relevance of dimerization, in particular considering the observation that the majority existed as a monomer, has not been experimentally established, but may contribute to inflammasome assembly. Another unique aspect of the NLRP3-PYD is the presence of disulfide bonds formed between the conserved Cys8 and Cys108, located in the α1 helix and the linker connecting the PYD to the NACHT domain. As NLRP3 inflammasome activation has been linked to reactive oxygen species (ROS) and redox signaling [54, 55], an intriguing possibility therefore is that the disulfide bond formation may have a role in linking redox signaling to conformational changes in the NLRP3-PYD and inflammasome assembly.
NLRP4-PYD
In contrast to the PYD structures determined for other PYD-containing proteins, NLRP4 does not contribute to inflammasome signaling, but has been reported as a negative regulator for autophagy, NF-κB signaling and production of type I interferons [56–58]. Accordingly, it did not interact with ASC [46]. As reported for the NLRP3-PYD, also the NLRP4-PYD forms a homodimer, as determined by crystallography, while it remains a monomer by NMR analysis [46]. Residues within the α1, α4, α5 and α6 helices form electrostatic and hydrophobic interactions with residues in the α5, α4, α1 and α6 helices, respectively. Several features are unique in the NLRP4-PYD. It possesses the shortest α1 helix, but the longest α2 and α6 helices. Also, the α3 helix is stabilized by eight hydrophobic residues - more than in any other known PYD [46]. Surprisingly, the electrostatic surface potential of the NLRP4-PYD is most similar to that of POP1 and ASC-PYD, showing a strong electrostatic dipole with extensive positively charged α2 and α3 helices and a weaker negatively charged α1 and α4 helical region. However, in spite of the similar arrangement of charged residues, the NLRP4-PYD did not interact with the ASC-PYD, further emphasizing that a complementary charged surface is not sufficient to promote PYD-PYD interactions [46]. While it is feasible that in vivo conditions may be more conducive for such an interaction, there is currently no supporting functional evidence available and this study was performed with a solubility enhancing tagged ASC-PYD. In addition, the NLRP4-PYD displays a second positively charged surface patch from residues of the α5 and α6 helices, which do not participate in the common type I binding mode, but have been reported for homotypic type II binding modes occurring in large protein platforms, such as the PIDDosome and the inflammasome [8, 9, 13, 59].
NLRP7-PYD
The NLRP7-PYD is similar to other PYDs, except NLRP1-PYD. In addition to the hydrophobic core a second hydrophobic surface patch is exposed, stabilizing the α2–α3 loop and the α3 helix. In the NLRP7-PYD this cluster is formed by six residues compared to the four and three residues in the ASC-PYD and POP1-PYD [18, 45]. However, also six residues form the second hydrophobic patch in the NLRP3-PYD and even eight are present in the NLRP4-PYD [18, 46]. Also, in the NLRP7-PYD, this feature is important for stabilizing the α3 helix and reducing flexibility in this region [45]. Contrary to above described PYDs, the NLRP7-PYD differs in the electrostatic surface, in spite of being most similar to POP1. The NLRP7-PYD lacks some of the positively charged surface in the α2 and α3 helices, which are replaced with hydrophobic residues. The negatively charged patches formed by the α1 and α4 helices are retained, as well as the surface exposed hydrophobic patches formed by the α2–α3 loop and the α4 helix. These residues are conserved among proteins interacting with the ASC-PYD, for example the NLRP3-PYD [18]. Due to reduced positive charge in the α2 and α3 helices, it has been suggested that the NLRP7-PYD may not interact with the ASC-PYD and this study failed to observe binding in vitro between both PYDs [45]. However, an interaction in vivo between NLRP7 and ASC and the formation of an NLRP7 inflammasome has been shown in macrophages [60], indicating that an ASC-PYD/NLRP7-PYD interaction indeed occurs. Although, a distinct binding mode may utilized compared to the binding of ASC/POP1- and ASC/NLRP3-PYDs, where negatively charged and hydrophobic residues are sufficient for this interaction. However, the in vitro conditions may not correctly reflect the conditions necessary for interaction, such as salt and in particular pH, as usually acidic pH solutions are required to purify soluble recombinant PYDs, due to their tendency to oligomerize and become insoluble. The crystal structure of the NLRP4-PYD for instance, revealed that PYD-PYD interaction is not solely dependent on complementary charged surfaces alone [46]. Since the negative charged α1 and α4 helices are conserved, this area may interact with the positive face of the ASC-PYD.
NLRP12-PYD
NLRP12 has been linked to inflammasome activation in response to Yersinia pestis infection [61]. However, there are no biochemical data available to indicate if PYD-mediated interactions with ASC are important for this function. Also, NLRP12 has been established as a negative regulator for pro-inflammatory cytokine production through inhibiting the non-canonical NF-κB activation [62–66]. The NLRP12-PYD is shorter compared to other PYDs and features an only 3 residue short 310 helix instead of an α3 helix. Also, the α2–α3 loop has a 2 residue deletion. Consequently, the α2–α3 loop in the NLRP12-PYD is slightly more flexible than in the NLRP7-PYD but more rigid than in the NLRP1-PYD, which lacks the α3 helix completely. In addition, instead of 6 hydrophobic residues stabilizing the α3 helix and the α2–α3 loop within the NLRP7-PYD, there are only three hydrophobic residues within the NLRP12-PYD [47]. The closest structural similarity is found with the POP1- and ASC-PYDs, which also show complementary basic and acidic patches in the α2/3 and α1/4 helices, and also display an exposed hydrophobic patch in the α3 helix. NMR spectroscopy did not observe any binding to the ASC-PYD [47], but an earlier study discovered binding of NLRP12 with the NF-κB, apoptosis and ubiquitination regulator FAF1 [67]. FAF1 contains a DD interacting domain (FID) forming a patch of three α helices, which interacts with residues within the α2 and α3 helices of the NLRP12-PYD [47]. Other non-homotypic PYD interactions have been determined by biochemical assays, including the ASC-PYD and ASC-CARD, ASC-PYD and Bax, ASC-PYD and caspase-8-DED, NLRP3-PYD and GBP5 [68–71]. However, this is the first non-homotypic PYD interaction supported by structural data, which may explain the NF-κB-inhibitory activity of NLRP12.
NLRP14-PYD
NLRP14 has not been linked to inflammasome activation and does not interact with ASC and its function is elusive. However, its crystal structure revealed a unique dimerization mechanism, which has not yet been observed for other PYDs. The NLRP14-PYD organization results from an asymmetric tetrameric arrangement of symmetric dimers [49]. Dimerization is a consequence of a fusion of α-helices 5 and 6, which are organized as an extended stem helix, which provides the dimerization interface of this novel interaction mode, referred to as open PYD conformation [49]. This open PYD conformation is stabilized by a hydrophobic motif within the stem helix, which also stabilizes the classical 6 α-helical closed PYD conformation, and has been referred to as hydrophobic core switching element [49]. Responsible for either conformation is a charge bridge forming a conformational regulation element (CRE) within α-helices 2 and 6 [49]. The presence of a positive charge bridging two opposing negative charges (intact charge bridge) has been predicted to promote the closed PYD conformation, while lack of a charge, as found in NLRP7, 10 and 14 promotes an open PYD conformation, although this has been experimentally validated only for the NLRP14-PYD [49]. Such a symmetric dimer would extend the acidic charged surface patch, compared to a monomer and may provide a novel interaction surface for PYD or other domains [49].
AIM2-PYD
PYHIN (ALR)-PYDs share many of the conserved PYD key residues, including primarily hydrophobic core residues, but contain a shorter α2–α3 loop and display unique motifs within the amino and carboxyl termini [50] (Figure 4). The crystal structure of the AIM2-PYD revealed that specific residues within the α2 helix contribute to the distinct surface charge and hydrophobicity of the PYD [50]. Furthermore, it has the longest α6 helix of all PYDs. Together these features may contribute to AIM2 specific functions. Most of the charged AIM2-PYD residues are surface exposed, but show a unique distribution. Acidic residues are dominant in α1–α2 helices and basic residues are present in α5–α6 helices [50]. Adjacent to the acidic α2 are surface exposed hydrophobic and non-charged residues (Phe27 and Phe28), some of which are unique to AIM2 proteins and conserved among AIM2 proteins from different species [50]. Intriguingly, a similar hydrophobic patch is necessary for the DED-mediated binding of FADD and pro-caspase-8 [72, 73]. Docking studies proposed that the acidic residues of the α2 helix and the adjacent hydrophobic residues of the AIM2-PYD are interacting with the ASC-PYD, which was confirmed by mutagenesis of the acidic and hydrophobic residues, which are both required for binding of the ASC and AIM2 PYDs in vitro [50]. Other interaction modes could occur between the acidic α1 helix of AIM2-PYD and the basic α2–α3 helices of the ASC-PYD and/or the basic α5 helix of the AIM2-PYD could interact with the acidic α4 of the ASC-PYD. Another important consideration is that AIM2 exists in an auto-inhibited conformation, where the acidic α2 helix of the PYD binds to the same basic patch on the HIN200 domain that binds DNA, implicating that DNA binding releases this auto-inhibited state of AIM2. Therefore, PYDs appear to not only trigger signaling through binding to adaptor proteins, but are also maintaining an auto-inhibited AIM2 conformation [74].
Structural differences within ASC-PYD interacting PYDs determined variations in the PYD-PYD interaction mechanism, which likely has implication for inflammasome assembly and consequently on innate immune responses. Although the importance of the PYD in inflammasome assembly is well established, the underlying molecular assembly mechanism for this ternary inflammasome complex has only been recently delineated, revealing a unique oligomerization mechanism responsible for the induced proximity activation of caspase-1.
Molecular Mechanism of PYD-mediated inflammasome formation
Over expression of PYD-containing proteins, in particular ASC, frequently showed a filamentous and punctate (speck) distribution within cells, reminiscent to death filaments reported earlier for DDFs. The punctate structures show a spherical organization with a hollow core, indicating that ASC-PYD self oligomerization occurs [75, 76]. It is also well established that interaction of the ASC-PYD with the PYD of ALRs and NLRs is required for inflammasome formation. As indicated above, the hydrophobic core is conserved and is essential for the overall fold of the PYD, which together with charged residues contributes to PYD-PYD interactions. In agreement with this concept, a comprehensive structure function analysis, utilizing ASC-PYD self polymerization into filaments as a readout, demonstrated that several conserved hydrophobic residues (I8, L12, L15, L20, F23, L27, L52, L56, L68, V72, M76), which are buried within the core and are required for its stabilization, are also necessary for ASC-PYD polymerization [77]. Furthermore, charged residues within α helices 2, 3 and 4 (K21, L25, K26, P40, R41, D48 and D51), which form the opposite charged surface patches, are required for ASC-PYD self polymerization. Interestingly, these did not include residues from α helix 1, thus excluding it from participating in ASC-PYD self polymerization [77]. However, a second study utilizing biochemical binding studies confirmed these residues, but also revealed that E13 within α helix 1 is necessary for the interaction [78]. Thus, ASC-PYD self polymerization requires the positive charged K21 (α2) and R41 (α3) on one molecule and E13 (α1), D48 (α3–α4 loop) and D51 (α4) on the other molecule (Figure 5) [78]. Moreover, helices α1 and α4 are perpendicular to helices α2 and α3, where the C-terminus of α1 and the N-terminus of α4 of one PYD are in contact with α2 and α3 of a second PYD and thus, the PYD homodimer is oriented so that the two binding motifs of each subunit are capable of engaging in additional interactions, thus explaining multi-protein complexes [78]. Docking studies revealed that ASC-PYDs can form an α-helical arrangement consisting of three molecules per turn, which can be polymerized into a filamentous structure, resembling structures observed upon over-expression of the ASC-PYD [78]. This model also predicted that the CARD of ASC is folded away from the PYD by its flexible linker and will therefore not interfere with the PYD-mediated ASC polymerization. Recently, ASC-PYD polymerization was analyzed from reconstituted inflammasomes in HEK293 cells by cryoelectron microscopy (EM) at near atomic resolution, which revealed that the filaments are actually comprised of helical polymers, as previously proposed [59]. Composite models of the NMR and EM structures revealed a model of an ASC-PYD filament with a hollow core and inner and outer diameters of approximately 20 and 90 angstroms, respectively [59].
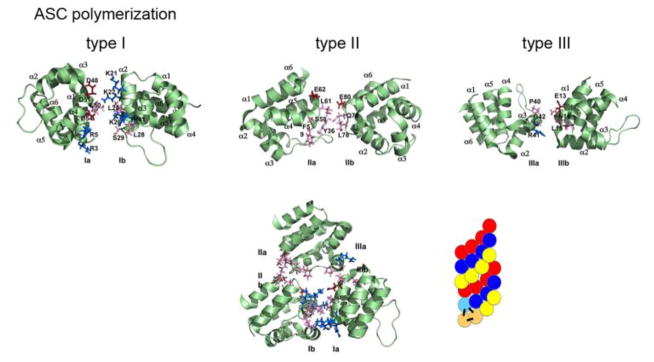
Figure 5.

The three types of ASC PYD interactions (type I, type II, and type III) [8, 59, 78, 107]. Negative charge is marked in red, positive charge is marked in blue, and neutral residues are marked in pink. The ASC-PYD polymer is comprised of three-start helical strands with C3 symmetry, resulting from type 1 interaction in each of the strands and type 2 and type 3 interactions between the strands [59, 78]. A model of the polymer is indicated, showing the interactions in the start helices on the bottom left [59].
Three electrostatic interaction modes have been described for DDFs [79]. Type I interaction of DDs, DEDs and CARDs [13, 72, 80, 81], and type II/III interaction for DDs [13, 80, 82]. The type I interaction is mediated by α1/α4 helices (type Ia) of one PYD with α2/α3 helices (type Ib) from a second PYD, as initially described above. The type II interaction is mediated by corners of the α4/α5 helices (type IIa) of one PYD with the corner of the α5/α6 helices (type IIb) of another PYD. The type III interaction is mediated by the corner of the α2/α3 helices (type IIIa) of one PYD with the corner of the α1/α2 helices (type IIIb) of another PYD. The ASC-PYD filament is comprised of three-start helical strands with a C3 symmetry with type I interactions in each of the strands and type II and type III interactions between the strands (Figure 5) [59]. Thus, remarkably, the short α3 helix found in PYDs together with the long flexible α2–α3 loop participates in all three interactions and not surprisingly, these regions showed conformational changes upon polymerization compared to monomeric ASC-PYD [59]. Mutation of key residues for each of the three interaction modes abrogated filament formation, further demonstrating that all three modes of interaction are required for ASC-PYD polymerization [59].
The key residues required for ASC-PYD polymerization are conserved in the PYDs of ASC and NLRP3, although the NLRP3-PYD has a lower positive surface charge, and indeed there is an overlapping binding surface mediating ASC-PYD self polymerization and binding to the NLRP3-PYD, as E13, K21, R41, D48, D51 were also necessary for binding to NLRP3-PYD [78]. However, based on the presence of two opposing binding motifs present in each PYD, ASC-PYD polymerization and interaction with the NLRP3-PYD (or other PYDs from NLRs and ALRs) are not mutually exclusive, and docking studies predicted a scenario of binding of an ASC-PYD homodimer with two NLRP3-PYDs [78]. Reconstituted inflammasomes from HEK293 cells were also used to dissect the ASC-PYD polymerization in the presence of the NLRP3-PYD and AIM2-PYD by electron microscopy, as these upstream PRRs are necessary to trigger inflammasome assembly. Immunogold labeling revealed that the AIM2-PYD and NLRP3-PYD localize at one end of the ASC-PYD-formed filaments and that the ASC-PYD forms the main part of the filament [59]. This result suggests that the AIM2-PYD and NLRP3-PYD nucleate polymerization of the ASC-PYD filaments. NLRP3 and AIM2 oligomerize upon activation, but their PYDs alone were less potent than a fragment of the PYD and the NACHT domain of NLRP3 (NLRP3ΔLRR) or full length NLRP3 (a ratio of ASC:NLRP3 of 15:1 showed the greatest effect). The presence of ATP, which causes oligomerization of the NACHT, further enhanced the polymerization efficiency. Transmission electron microscopy using in vitro reconstituted NLRP1 inflammasomes earlier revealed a pentameric and heptameric NLRP complex upon activation [53]. Similarly, dsDNA, which is the ligand for AIM2, enhanced the polymerization efficiency of AIM2. Thus, activation and subsequent oligomerization of upstream NLRs and ALRs dramatically increases the nucleation efficiency for ASC-PYD polymerization. Furthermore, activation of a limited number of receptor molecules promotes the nucleation of many more ASC molecules, thus dramatically amplifying the response (Figure 6a). In fact, one molecule of NLRP3ΔLRR could polymerize 1600 ASC-PYD molecules. A higher molar ratio of NLRP3 was required to achieve the same level of oligomerization compared to NLRP3ΔLRR, likely due to the autoinhibition of NLRP3 by its LRRs [59]. This model can likely be adapted to other ASC-interacting oligomerized PYDs, such as IFI16, which also forms filaments upon binding dsDNA [83].
Figure 6.
Model of inflammasome assembly. NLRP3 or AIM2 (not shown) nucleate ASC-PYD polymerization. (a) The polymeric assembly of ASC and caspase-1 and (b) a cross section of the polymer has been modified from Lu and colleagues [59]. (c) An alternative model, observed in macrophages after Salmonella infection, modified from Man and colleagues [93]. In this model, the sensors are composed of a complex of NLRP3 and NLRC4 (not shown).
Studies with reconstituted inflammasomes in yeast revealed the mechanism for ASC-PYD polymerization, which is similar to MAVS-CARD polymerization [84], since they both functions as a prion. A defining property of prions is their ability to convert monomeric proteins into self-perpetuating polymerized forms, which adopt an SDS-resistant oligomeric fibrous state [85–87]. Nucleation is necessary as self-polymerization is prevented by high-energy barrier. Once nucleated, monomeric prions are energetically less stable and therefore the whole cell has to either commit to the monomeric or the non-reversible polymerized prion form. ASC-PYD prion conversion is nucleated by upstream PYDs, including the NLRP3- and AIM2-PYDs. Once the prion switch was achieved in yeast, ASC converted into large particles. Furthermore, polymerized ASC-PYD could induce monomeric ASC into a self-perpetuating polymer and the prion activity would explain why macrophages usually contain one large inflammasome with all ASC present in the aggregated form [75]. Furthermore, a functional prion domain could replace the ASC-PYD in biochemical assays for caspase-1 activation, and mutants that prevent ASC polymerization also prevented its prion activity and failed to activate caspase-1 [84]. Interestingly, ASC prion formation constitutes a gain of function rather than a loss of function, which is usually observed for prions. Due to the self-amplification of a signal, the prion-aggregation mechanism enables a highly sensitive responsiveness of inflammasome assembly. Recently, the biological relevance for prion-like ASC particles was also uncovered. Subsequent to inflammasome activation polymerized ASC particles are released through pyroptosis. They are phagocytized by neighboring cells where they trigger polymerization of endogenous ASC, which results in inflammasome activation in the absence of a specific trigger and an upstream sensor [88, 89]. Hence, these particles act as danger signals, which perpetuate inflammasome responses [90]. In fact, prion-like ASC particles have been found in the serum of human Cryopyrinopathy patients, who harbor a constitutive active NLRP3 inflammasome and suffer from systemic hyper-inflammation and in the bronchoalveolar lavage fluid of patients with Chronic obstructive pulmonary disease (COPD) and pneumonia [88, 89].
The ASC-PYD is connected by a flexible linker to the ASC-CARD and analysis of the NMR structure of ASC together with electron microscopy of ASC-PYD filaments revealed that the CARD is arranged at the outside of the filaments, thus making it easily accessible for pro-caspase-1 [41, 59]. This was confirmed by reconstitution of the ternary complex and further revealed that this complex is star shaped, with a shorter ASC filament than that observed for the ASC-PYD. However, these ASC filaments acted as activation platforms for pro-caspase-1 and were able to nucleate branched filaments of pro-caspase-1 through their CARDs (Figure 6b) [59]. Similar to NLR- and ALR-mediated nucleation of PYD filaments, ASC was able to nucleate pro-caspase-1 at sub-stoichiometric ratios of 1:20, thus amplifying this response even further. The shorter ASC filaments observed in the ternary complex may be caused by the rapid nucleation of pro-caspase-1, which could draw all ASC in a complex with pro-caspase-1 [59]. As pro-caspase-1 is activated by induced proximity and trans processing upon dimerization, such pro-caspase-1 filaments provide the platform for its efficient self-activation [91]. This sequential amplification was also observed in MSU-activated macrophages. MSU initiates the assembly of the NLRP3 inflammasome and quantitative immunoblot analysis of this complex also revealed an ASC to caspase-1 ratio of at least 1:3.5 [59]. Purification of this complex revealed filamentous as well as barn of yarn-like globular structures of about 2 μm in diameter. These globular structures are reminiscent to the punctate aggregates formed after inflammasome activation in macrophages and present a hollow core [75, 76, 92]. This may be a consequence of shorter endogenous caspase-1 filaments, rather than the long filaments caused by over expression of caspase-1, which may therefore preferentially assemble punctate structures. Furthermore, cofactors and other inflammasome-associated proteins expressed in macrophages as well as posttranslational modification of endogenous inflammasome components may further promote the punctate structures. Imaging of endogenous inflammasome components in macrophages showed that all three components of this ternary complex localize to these aggregates upon inflammasome activation, which are usually present as a single punctate structure, referred to as speck or pyroptosome [75, 92]. These aggregates, rather than elongated filaments have now been shown for various inflammasome activating stimuli. Super resolution fluorescence imaging of the Salmonella-induced endogenous inflammasome structure in macrophages further suggested a concentric spatial organization, where ASC surrounded a layer of oligomerized NLRs, with a core containing caspase-1 [93]. Oligomerized NLRs act as a nucleation point for ASC inflammasome formation also in this model. However, rather than linear ASC polymerization, the consequence is a spherical structure with a core of caspase-1, implying an inverted structure (Figure 6c). The difference in these structures likely results from the over expressed ternary complex forming filaments or spherical higher order structures, while the endogenous native complex assembles into spheres. This study further proposed a highly dynamic and more complex protein platform, which also contained caspase-8 in the core and the NLR layer was composed of distinct NLRP3 and NLRC4 layers, which are both involved in sensing Salmonella [93]. Thus, the localization of caspase-1 to the core in Salmonella-induced inflammasome structures may be due to its proximity to the inner layer composed of NLRC4, which can directly bind to caspase-1 through CARD-CARD interactions.
Although, this mechanism has been shown for AIM2 and NLRP3, not all NLRPs contain the critical residues that mediate the interaction of the NLRP3-PYD with the ASC-PYD, and, they also vary in the distribution of positively and negatively charged surface patches. The charged patches within the PYDs of NLRP7, NLRP10 and NLRP12 are in similar locations than in NLRP3 and AIM2, but are less prominent and in NLRP7 and NLRP12 the positively charged cluster is interrupted by a hydrophobic patch. The NLRP1-PYD has a different structure as most NLRPs as it lacks the α3 helix, and consequently has an electrostatic surface that is considerably different. However, it still interacts with ASC [14]. Similarly, NLRP7 forms an inflammasome with ASC, suggesting a certain level of flexibility for PYD-PYD interactions [60]. Such a variation is further supported by the proposed binding mode of the NLRP14-PYD, which generates a novel binding surface upon dimerization [49].
Regulation of PYRIN domain interactions
The PYD is essential for inflammasome assembly following activation of NLRP3 and other PYD-containing PRRs and disassembly of this ternary signalling complex is required for resolution of these responses. Besides inflammasome-activating PYD containing proteins, humans also encode a family of PYD-only proteins (POPs), which are also referred to as PYD containing (PYDC) [21, 94, 95]. POPs are small, 10 to 13 kDa proteins encoded in humans, which entirely consist of a single PYD and specifically bind to other PYD proteins to interfere with essential PYD-PYD interactions. Phylogenetic tree analysis highlights the presence of a POP for each of the major human PYD branches, while these proteins are absent from mice, emphasizing species differences in the mechanism for regulating PYD-formed protein complexes (Figure 7). Moreover, differential splicing contributes to the expression of proteins with essentially only a PYD, which provides yet another layer of regulation [96].
Figure 7.
Phylogenetic tree analysis of (a) human and (b) mouse PYDs, showing POPs for each of the major PYD cluster in humans and lack of these proteins in mice. PYDs linked to inflammasome activation are highlighted in green and the POPs in red. Method: Neighbor Joining; Best Tree; tie breaking = Systematic Distance: Uncorrected (“p”), Gaps distributed proportionally.
POP1
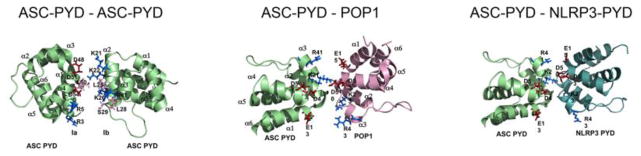
The first PYRIN-only protein, POP1, was discovered in silico due to its high identity (64%) with the ASC-PYD [21]. POP1 encodes only a PYD and is predicted to function as a negative regulator, which however, has not yet been demonstrated. The POP1 gene is located in close proximity to ASC and likely originated by exon duplication. It is highly similar to the PYD of ASC and presents 64% sequence identity. As expected from this high degree of sequence similarity, POP1 follows the typical structure of a PYD and shows almost identical electrostatic surface patches as the ASC-PYD [44]. Similar to the ASC-PYD, the electrostatic potential surface patches are divided into two sub-patches by intermediate hydrophobic amino acid residues (Figure 3) [97]. POP1 also contains the extended loop between the α2 and α3 helices, but this loop and the α3 helix revealed a greater degree of disorder than the other helices [44]. A separate study obtained a structure that differed markedly within the α2–α4 region, where the α2 and α4 helices displayed differences in orientation with an unstructured α3, reminiscent of the NLRP1-PYD [98, 99]. However, in consideration of the high sequence identity of POP1 to the ASC-PYD and its origination from ASC by exon duplication [21], this structure is less likely [44]. Based on the high structural similarity of POP1 with the ASC-PYD, they should interact with each other, similar to ASC-PYD oligomers [21]. The positive surface formed by the α2 and α3 helices of POP1 is predicted to interact with the negative surface formed by the α1 and α4 helices of the ASC-PYD. Mutagenesis experiments confirmed that the negatively charged residues of the ASC-PYD (D6, E13, D48, D54) and the positively charged residues of POP1 (K21, R41) are essential for this interaction [97]. In addition, the negative surface on POP1 interacts with the positive surface of the ASC-PYD, similar to the self polymerization of ASC-PYD [78]. The interaction between POP1 and ASC-PYD resembles a type I binding mode, similar to ASC self-association and the ASC-NLRP3-PYD interaction (Figure 8). Site directed mutagenesis of these residues abolished the interaction between POP1 and ASC PYD [78, 97]. Thus, POP1 binding and ASC self-association may be competitive, suggesting that POP1 may block the polymerization of the ASC-PYD (Figure 9a). The five charged residues critical for the interaction between the ASC-PYD and POP1 are also conserved in the NLRP3-PYD. Hence, it is possible that POP1 also binds to the NLRP3-PYD via a similar interaction mode, which may affect the nucleation of ASC-PYD polymerization (Figure 9b). However, in vitro POP1 enhanced inflammasome activity, which may be a consequence of participating in ASC-PYD polymers, since opposing negative and positive surface interaction motifs can be utilized [21]. Also, POP1 inhibited NF-κB activity by interacting with IKKα and IKKβ [21].
Figure 8.

Type I interaction between two ASC-PYDs, ASC-PYD - POP1 and ASC-PYD - NLRP3-PYD, showing the overlapping interaction motif as identified by mutagenesis [78, 97].
Figure 9.

Proposed mechanism for the function of POPs, which either may bind to (a) the ASC-PYD, as demonstrated for POP1 and POP2 [21, 100, 102], or to (b) the PYD of upstream sensors, as shown for POP2 and POP3 [100, 103].
POP2
The second POP, POP2, was discovered by two independent groups [100, 101]. POP2 is a single exon gene and encodes a 97 amino acid PYD with high similarly to the PYDs of NLRP2 and NLRP7 [100]. Also POP2 affects NF-κB and the inflammasome, the latter by binding to ASC, reminiscent of POP1 [100, 101]. While the inhibition of NF-κB occurs by a mechanism that is independent of ASC, POP2 prevents the binding of ASC to the NLRP3-PYD and NLRP1-PYD [100, 102]. Although the POP2 structure is not solved, the α1 helix was necessary and sufficient to inhibit NF-κB activation and diminished inflammasome assembly [102]. The interaction of POP2 with the ASC-PYD and inflammasome inhibition depended on three acidic residues (E6, D8, E16). However, these residues did not affect the NF-κB inhibitory function of POP2. Hence, the function of the α1 helix in inflammasome and NF-κB inhibition are mechanistically uncoupled [102].
POP3
Also POP3 is a single exon gene. It is located in the HIN-200 gene cluster between IFI16 and PYHIN1, which also encodes AIM2 and MNDA [103]. POP3 is the largest of the three POPs and contains a PYD with 5 α-helices instead of 6 predicted α-helices. POP3 is most identical to the PYD of AIM2 as well as other members of the HIN-200 family, and shares the unique sequence motifs found in HIN-200 PYDs (Figure 4) [103]. Consequently, POP3 binds to the PYDs of AIM2 and IFI16 and prevents their interaction with ASC. However, unlike POP1 and POP2, POP3 does not interact with the ASC-PYD [103]. Thus, it inhibits ALR inflammasomes by binding to the sensor, rather than the adaptor (Figure 9b). POP3 is so far also the only POP for which an inflammasome inhibitory role has been demonstrated on an endogenous level and in vivo [103].
POP4
Recently, a fourth POP, POP4, has been predicted, which is encoded by the pseudogene NLRP2P and shares 80% identity with POP2 [104]. Although the α1 helix of POP4 is highly similar to the one in POP2, it lacks the acidic residues at positions 6 and 16 that are present in POP2, which prevents POP4 from binding to ASC and inhibiting inflammasomes [104]. However, it retained its inhibitory effect on NF-κB activation. POP4 is significantly shorter than other POPs and with only 45 amino acids, it contains only the α1 and α2 helices and thus, is unlikely to adopt the characteristic PYD fold. Structural studies will be required to investigate if this truncated protein can adopt a partial PYD fold.
Molecular mechanism for inflammatory disease
As discussed above, the NLRP1-PYD displays an unstructured flexible loop replacing the α3 helix found in other PYDs and DDFs [42]. Familial Mediterranean fever (FMF) is the prototypical autoinflammatory disease and is caused by hereditary mutations in Pyrin [39, 40]. One of these mutations, R42W, is found inside the PYD and this residue corresponds to A43 in the NLRP1-PYD and is located within the flexible loop that normally forms the α3 helix. This residue correlates to residue R41 in ASC and R41W mutation results in deficient ASC-PYD filament formation, consistently with the notion that the α3 helix is involved in all three PYD interaction types [77]. Similarly, a single point mutation inside the Fas-DD, V238N, results in the hyperproliferative mutation (Faslpr). V238 is located in the linker connecting the α2–α3 helices and is required for stabilizing the α3 helix through hydrophobic interactions. Hence, mutation of V238N caused the unfolding of the α3 helix and the loss of FADD binding [105]. The subsequent defect in apoptosis of this mutation caused systemic autoimmune disease in mice [106]. Thus, mutations that abrogate or alter PYD-PYD interactions likely cause excessive or impaired responses and are linked to human disease, further emphasizing the importance of well balanced PYD signaling for the maintenance of homeostasis.
Future Perspectives
Although the PYD and inflammasomes have been identified more than a decade ago, the molecular mechanism responsible for the PYD-mediated formation of high molecular inflammasome complexes has only recently been elucidated, and provided evidence for a unique prion-like polymerization mechanism for inflammasome assembly. These data support a model where NLRs and ALRs are responsible for the initial nucleation of ASC, but the actual activation of caspase-1 is then mediated by oligomerized ASC. However, alternative models have also been proposed. Self-oligomerization of the ASC-PYD may rather act to prevent interaction with upstream PRRs and thereby inhibit inflammasome activity [89]. The models of filamentous inflammasome assembly are based on over expression studies and therefore more in depth studies are needed to consolidate these findings with the spherical inflammasomes (“specks”) observed on endogenous level in macrophages, similar to the structures induced by Salmonella. The hypothesis that such spheres indeed represent higher order - or more organized filamentous structures is a possible explanation, but additional endogenous inflammasomes will need to be imaged. Furthermore, accessory proteins present in endogenous inflammasomes, may affect the overall structure. Co-crystal structures of ASC with NLRs or ALRs or of ternary complexes with caspase-1 will ultimately be necessary to support and refine current models of inflammasome assembly. In addition quantitative co-purification of endogenous inflammasome proteins is required to solve the underlying stoichiometry. Such studies will also provide insights into the mechanism by which POPs can inhibit this PYD-driven process. Lastly, although inflammasome assembly is the best understood process for the PYD, not all PYD containing proteins are involved in inflammasome formation, but have been linked to signaling affecting NF-κB, type I interferons, autophagy and apoptosis. However, the precise role of homotypic and potentially heterotypic PYD interactions in these processes, including any prion-like PYD polymerization, is largely unknown.
Acknowledgments
This work was supported by the National Institutes of Health (GM071723, AI099009 and AR064349 to C.S., and AR057532 to A.D.) and the American Heart Association (12GRNT12080035 to C.S).
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Ashkenazi A, Salvesen G. Regulated Cell Death: Signaling and Mechanisms. Annu Rev Cell Dev Biol. 2014;30:337–356. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- 2.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Current Opinion in Cell Biology. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Chang DW, Ditsworth D, Liu H, Srinivasula SM, Alnemri ES, Yang X. Oligomerization is a general mechanism for the activation of apoptosis initiator and inflammatory procaspases. J Biol Chem. 2003;278:16466–16469. doi: 10.1074/jbc.C300089200. [DOI] [PubMed] [Google Scholar]
- 6.Park HH. Structural features of caspase-activating complexes. Int J Mol Sci. 2012;13:4807–4818. doi: 10.3390/ijms13044807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park HH, Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber CH, Vincenz C. The death domain superfamily: a tale of two interfaces? Trends Biochem Sci. 2001;26:475–481. doi: 10.1016/s0968-0004(01)01905-3. [DOI] [PubMed] [Google Scholar]
- 9.Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci. 2011;36:541–552. doi: 10.1016/j.tibs.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y. Apoptosome: The Cellular Engine for the Activation of Caspase-9. Structure. 2002;10:285–288. doi: 10.1016/s0969-2126(02)00732-3. [DOI] [PubMed] [Google Scholar]
- 11.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–432. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 12.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 13.Park HH, Logette E, Raunser S, Cuenin S, Walz T, et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinon F, Burns K, Tschopp J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-1b. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 15.Shi J, Zhao Y, Wang Y, Gao W, Ding J, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014 doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 16.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 18.Bae JY, Park HH. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J Biol Chem. 2011;286:39528–39536. doi: 10.1074/jbc.M111.278812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin BN, Wang C, Willette-Brown J, Herjan T, Gulen MF, et al. IKKα negatively regulates ASC-dependent inflammasome activation. Nat Commun. 2014;5:4977. doi: 10.1038/ncomms5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang T-H, Park JH, Park HH. Novel disulfide bond-mediated dimerization of the CARD domain was revealed by the crystal structure of CARMA1 CARD. PLoS One. 2013;8:e79778. doi: 10.1371/journal.pone.0079778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stehlik C, Krajewska M, Welsh K, Krajewski S, Godzik A, Reed JC. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated NF-kB and pro-Caspase-1 regulation. Biochem J. 2003;373:101–113. doi: 10.1042/BJ20030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castanier C, Zemirli N, Portier A, Garcin D, Bidère N, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ver Heul AM, Fowler CA, Ramaswamy S, Piper RC. Ubiquitin regulates caspase recruitment domain-mediated signaling by nucleotide-binding oligomerization domain-containing proteins NOD1 and NOD2. J Biol Chem. 2013;288:6890–6902. doi: 10.1074/jbc.M112.413781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ver Heul AM, Gakhar L, Piper RC, Subramanian R. Crystal structure of a complex of NOD1 CARD and ubiquitin. PLoS One. 2014;9:e104017. doi: 10.1371/journal.pone.0104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng W, Sun L, Jiang X, Chen X, Hou F, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinon F, Hofmann K, Tschopp J. Curr Biol. 2001;11:R118–R120. doi: 10.1016/s0960-9822(01)00056-2. [DOI] [PubMed] [Google Scholar]
- 28.Bertin J, DiStefano PS. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death Differ. 2000;7:1273–1274. doi: 10.1038/sj.cdd.4400774. [DOI] [PubMed] [Google Scholar]
- 29.Pawłowski K, Pio F, Chu Z, Reed JC, Godzik A. PAAD - a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem Sci. 2001;26:85–87. doi: 10.1016/s0968-0004(00)01729-1. [DOI] [PubMed] [Google Scholar]
- 30.Staub E, Dahl E, Rosenthal A. The DAPIN family: a novel domain links apoptotic and interferon response proteins. Trends Biochem Sci. 2001;26:83–85. doi: 10.1016/s0968-0004(00)01717-5. [DOI] [PubMed] [Google Scholar]
- 31.Fairbrother WJ, Gordon NC, Humke EW, O’Rourke KM, Starovasnik MA, et al. The PYRIN domain: a member of the death domain-fold superfamily. Protein Sci. 2001;10:1911–1918. doi: 10.1110/ps.13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 33.Nadiri A, Wolinski MK, Saleh M. The inflammatory caspases: key players in the host response to pathogenic invasion and sepsis. J Immunol. 2006;177:4239–4245. doi: 10.4049/jimmunol.177.7.4239. [DOI] [PubMed] [Google Scholar]
- 34.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 37.Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, et al. A Critical Role for Human Caspase-4 in Endotoxin Sensitivity. J Immunol. 2014;193:335–343. doi: 10.4049/jimmunol.1303424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer HD. Caspase-4 Is Required for Activation of Inflammasomes. The Journal of Immunology. 2012;188:1992–2000. doi: 10.4049/jimmunol.1101620. [DOI] [PubMed] [Google Scholar]
- 39.Consortium TFF. A candidate gene for familial mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 40.Consortium TIF. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 41.de Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC) J Biol Chem. 2009;284:32932–32941. doi: 10.1074/jbc.M109.024273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiller S, Kohl A, Fiorito F, Herrmann T, Wider G, et al. NMR Structure of the Apoptosis- and Inflammation-Related NALP1 Pyrin Domain. Structure. 2003;11:1199–1205. doi: 10.1016/j.str.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Liepinsh E, Barbals R, Dahl E, Sharipo A, Staub E, Otting G. The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J Mol Biol. 2003;332:1155–1163. doi: 10.1016/j.jmb.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 44.Natarajan A, Ghose R, Hill JM. Structure and dynamics of ASC2, a pyrin domain-only protein that regulates inflammatory signaling. J Biol Chem. 2006;281:31863–31875. doi: 10.1074/jbc.M605458200. [DOI] [PubMed] [Google Scholar]
- 45.Pinheiro AS, Proell M, Eibl C, Page R, Schwarzenbacher R, Peti W. Three-dimensional Structure of the NLRP7 Pyrin Domain: Insight into PYRIN-PYRIN-mediated effector domain signaling in innate immunity. Journal of Biological Chemistry. 2010;285:27402–27410. doi: 10.1074/jbc.M110.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eibl C, Grigoriu S, Hessenberger M, Wenger J, Puehringer S, et al. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry. 2012;51:7330–7341. doi: 10.1021/bi3007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pinheiro AS, Eibl C, Ekman-Vural Z, Schwarzenbacher R, Peti W. The NLRP12 pyrin domain: structure, dynamics, and functional insights. J Mol Biol. 2011;413:790–803. doi: 10.1016/j.jmb.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Do KH, Park HH. Crystallization and preliminary X-ray crystallographic studies of cPOP1. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69:292–294. doi: 10.1107/S1744309113002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eibl C, Hessenberger M, Wenger J, Brandstetter H. Structures of the NLRP14 pyrin domain reveal a conformational switch mechanism regulating its molecular interactions. Acta Crystallogr D Biol Crystallogr. 2014;70:2007–2018. doi: 10.1107/S1399004714010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the Absent in Melanoma 2 (AIM2) Pyrin Domain Provides Insights into the Mechanisms of AIM2 Autoinhibition and Inflammasome Assembly. J Biol Chem. 2013;288:13225–13235. doi: 10.1074/jbc.M113.468033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012;37:1009–1023. doi: 10.1016/j.immuni.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti T-D, Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 54.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 55.Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. J Leukoc Biol. 2012;92:951–958. doi: 10.1189/jlb.0512265. [DOI] [PubMed] [Google Scholar]
- 56.Cui J, Li Y, Zhu L, Liu D, Songyang Z, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13:387–395. doi: 10.1038/ni.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fiorentino L, Stehlik C, Oliveira V, Ariza ME, Godzik A, Reed JC. A novel PAAD-containing protein that modulates NF-kappa B induction by cytokines tumor necrosis factor-alpha and interleukin-1beta. J Biol Chem. 2002;277:35333–35340. doi: 10.1074/jbc.M200446200. [DOI] [PubMed] [Google Scholar]
- 58.Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011;186:1646–1655. doi: 10.4049/jimmunol.1001654. [DOI] [PubMed] [Google Scholar]
- 59.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, et al. An NLRP7-Containing Inflammasome Mediates Recognition of Microbial Lipopeptides in Human Macrophages. Immunity. 2012;36:464–476. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vladimer GI, Weng D, Paquette SWM, Vanaja SK, Rathinam VAK, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol. 2007;178:1256–1260. doi: 10.4049/jimmunol.178.3.1256. [DOI] [PubMed] [Google Scholar]
- 63.Williams KL, Taxman DJ, Linhoff MW, Reed W, Ting JP-Y. Cutting edge: Monarch-1: a pyrin/nucleotide-binding domain/leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J Immunol. 2003;170:5354–5358. doi: 10.4049/jimmunol.170.11.5354. [DOI] [PubMed] [Google Scholar]
- 64.Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem. 2005;280:39914–39924. doi: 10.1074/jbc.M502820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaki MH, Vogel P, Malireddi RKS, Body-Malapel M, Anand PK, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. 2011;20:649–660. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, et al. NLRP12 Suppresses Colon Inflammation and Tumorigenesis through the Negative Regulation of Noncanonical NF-κB Signaling. Immunity. 2012;36:742–754. doi: 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kinoshita T, Kondoh C, Hasegawa M, Imamura R, Suda T. Fas-associated factor 1 is a negative regulator of PYRIN-containing Apaf-1-like protein 1. International Immunology. 2006;18:1701–1706. doi: 10.1093/intimm/dxl104. [DOI] [PubMed] [Google Scholar]
- 68.Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, et al. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell Biol. 2004;6:121–128. doi: 10.1038/ncb1087. [DOI] [PubMed] [Google Scholar]
- 69.Masumoto J, Dowds TA, Schaner P, Chen FF, Ogura Y, et al. ASC is an activating adaptor for NF-kappaB and caspase-8-dependent apoptosis. Biochem Biophys Res Commun. 2003;303:69–73. doi: 10.1016/s0006-291x(03)00309-7. [DOI] [PubMed] [Google Scholar]
- 70.Masumoto J, Taniguchi S, Sagara J. Pyrin N-terminal homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem Biophysical Res Commun. 2001;280:652–655. doi: 10.1006/bbrc.2000.4190. [DOI] [PubMed] [Google Scholar]
- 71.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang JK, Wang L, Zheng L, Wan F, Ahmed M, et al. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell. 2005;20:939–949. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaufmann M, Bozic D, Briand C, Bodmer J-L, Zerbe O, et al. Identification of a basic surface area of the FADD death effector domain critical for apoptotic signaling. FEBS Lett. 2002;527:250–254. doi: 10.1016/s0014-5793(02)03146-0. [DOI] [PubMed] [Google Scholar]
- 74.Jin T, Perry A, Jiang J, Smith P, Curry J, et al. Structures of the HIN Domain: DNA Complexes Reveal Ligand Binding and Activation Mechanisms of the AIM2 Inflammasome and IFI16 Receptor. Immunity. 2012;36:561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol. 2009;182:3173–3182. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 77.Moriya M, Taniguchi S, Wu P, Liepinsh E, Otting G, Sagara J. Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry. 2005;44:575–583. doi: 10.1021/bi048374i. [DOI] [PubMed] [Google Scholar]
- 78.Vajjhala PR, Mirams RE, Hill JM. Multiple binding sites on the ASC pyrin domain allow self-association and interaction with NLRP3. J Biol Chem. 2012;287:41732–41743. doi: 10.1074/jbc.M112.381228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weber CH, Vincenz C. A docking model of key components of the DISC complex: death domain superfamily interactions redefined. FEBS Lett. 2001;492:171–176. doi: 10.1016/s0014-5793(01)02162-7. [DOI] [PubMed] [Google Scholar]
- 80.Lin S-C, Lo Y-C, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399:549–557. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 82.Xiao T, Towb P, Wasserman SA, Sprang SR. Three-Dimensional Structure of a Complex between the Death Domains of Pelle and Tube. Cell. 1999;99:545–555. doi: 10.1016/s0092-8674(00)81542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci U S A. 2014;111:E62–E71. doi: 10.1073/pnas.1313577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cai X, Chen J, Xu H, Liu S, Jiang Q-X, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156:1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Halfmann R, Lindquist S. Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J Vis Exp. 2008;17 doi: 10.3791/838. pii: 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hou F, Sun L, Zheng H, Skaug B, Jiang Q-X, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 88.Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 89.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Broderick L, Hoffman HM. cASCading specks. Nat Immunol. 2014;15:698–700. doi: 10.1038/ni.2942. [DOI] [PubMed] [Google Scholar]
- 91.Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem. 2009;284:6546–6553. doi: 10.1074/jbc.M806121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Man SM, Hopkins LJ, Nugent E, Cox S, Glück IM, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014;111:7403–7408. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stehlik C, Dorfleutner A. COPs and POPs: Modulators of Inflammasome Activity. J Immunol. 2007;179:7993–7998. doi: 10.4049/jimmunol.179.12.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Le HT, Harton JA. Pyrin- and CARD-only Proteins as Regulators of NLR Functions. Front Immunol. 2013;4:275. doi: 10.3389/fimmu.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bryan NB, Dorfleutner A, Kramer SJ, Yun C, Rojanasakul Y, Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J Inflamm. 2010;7:23. doi: 10.1186/1476-9255-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Srimathi T, Robins SL, Dubas RL, Chang H, Cheng H, et al. Mapping of POP1-binding site on pyrin domain of ASC. J Biol Chem. 2008;283:15390–15398. doi: 10.1074/jbc.M801589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Espejo F, Green M, Preece NE, Assa-Munt N. J Biomol NMR. 2002;23:151–152. doi: 10.1023/a:1016398403157. [DOI] [PubMed] [Google Scholar]
- 99.Espejo F, Patarroyo ME. Determining the 3D structure of human ASC2 protein involved in apoptosis and inflammation. Biochem Biophys Res Commun. 2006;340:860–864. doi: 10.1016/j.bbrc.2005.12.087. [DOI] [PubMed] [Google Scholar]
- 100.Dorfleutner A, Bryan NB, Talbott SJ, Funya KN, Rellick SL, et al. Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect Immun. 2007;75:1484–1492. doi: 10.1128/IAI.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bedoya F, Sandler LL, Harton JA. Pyrin-only protein 2 modulates NF-kappaB and disrupts ASC:CLR interactions. J Immunol. 2007;178:3837–3845. doi: 10.4049/jimmunol.178.6.3837. [DOI] [PubMed] [Google Scholar]
- 102.Atianand MK, Harton JA. Uncoupling of Pyrin-only protein 2 (POP2)-mediated dual regulation of NF-κB and the inflammasome. J Biol Chem. 2011;286:40536–40547. doi: 10.1074/jbc.M111.274290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Khare S, Ratsimandresy RA, de Almeida L, Cuda CM, Rellick SL, et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol. 2014;15:343–353. doi: 10.1038/ni.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Porter KA, Duffy EB, Nyland P, Atianand MK, Sharifi H, Harton JA. The CLRX. 1/NOD24 (NLRP2P) pseudogene codes a functional negative regulator of NF-κB, pyrin-only protein 4. Genes Immun. 2014;15:392–403. doi: 10.1038/gene.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eberstadt M, Huang B, Olejniczak ET, Fesik SW. The lymphoproliferation mutation in Fas locally unfolds the Fas death domain. Nature Structural Biology. 1997;4:983–985. doi: 10.1038/nsb1297-983. [DOI] [PubMed] [Google Scholar]
- 106.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 107.Park HH. Structural analyses of death domains and their interactions. Apoptosis. 2011;16:209–220. doi: 10.1007/s10495-010-0571-z. [DOI] [PubMed] [Google Scholar]