

Summary
The host immune system functions constantly to maintain chronic commensal and pathogenic organisms in check. The consequences of these immune responses on host physiology are as yet unexplored, and may have long-term implications in health and disease. We show that chronic viral infection increased epithelial turnover in multiple tissues, and the antiviral cytokines Type I interferons (IFNs) mediates this response. Using a murine model with persistently elevated Type I IFNs in the absence of exogenous viral infection, the Irgm1-/- mouse, we demonstrate that Type I IFNs act through non-epithelial cells, including macrophages, to promote increased epithelial turnover and wound repair. Downstream of Type I IFN signaling, the highly related IFN-stimulated genes Apolipoprotein L9a and b activate epithelial proliferation through ERK activation. Our findings demonstrate that the host immune response to chronic viral infection has systemic effects on epithelial turnover through a myeloid-epithelial circuit.
Introduction
Epithelial cells create lining and duct structures that are associated with many organs in the body (Blanpain et al., 2007). One major function of these structures is to provide a first-line barrier against the environment (Ashida et al., 2012). A central component of this protection is cellular turnover, a highly regulated process of shedding and regeneration of differentiated cells that at the same time maintains barrier integrity. Loss of balance in this process results in the eventual loss of barrier function. This concept is most obvious in the intestine, which normally has a high epithelial turnover rate (Kuhnert et al., 2004; Lee et al., 2009). At homeostasis, each distinct epithelial structure has a different rate of turnover, but the determinants of these unique rates are poorly understood (Pellettieri and Sanchez Alvarado, 2007).
Turnover rates must also be capable of modulation in response to injury so that wound repair can efficiently occur. Damaged epithelial cells must be shed and rapidly replaced with new cells generated by self-renewing stem cells (Blanpain et al., 2007). Examples of damage that can alter turnover rates include irradiation, malnutrition, and bacterial and parasitic infection (Cliffe et al., 2005; Creamer, 1967; Luperchio and Schauer, 2001; Rijke et al., 1975).
The microbiome of the host is a key component of the environment that is involved in homeostasis and injury response (Packey and Ciorba, 2010; Pfefferle and Renz, 2014; Scales and Huffnagle, 2013). The virome is a relatively unexplored component of the microbiome and is the complex collection of chronic viruses within a given host (Virgin, 2014; Virgin et al., 2009). The role of these chronic viral infections in epithelial cellular turnover has not been previously addressed.
Type I interferons (IFNs) are a candidate for mediating systemic alterations in response to viruses. They are a family of innate immune cytokines that are produced as a result of viral and other infections (Muller et al., 1994). They include multiple IFNαs, IFNβ, and other subtypes (Pestka et al., 2004). Once expressed and secreted from the cell, Type I IFNs all bind to a common Type I IFN receptor, IFNAR, which is expressed on most cell types (de Weerd et al., 2007). Despite sharing a single receptor, Type I IFNs can have different cellular effects depending on the IFN subtype, the cell type, and the context (i.e. additional cytokine signals) (Ivashkiv and Donlin, 2014; Thomas et al., 2011). Upon ligand binding, Janus kinases (JAKs) that are constitutively associated with IFNAR phosphorylate the receptor and the signaling transducers and activators of transcription (STATs) molecules (de Weerd et al., 2007). Upon phosphorylation, STATs form complexes that translocate to the nucleus to induce the expression of hundreds of interferon stimulated genes (ISGs).
ISGs can be involved in a plethora of cellular processes including apoptosis, transcriptional activation and repression, modulation of immune cells and cytokine expression, protein degradation, and post-transcriptional regulation of gene expression (de Veer et al., 2001). However, the functions of many ISGs are yet to be discovered, as it is difficult to use viral infection models to distinguish the effects of Type I IFNs and ISGs on host physiology from the effects of viral infection itself. To definitively show that Type I IFNs mediate an effect, loss of function studies are required, but deficiency of Type I IFNs during viral infection typically results in unhindered viral replication, alterations in the immune response, and significant morbidity and mortality. For example, loss of Type I IFN signaling during infection with murine homologues of ubiquitous chronic viruses such as herpesvirus and cytomegalovirus results in increased viral titers and mortality (Barton et al., 2005; Chong et al., 1983; Dutia et al., 1999). Here, we used two in vivo models to study the impact of Type I IFNs on host physiology: i) the Irgm1-/- mouse, that we found has persistently elevated Type I IFNs in the absence of pathogenic viral infection, and ii) injection of polyinosinic:polycytidylic acid (polyI:C), a synthetic double-stranded RNA that stimulates Type I IFN induction. We showed that chronic viral infection promoted epithelial turnover in multiple organs. We then demonstrated that Type I IFN signaling through macrophages promoted epithelial proliferation and enhanced injury repair via the ISGs Apolipoprotein L9a and b.
Results
Chronic viral infection promoted turnover of multiple epithelial organs
To test the role of chronic viral infection in the modulation of epithelial turnover, we infected C57BL/6 wild-type (WT) mice with murine cytomegalovirus (MCMV), a DNA herpesvirus homologous to highly prevalent human CMV (Virgin et al., 2009). Seven days post-infection with MCMV (commencement of the chronic phase) (Munks et al., 2006), we observed increased epithelial cell proliferation and cell death within normally low-turnover organs (i.e. kidney, liver, salivary gland, and pancreas; Figure 1A-C) (Pellettieri and Sanchez Alvarado, 2007). The continued local presence of infectious virus was not required for increased turnover, as only the salivary gland had detectable titers at this time, consistent with the known kinetics of MCMV in C57BL/6 mice (Figure S1A) (Munks et al., 2006; Sumaria et al., 2009). A homologue of chronic human herpesvirus, murine gammaherpesvirus 68 (MHV68; (Virgin et al., 2009)), also promoted epithelial proliferation 16 days post-infection in the absence of detectable titers in target tissues (Figure S1B-C). At this time point, MHV68 is entering its chronic latent phase (Tibbetts et al., 2003). However, in the small intestine, we found that while MCMV stimulated an increase in proliferation (Figure 1D-E), infection with MHV68 or a persistent strain of enteric murine norovirus (Nice et al., 2013) did not (Figure S1D-G). These results suggested that certain chronic viral infections can promote epithelial turnover in multiple organs.
Figure 1. MCMV infection promoted epithelial turnover in multiple organs.

A) Representative immunohistochemistry for Ki67 in kidney, liver, salivary gland (SG), and exocrine pancreas of mock-infected mice or mice infected with 5×104 pfu MCMV (Smith strain), 7 days (d7) after intraperitoneal (i.p.) infection. Blue bars =100μm, red bars = 50μm. Black boxes indicate insets (right-most column). Black arrowheads show examples of enumerated Ki67+ epithelial cells. B-C) Graph of the average number ± standard error of the mean (SEM) of B) Ki67+ epithelial cells per high power field (hpf; 400×) and C) TUNEL+ bodies/hpf relative to mock-infected animals. For B), approximately 25 hpf were quantified per organ and only epithelial cells were counted, and for C), approximately 50 hpf per organ were quantified. Graph of D) average crypt height and E) average number of mitotic figures ± SEM of 100 crypts in mock- and MCMV-infected mice. Lengths of crypts were measured from histological slides using ImageJ software. Mitotic figures were counted manually. For B) to E), mock-infected n=6 mice, MCMV-infected n=5 mice from 2 independent experiments (ind. expts.). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 by Student's t test for each organ. See also Figure S1.
Mice deficient in Irgm1 provided a useful model to study the effects of chronic elevated Type I IFNs
We hypothesized that Type I IFNs induced by viral infection stimulated epithelial turnover. However, we could not test this hypothesis in mice lacking the Type I interferon response because these cytokines are required for survival after infection with MCMV and MHV68 (Barton et al., 2005; Chong et al., 1983; Dutia et al., 1999). Therefore, we used a mouse model that we discovered has chronic elevated Type I IFN levels in the absence of infection, the Irgm1-/- mouse. Irgm1 is a p47 GTPase initially studied for its role in host defense against intracellular protozoans and bacteria and in autophagy (Collazo et al., 2001; Feng et al., 2004; Liu et al., 2013). Previous studies showed that Irgm1-/- mice have elevated serum Type II IFN (IFNγ) (King et al., 2011). However, Irgm1-/- mice bred in our enhanced barrier facility (Cadwell et al., 2010) did not have detectable serum IFNγ, and organ levels of IFNγ did not differ between WT and Irgm1-/- mice (Figure S2A). Instead, using a highly sensitive bioassay system (Newby et al., 2007), Type I IFNs were elevated in the serum of uninfected Irgm1-/- but not Irgm1+/- mice. Blocking antibodies specific for the Type I IFN receptor chain IFNAR1 and a pan-anti-IFNα antibody effectively blocked the anti-viral activity found in Irgm1-/- serum (Figures 2A-B and S2B-D). Type I IFNs were elevated in Irgm1-/- mice from post-natal day 7 to as late as one year (Figure S2E-F). The increased Type I IFNs were functional as ISGs such as Oas2 and Mx2 were elevated in numerous tissues of Irgm1-/- mice (Figure 2C-D). The systemic elevation of Type I IFNs in Irgm1-/- mice has not been previously reported, and provided us with a useful model to study the role of these cytokines on host physiology.
Figure 2. Irgm1-/- mice had elevated functional circulating Type I IFNs.

A) Graph of IFN activity measured in serum using a bioassay system (described in Figure S2C). IFN activity was calculated using a standard curve from recombinant murine IFNαA run concomitantly with assay samples. WT n=22 mice, Irgm1-/- n=28 mice from 5 ind. expts. B) Bars show anti-viral activity of WT and Irgm1-/- serum after administration of anti-Ifnar1, anti-pan-IFNα, and anti-IFNβ1 antibodies during the serum bioassay. WT n=4 mice, Irgm1-/- n=6 mice. C-D) Graphs of mean fold change ± SEM of D) Oas2 and E) Mx2 mRNA in Irgm1-/- tissues compared to WT tissues. N=2-9 mice per genotype per organ. *p<0.05; ** p < 0.01, ****p<0.0001 by Student's t test. See also Figure S2.
Type I IFNs promoted epithelial proliferation and turnover
To determine if Type I IFNs increased epithelial turnover, we compared WT, Irgm1-/-, and Irgm1-/-Ifnar-/- mice that lacked responsiveness to Type I IFNs but retained elevated Type I IFN serum levels (Figure S3A). We found increased epithelial proliferation and cell death in the kidney, pancreas, liver, and salivary gland of Irgm1-/- mice but not Irgm1-/-Ifnar-/- mice compared to WT mice (Figures 3A-B). The effects of Type I IFNs were not global, as proliferation levels in skeletal muscle, lung alveolar cells, and thyroid gland epithelial cells in Irgm1-/- mice were similar to controls (Figure S3B).
Figure 3. Elevated Type I IFNs in Irgm1-/- mice promoted enhanced epithelial turnover.

A) Quantification of the average number of Ki67+cells/hpf ± SEM. n=6-11 mice per genotype. B) Quantification of average number of TUNEL+ bodies per hpf ± SEM. n=5-10 mice per genotype. For A) and B), approximately 50 hpfs were examined per organ. For each organ, means with different letters are significantly different (p<0.05) by Tukey's multiple comparisons test. C) Representative histology of small intestinal crypts of WT, Irgm1-/- and Irgm1-/-Ifnar-/- mice. Red bar = 50μm. Yellow bars delineate the height of a crypt; red arrowheads mark mitotic figures. Graph of D) average crypt height and E) average number of mitotic figures (MF) ± SEM of 100 crypts in WT (n=23 mice), Irgm1-/- (n=18 mice), Irgm1-/-Ifnar-/- (n=18 mice) and Ifnar-/- (n=10 mice) mice compiled from 4 ind. expts. Bars with different letters have significantly different means (p<0.05) by Tukey's multiple comparisons test. F) Quantification of the average number ± SEM of BrdU+ cells per crypt. Mice were injected with BrdU 1h before sacrifice. The number of immunofluorescently-stained BrdU+ cells was counted over an average of 50 crypts. WT n=3 mice, Irgm1-/- n=3 mice, Irgm1-/-Ifnar-/- n=2 mice. G) Graph of the average migration rate ± SEM of small intestinal epithelial cells calculated relative to WT controls. n=100 villi/mouse evaluated in a 2-color thymidine analog experiments. WT n=11 mice, Irgm1-/- n=17 mice, Irgm1-/-Ifnar-/- n=9 mice from 4 ind. expts. For F) to G), **p<0.01, ****p<0.0001 by Tukey's multiple comparisons test. See also Figure S3.
We further examined the effect of Type I IFNs on proliferation in the intestine, where proliferation, cell death, and cell migration occur in defined areas (Creamer, 1967; Potten, 1998). Irgm1-/- but not Irgm1-/-Ifnar-/- intestines showed increased epithelial proliferation compared to controls, as assessed by crypt height, and BrdU incorporation and mitotic figure counts (Figure 3C-F). Increased proliferation was observed throughout Irgm1-/- small intestinal crypts, including regions enriched for stem cells (crypt base) and regions enriched for transit-amplifying cells (upper crypt) (Figure S3C). As an additional control, we found that intestinal epithelial proliferation in Ifnar-/- mice was not independently altered compared to WT controls (Figure 3D-E).
Turnover in the small intestine takes place, in part, via cell migration from the base of crypts that culminates with apoptosis and shedding of differentiated cells at the tips of villi (Creamer, 1967). Using a double thymidine analog labeling method (Mahoney et al., 2008), we found that epithelial cells migrated faster in Irgm1-/- mice compared to WT or Irgm1-/-Ifnar-/- mice (Figures 3G and S3D). Notably, no excess cell death of progenitor intestinal epithelial cells occurred in Irgm1-/- mouse crypts (Figure S3E). In addition, recombinant murine IFNαA did not directly induce cell death in primary intestinal epithelial progenitor cells (Miyoshi et al., 2012; Miyoshi and Stappenbeck, 2013) derived from WT, Ifnar-/- and Irgm1-/- mice, despite the capacity of these cells to respond to type I IFNs by increasing ISG expression (Figure S3F-G). These results suggested that Type I IFNs did not directly induce cell death in epithelial progenitor cells. However, it is likely that turnover-associated death of differentiated cells at villus tips was accelerated.
Elevated Type I IFNs in Irgm1-/- mice enhanced wound healing
To gauge the physiologic importance of Type I IFN-driven epithelial turnover, we evaluated wound repair. We first administered diclofenac, a non-steroidal anti-inflammatory drug (NSAID), which induces small intestinal ulcerations within 24 hours after a single i.p. dose of 60mg/kg (Ramirez-Alcantara et al., 2009). While ulcers were formed in all mice after 18 hrs, Irgm1-/- mice had fewer detectable intestinal ulcers compared to WT and Irgm1-/-Ifnar-/- mice at day 2 and 4 post-injection (Figures 4A-B and S4A), indicating that Irgm1-/- mice healed ulcerations more rapidly than controls. We also investigated colonic repair using a biopsy injury model (Seno et al., 2009). In this model, wound-associated epithelial (WAE) cells migrate over the wound bed surface to cover the injury. The source of WAE cells are wound-adjacent crypts, and decreased epithelial cell proliferation in these crypts is associated with slowed and incomplete wound healing (Miyoshi et al., 2012; Seno et al., 2009). We observed a greater distance covered by WAE cells at day 4 post-injury in wounds of Irgm1-/- mice that correlated with the percentage of proliferating cells in wound-adjacent crypts (Figure 4C-D, S4B). Taken together, these findings demonstrated that elevated epithelial proliferation in response to Type I IFNs enhanced healing after epithelial damage.
Figure 4. Irgm1-/- mice had accelerated wound healing due to Type I IFNs.

A) Graph of the average number ± SEM of ulcers per small intestine 18h, 2 days (D2), and 4 days (D4) after i.p. injection of 60mg/kg dicloflenac sodium salt in sterile PBS. N=3-8 mice per genotype per day; from 5 ind. expts. *p<0.05 by Tukey's multiple comparisons test. B) Representative whole mount images of formalin-fixed small intestinal wounds of WT, Irgm1-/- and Irgm1-/-Ifnar-/- mice 4 days after diclofenac injection. Dotted black lines outline the wound. All images at 32×. C) Graph of the average distance covered by WAE cells 4 days after colonic biopsy wounding. Quantification was performed blinded on whole mount images of formalin-fixed wounds using Image J software. 10 measurements per wound were averaged. WT n=8 wounds in 4 mice, Irgm1-/- n=12 wounds in 4 mice, Irgm1-/-Ifnar-/- n=9 wounds in 3 mice. D) Representative whole mount images at 90× magnification of colonic biopsy wounds 4 days after wounding. Black arrows delineate the distance migrated by WAE cells. See also Figure S4.
Type I IFNs signaled through macrophages to promote epithelial proliferation
We further extended our findings by using an additional model of Type I IFN production: injection of polyI:C, a synthetic dsRNA (Figures S5A-B). Daily low dose polyI:C injection for 4 days stimulated Type I IFN production and increased epithelial proliferation in WT but not Ifnar-/- mice (Figures 5A-F and S5A-B). This data demonstrates that exogenous Type I IFNs can increase epithelial proliferation.
Figure 5. Type I IFNs signaled through macrophages to indirectly promote epithelial turnover.

A) Graph of A) the average crypt height and B) average number of MF/100 crypts ± SEM in WT and Ifnar-/- mice injected i.p. with saline or 5mg/kg polyI:C for 4 days. WT n=13 mice for each treatment; Ifnar-/- n=5 mice for each treatment. C-F) Graphs of the average number of Ki67+ cells/hpf ± SEM relative to saline-treated mice in submandibular salivary gland, exocrine pancreas, liver, and kidney. WT n=7-8 mice each treatment, Ifnar-/- n=3 mice each treatment. For A-F), orange bars represent saline-treated mice, and purple bars polyI:C-treated mice. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 by Tukey's multiple comparisons test. Graph of G) the average crypt height and the average number of MF of 100 crypts ± SEM in control (n=9 mice), Irgm1-/- (n=8 mice), Irgm1-/-IfnarVC (n=7 mice), and IfnarVC (n=6 mice) mice. Control mice were Irgm1+/+Ifnarf/f-VillinCre negative. Means with different letters are significantly different by Tukey's multiple comparisons test. H) Graph of the average crypt height and average number of MF/100 crypts ± SEM in control and IfnarLysM mice injected i.p. 5mg/kg polyI:C for 4 days. Control: saline n=9 mice, polyI:C n=11 mice. IfnarLysM: saline n=7 mice, polyI:C n=8 mice. Control mice were Ifnarf/f-LysMCre negative. Results compiled from 3 ind. expts. I) Graph of the average crypt height and average number of MF/100 crypts ± SEM in WT mice treated with isotype control (saline n=5 mice, polyI:C n=6 mice) or anti-Ly6G (saline n=6 mice, polyI:C n=6 mice). Results compiled from 2 ind. expts. For H-I), the averages ± SEM relative to saline-treated mice are shown. *p<0.05; ****p<0.0001 by Student's t test. See also Figure S5.
We next tested whether Type I IFNs act directly on the epithelium to promote proliferation by crossing Irgm1-/- mice with mice lacking Ifnar solely in the intestinal epithelium (Irgm1-/-Ifnarf/f-Villin-Cre; Irgm1-/- IfnarVC mice; Figure S5C (el Marjou et al., 2004)). Irgm1-/-IfnarVC mice had elevated levels of serum Type I IFNs and increased intestinal epithelial proliferation comparable to Irgm1-/- mice (Figures 5G and S3A). This finding suggested that non-epithelial cell type(s) must be responding to Type I IFNs to stimulate epithelial cell proliferation. We verified this finding in vitro: addition of IFNα did not directly affect proliferation of primary intestinal epithelial cells (Figure S5D).
Next, to identify the cell type required for epithelial hyperproliferation in response to type I IFN, we assessed the effects of polyI:C injection in mice deficient in Ifnar expression in different cell types. PolyI:C treatment of IfnarVC mice showed augmented intestinal epithelial turnover (Figure S5E-F). However, polyI:C treatment of mice lacking Ifnar on macrophages and granulocytes (Ifnarf/f-LysM-Cre; IfnarLysM mice (Prinz et al., 2008)) did not induce epithelial proliferation to the same extent as controls (Figure 5H), even though Type I IFN levels were induced at similar levels (Figure S5G). Endogenous LysM is highly expressed on both neutrophils and macrophages (Cross et al., 1988). However, neutrophil depletion did not diminish epithelial hyperproliferation induced by polyI:C injection, consistent with macrophages mediating this effect (Figures 5I and S5H-I).
The ISGs Apol9a/b were candidate factors to mediate proliferation in Irgm1-/- mice
We next determined the factors downstream of IFN signaling that augmented epithelial turnover. Serum from Irgm1-/-, WT, polyI:C-treated and saline-treated mice showed no elevation in several established pro-proliferative factors (Figure S6A-B). We therefore performed microarray analysis of whole genome RNA of salivary gland, small intestine and isolated colonic macrophages from Irgm1-/- and WT mice (Figure S6C). Apol9a and Apol9b, highly homologous apolipoproteins (98% by nBLAST analysis), were two factors with enhanced gene expression in Irgm1-/- mice that were also potentially secreted (Figure S6C and Table S1). These apolipoproteins are ISGs (Rusinova et al., 2013); however, their exact function is unknown. We verified enriched gene expression of Apol9a/b in Irgm1-/- mice using qRT-PCR (Figure 6A). Apol9a/b gene expression was also elevated in MCMV-infected tissues compared to mock-infected animals (Figure 6B). In situ hybridization for Apol9a showed expression in discrete foci within the salivary gland and small intestine in Irgm1-/- mice (Figure 6C-D). Within these foci, most cells were positive for Apol9a, including macrophages (Figure S6D), other stromal cells, and epithelial cells themselves (Figure 6C-D, insets).
Figure 6. The ISGs Apol9a and b were elevated in stromal and epithelial cells of Irgm1-/- and MCMV-infected mice.
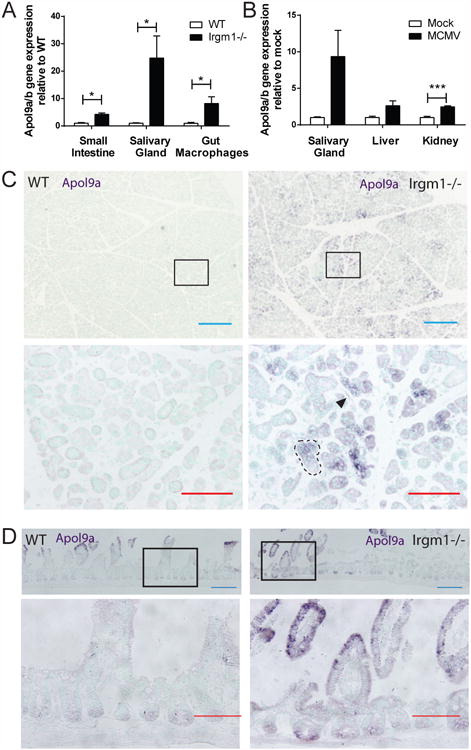
A) Quantification of the average expression ± SEM of Apol9a/b mRNA in Irgm1-/- small intestine, salivary gland and isolated colonic macrophages relative to WT tissue as measured by qRT-PCR. n=3 mice per genotype. Primers were designed to amplify both Apol9a and b. *p<0.05 by Student's t test for each organ. B) Quantification of the average expression ± SEM of Apol9a/b mRNA in MCMV-infected salivary gland, liver and kidney relative to mock-infected tissue as measured by qRT-PCR. Mock-infected n=3 mice, MCMV-infected n=5 mice. ***p<0.001 by Student's t test for each organ. C-D) Representative in situ hybridization images for Apol9a in WT and Irgm1-/- C) salivary gland and D) small intestine. Apol9a expression is indicated by dark purple staining. Lower images are insets of upper images showing foci of Apol9a expression. The black arrowhead indicates an Apol9a-positive stromal cell and dashed black lines outline an Apol9a-positive epithelial gland. Blue bar=200μm, red bar=100μm. See also Figure S6.
Apolipoprotein L9 promoted epithelial proliferation through ERK activation
Notably, the focal pattern of Apol9a gene expression within the salivary gland was associated with areas of high epithelial proliferation (Figure S7A). To determine whether Apol9a could promote proliferation, we used a bioluminescence-based assay to quantify proliferation of Apol9a-expressing epithelial cells. This method utilized knock-in mice expressing a fusion protein between Cdc25A and click beetle red luciferase (CBRLuc) in the endogenous Cdc25a locus (Figure 7A-C). Cdc25A is a cell cycle phosphatase with peak protein levels during mitosis (Boutros et al., 2006). Luminescence from Cdc25A-CBRLuc-expressing cells upon addition of D-luciferin substrate thus provided a direct readout of mitotic activity. Imaging of Cdc25A-CBRLuc colonic epithelial spheroid cultures revealed an increase in luminescence over a 48h culture period (Figures 7C and S7B), which correlated with the number of viable cells (Figure S7C).
Figure 7. Apol9a augmented epithelial proliferation through ERK1/2 activation.
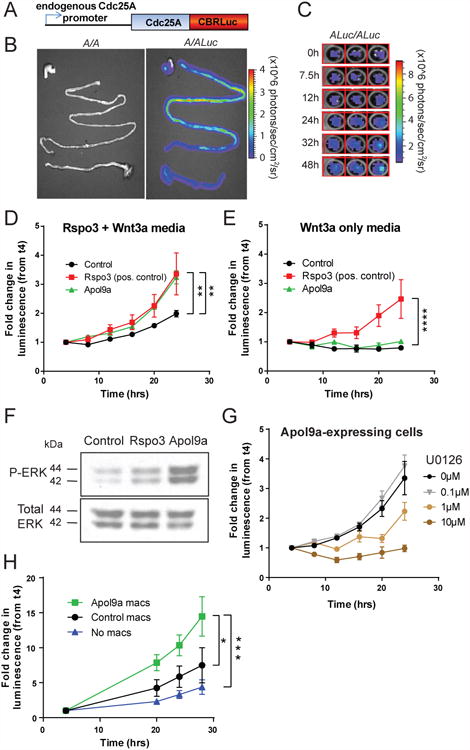
A) Schematic representation of Cdc25A-CBRLuc fusion protein expressed in the endogenous Cdc25A locus. B) Gray scale photograph and bioluminescence image of gastrointestinal tract of WT mice (A/A) and knock-in mice heterozygous (A/Luc) for the Cdc25A-CBRLuc fusion protein. Mice were injected i.p. with D-luciferin, sacrificed and the labeled organs were subjected to bioluminescence imaging ex vivo. Photon flux is indicated by the pseudocolored heatmap. C) Bioluminescence image of Cdc25A-CBRLuc-expressing colonic epithelial cell growth over time. Colonic epithelial spheroid cultures were established from homozygous (ALuc/ALuc) knock-in mice. D-luciferin was added (t = 0h) and bioluminescence was measured, and fresh media with D-luciferin replaced at 24h. D-E) Graph of the fold change in luminescence during a 24h period after addition of D-luciferin in Cdc25A-CBRLuc intestinal spheroid cultures cultured with D) media containing Rspo3 and Wnt3a or E) Wnt3a only. Cells express Rspo3, Apol9a, or a negative control. The average fold change in luminescence relative to luminescence measured at 4h, ± SEM, after background subtraction, of 3 ind. expts. are shown, with 12-18 replicates per cell line per experiment. D) p=0.0389 and E) p= 0.0574 by repeated measures 2-way ANOVA. Asterisks indicate **p<0.01 and ****p<0.0001at 24h only by Dunnett's multiple comparisons test with comparison to control cells. F) Western blot of phosphorylated and total ERK1/2 (p42 and p44) of cells expressing a negative control, Rspo3, or Apol9a. Background-subtracted representative image from 3 ind. expts. is shown. G) Graph of the fold change in luminescence measured during a 24h period after addition of D-luciferin and differing concentrations of MEK inhibitor U0126 in Apol9a-expressing Cdc25A-CBRLuc intestinal spheroid cultures. The average fold change in luminescence relative to luminescence measured at 4h, ± SEM, of 2 ind. expts. is shown, with 3 replicates per dose per experiment. P=0.0066 by repeated measures 2-way ANOVA. H) Graph of the fold change in luminescence measured during a 28h period after addition of D-luciferin of Cdc25A-CBRLuc intestinal spheroid cultures cultured with WT macrophages expressing Apol9a or a control construct, or without macrophages. The average fold change in luminescence relative to luminescence measured at 4h, ± SEM, of 3 ind. expts. is shown, with 4-5 technical replicates per experiment. P=0.0297 by repeated measures 2-way ANOVA. *p<0.05, ***p<0.001 at 24hrs only by Sidak's multiple comparisons test. See also Figure S7.
We assessed luminescence levels of Cdc25A-CBRLuc intestinal epithelial cells expressing either Apol9a, a positive control R-spondin3 (Rspo3), or a negative control construct for 24 hours (Figures 7D and S7B). R-spondins enhance Wnt signaling to promote proliferation and survival of tissue stem cells (Haegebarth and Clevers, 2009). Both R-spondin3 and Wnt3a are required in the normal growth media for primary intestinal epithelial spheroids (Miyoshi et al., 2012; Miyoshi and Stappenbeck, 2013). Under these conditions, small intestinal epithelial cells expressing Apol9a and the positive control Rspo3 had greater proliferation compared to control cells (Figure 7D). However, in media with Wnt3a alone, only epithelial cells expressing Rspo3 were able to proliferate, suggesting that Apol9a cannot substitute for Rspo3 to augment Wnt signaling (Figure 7E). Indeed, no significant enhancement of Wnt signaling was observed in vivo in Irgm1-/- mice (Figure S7D-E). Similar results were obtained in WT cells expressing Apol9a and Rspo3 using an EdU incorporation assay (Figure S7F). These results suggested that Apol9a expression within epithelial cells was able to stimulate epithelial proliferation.
Next, to discover potential pathways downstream of Apol9a expression, we analyzed cell lysates of control, Rspo3, and Apol9a-expressing intestinal epithelial cells using a phospho-kinase dot blot array. Apol9a-expressing epithelial cells showed elevations in phospho-ERK1/2 compared to control cells (Figure S7G), which we verified by immunoblotting (Figure 7F). ERK kinases are the downstream signaling component of the MAPK pathway, and have been implicated in promoting cell cycle progression and proliferation (Rubinfeld and Seger, 2005). Addition of the MEK inhibitor U0126 inhibited ERK1/2 phosphorylation and reduced proliferation of Apol9a-expressing intestinal epithelial cells, as well as control cells, in a dose-dependent manner (Figures 7G and S7H-I). These findings implicated activation of the ERK pathway, which was important for epithelial cell proliferation, downstream of Apol9a expression.
Macrophages expressing Apol9a were able to promote epithelial proliferation in trans
Finally, we investigated the role of Type I IFN signaling in macrophages on Apol9a-mediated epithelial proliferation. Based on the localization of Apol9a on macrophages by in situ hybridization (Figure S6D), we determined whether Type I IFN signaling could induce Apol9a/b in macrophages and stimulate proliferation in trans. First, we found that Apol9a/b expression was induced by IFNα in WT but not Ifnar-/- macrophages (Figure S7J). We then transfected 293FT cells with Flag-tagged Apol9a and detected Flag-tagged Apol9a protein within the supernatant, suggesting that Apol9a can be secreted (Figure S7K). Next, we expressed Apol9a or a control construct in WT bone marrow-derived macrophages and co-cultured these cells with Cdc25A-CBRLuc intestinal epithelial cells. Macrophages expressing Apol9a enhanced proliferation of epithelial cells greater than control macrophages or epithelial cultures alone (Figure 7H). These results suggested that macrophages promoted epithelial proliferation in trans via Apol9a expression.
Discussion
Taken together, these findings uncover a pathway linking viral infection with enhanced epithelial turnover via Type I IFNs. Specifically, we showed that elevated Type I IFNs can signal through macrophages to enhance epithelial proliferation and injury repair. We demonstrated that the highly related ISGs Apol9a/b promoted epithelial proliferation. These results redefine the current view of Type I IFN effects on proliferation in vivo and enhance our understanding of conditions with elevations in these cytokines, such as viral infection and autoimmunity.
The indirect effect on epithelial proliferation driven by viral infection has not been previously described. The viruses that stimulated epithelial proliferation are murine homologues of highly prevalent human chronic viruses, CMV and herpesvirus, both systemic DNA viruses (Virgin et al., 2009). In contrast, murine norovirus, an RNA virus, did not stimulate proliferation in the intestine, where it is tropic (Nice et al., 2013). These results suggest that not all viruses can stimulate enhanced epithelial proliferation, perhaps due to differences in tissue tropism, viral pathogenesis and/or virus-encoded proteins. In humans, host factors may also play a role in the extent of induced proliferation, as genetic differences can affect immune responses to pathogens, including the Type I IFN response (Lee et al., 2014). Since chronic viral infection clearly has a systemic effect on host physiology, and all individuals possess a collection of persistent viruses as their virome, the use of mouse models with chronic viral infections may uncover new biology.
We found that the effects of Type I IFN on epithelial proliferation are indirect through an additional cell type that includes macrophages. We propose that macrophages can act as a relay. Because of their widespread distribution, these cells are poised to synthesize information from multiple local and remote sources and then in turn instruct proximal epithelial cells. The use of an integrative relay enables the separation of signal from noise and allows for the amplification of additive weak signals. The pattern of foci of Apol9a expression observed in the salivary gland and small intestine supports the concept of a central cell that spreads and amplifies signals to neighboring cells. Our data suggest a model in which Type I IFNs signal on macrophages to promote expression of Apol9a/b, which can stimulate proliferation in nearby epithelial cells. One possibility is that Apol9a/b is secreted from macrophages and then stimulates its own expression in epithelial cells, where it activates ERK to promote cell cycle progression. It is also possible that other ISGs expressed in macrophages can promote Apol9a/b expression in epithelial cells as well. Our work extends a proposed role for macrophages in epithelial maintenance and repair by implicating Type I IFNs in this pathway (Farache et al., 2013). This myeloid-epithelial circuit could be used by other cytokine signaling systems to modulate diverse epithelial functions.
We propose that Apol9a/b is one of the mediators of the Type I IFN response directed towards epithelial progenitors. Relatively little is known about the ApoLfamily. In humans, there are 6 known ApoLs, 5 of which are thought to be interferon-stimulated (Rusinova et al., 2013; Smith and Malik, 2009). In mice, there are as many as 14 apolipoprotein L members (Vanhollebeke and Pays, 2006). In contrast to other apolipoprotein families, only APOL1 has been implicated in lipid transport and metabolism to date (Albert et al., 2005; Duchateau et al., 2000). Interestingly, increased expression of ApoLs has been observed in cervical and breast cancer (Ahn et al., 2004; Jung et al., 2005). Apol9b has been implicated in anti-viral protection in L929 cells and primary neurons, but the extent and mechanism of such protection was unclear (Kreit et al., 2014). In this report, we show that Apol9a can be secreted and promotes epithelial proliferation. Since there are hundreds of ISGs that can be induced in a given cell type, it is unlikely that Apol9a and b are the sole mediators of Type I IFN-dependent epithelial proliferation. Other ISGs may have redundant functions. Nonetheless, Apol9a/b and the ApoL family are ISGs that warrant further study in the context of homeostasis and during viral infection.
We show that Apol9a expression can enhance activation of the ERK pathway and in turn proliferation. The connection between the ERK pathway and proliferation is well supported by previous studies, which show that ERK activation promotes cell cycle progression and cellular differentiation (Rubinfeld and Seger, 2005; Zhang and Liu, 2002). Sustained ERK activation activates cyclin D1 expression and decreases CDK inhibitor levels to allow cells to pass the G1 restriction point and enter the S phase of the cell cycle (Rubinfeld and Seger, 2005). Thus, an increase in ERK activation would be able to increase the rate of cell cycling.
Modulation of the ERK pathway would allow for an additional level of control of proliferation in epithelial organs beyond Wnt signaling, which is required for the maintenance of stem and progenitor cells (Haegebarth and Clevers, 2009; Reya and Clevers, 2005). Wnt ligands bind to their receptor complex and trigger the localization of β-catenin to the nucleus, where it associates with transcription factors that activate multiple target genes to promote a baseline rate of self-renewal and proliferation (Reya and Clevers, 2005). The source of Wnts is thought to be local non-epithelial mesenchymal cells and, in the intestine, also Paneth cells (Farin et al., 2012). In contrast, Type I IFN signaling can generate a signal at the systemic level. Downstream activation of the ERK pathway could then increase the normal rate of cell cycling in stem and progenitor cells. We propose that Type I IFNs provide additional systemic control of epithelial proliferation and healing under non-homeostatic conditions of infection and injury.
Finally, many human conditions are associated with elevated Type I IFN signatures, similar to our observations in the Irgm1-/- mouse. For example, upregulation of Type I IFNs is associated with the Type I Interferonopathies, a group of Mendelian disorders (Crow, 2011; Rice et al., 2014), as well as Systemic Lupus Erythematosus (SLE) (Di Domizio and Cao, 2013). These diseases, which include Aicardi-Goutieres Syndrome (AGS) and spondyloenchondrodysplasia (SPENCD) are proposed to arise from either i) inappropriate activation of the Type I IFN response or ii) inadequate negative regulation of Type I IFN production (Crow, 2011). While the molecular bases of some cases of Type I Interferonopathy are known, many cases remain genetically uncharacterized. It would be interesting to determine whether there are gene changes in human IRGM in these cases that are linked with elevated Type I IFN. In addition, we have only limited understanding of the role of Type I IFNs on disease pathogenesis and progression. Our findings here suggest that elevated Type I IFNs may not be sufficient to cause the severe autoimmunity seen in SLE, AGS, and SPENCD. We did not observe the abnormal skin, brain, or bone dysplasias typically seen in these conditions in Irgm1-deficient mice (results not shown). Instead, we observed increased epithelial turnover, with both augmented cell proliferation and cell death. It is possible that this elevated turnover could increase the risk for exposure of cell contents, such as nucleic acids, to the immune system. Additional genetic or environmental contributions could then trigger autoimmunity to the cellular components. Our findings show that constitutively elevated Type I IFNs can promote systemic epithelial proliferation and cell death, thus expanding our knowledge of the host response during infection and autoimmunity.
Experimental Procedures
Mice
All experimental procedures were performed under approval by Washington University's Animal Studies Committee. For experiments with knockout mouse lines, heterozygote breeding pairs were used to obtain WT controls. For experiments using lineage specific knockouts, heterozygous crosses were used to generate Cre-negative controls. Cdc25A-CBRLuc reporter mice were generated on a B6(Cg)-Tyrc-2J/J (B6-albino) background. A targeting construct containing DNA encoding a fusion protein of Cdc25A and click beetle red luciferase (CBRLuc) was knocked-in to the Cdc25A locus by homologous recombination.
In vivo treatment of mice
For MCMV infection, 6-8 week-old male and female mice were injected i.p. with 5×104 plaque-forming units (pfu) MCMV Smith strain (salivary gland-derived). Mice were sacrificed 7 days post-infection. For MHV68 infection, 6-8 week-old male and female mice were injected i.p. with 1×106 pfu virus. Mice were sacrificed 7 days post-infection. Titer determinations for both MCMV and MHV68 were performed on NIH 3T12 fibroblasts with methylcellulose overlay as previously described (Canny et al., 2014). The limit of detection of the MCMV plaque assays was 40PFU/organ, while the limit of detection of the MHV68 assay was 100PFU/organ. For polyI:C experiments, 5mg/kg polyI:C (GE Healthcare Life Sciences) diluted in sterile saline or saline alone was injected i.p. once every 24h for 4 days. Mice were sacrificed on day 5. For neutrophil-depletion experiments, 500μg/mouse of monoclonal anti-Ly6G antibody (Bio×Cell) or rat IgG2a isotype control (Bio×Cell) diluted in 250μL sterile saline was injected i.p. one day prior and 2 days following the onset of polyI:C injections.
Type I IFN bioassay
Type I IFN bioassay was performed as previously described (Newby et al., 2007), with further details provided in the Extended Experimental Procedures.
Tissue Histology and Immunohistochemistry
Detailed methods are described in Extended Experimental Procedures. Epithelial mitotic figures were quantified from H&E-stained sections of Bouin's fixed small intestines. Crypt depth was measured from the same sections by ImageJ software. All slides were blinded prior to quantification.
Primary intestinal spheroid epithelial culture
Colonic and small intestinal crypts were isolated from mice and cultured in a 3D-Matrigel as previously described (Miyoshi et al., 2012; Miyoshi and Stappenbeck, 2013).
Bioluminescence imaging of whole organs and intestinal epithelial spheroids
Detailed methods are further described in Extended Experimental Procedures. Colonic crypts were isolated from knock-in mice that were homozygous (ALuc/ALuc) for the Cdc25A-CBRLuc fusion protein and intestinal spheroids were cultured in conditioned media as described above. For luminescence experiments with lentivirus construct-expressing cells, spheroids were trypsinized and seeded onto 96-well plates. Media containing 200μg/mL D-luciferin (Biosynth) was added to each well, and cells were imaged at 37°C in an Envision Multilabel Plate reader (Perkin Elmer) with a 1s exposure per well. Luminescence was measured every 4h for 24h and the fold change in luminescence relative to luminescence measured at 4h was plotted. This time point was chosen because the measurements at t0 are close to background, as it takes some time after the addition of D-luciferin for this substrate to be metabolized by the luciferase.
Statistics
GraphPad Prism software (version 6) was used to perform all statistical analyses unless otherwise specified.
Supplementary Material
Acknowledgments
This work was supported by the NIH (DK071619 (T.S.S.) AI08488702 (H.W.V.), P50CA94056 (D. P.-W., T.S.S.), AI080672 (D.J.L), and the CCFA Genetics Consortium (T.S.S., H.W.V.). L.S. was supported by The Shawn Hu and Angela Zeng Graduate Fellowship. The Washington University Digestive Disease Research Core Center is supported by a grant from the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) (P30DK052574). We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital, for the use of the High-Throughput Screening Core. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30CA91842. Additionally, we thank K. Sheehan and R. Schreiber for the anti-pan-IFNα and anti-IFNβ antibodies (this work supported by the NIH; AR048335 and AI056160), and D. Kreamalmeyer for expertise in animal care.
Footnotes
Author Contributions: L.S. and T.S.S. designed experiments, analyzed results and prepared the manuscript. H.M. designed experiments and analyzed results. S.O., D.P.W. and H.P.W. generated the Cdc25A-CBR-Luc knock-in mouse. A.L.S., T.J.N., L.A.F, A.R.F. and H.W.V. aided in viral infections, A.C.B. conducted double thymidine analog experiments, and N.A.M. performed wound biopsy experiments. L.S. conducted all other experiments. H.W.V. and D.J.L. contributed to experimental design and manuscript preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn WS, Bae SM, Lee JM, Namkoong SE, Han SJ, Cho YL, Nam GH, Seo JS, Kim CK, Kim YW. Searching for pathogenic gene functions to cervical cancer. Gynecol Oncol. 2004;93:41–48. doi: 10.1016/j.ygyno.2003.11.031. [DOI] [PubMed] [Google Scholar]
- Albert TS, Duchateau PN, Deeb SS, Pullinger CR, Cho MH, Heilbron DC, Malloy MJ, Kane JP, Brown BG. Apolipoprotein L-I is positively associated with hyperglycemia and plasma triglycerides in CAD patients with low HDL. J Lipid Res. 2005;46:469–474. doi: 10.1194/jlr.M400304-JLR200. [DOI] [PubMed] [Google Scholar]
- Ashida H, Ogawa M, Kim M, Mimuro H, Sasakawa C. Bacteria and host interactions in the gut epithelial barrier. Nat Chem Biol. 2012;8:36–45. doi: 10.1038/nchembio.741. [DOI] [PubMed] [Google Scholar]
- Barton ES, Lutzke ML, Rochford R, Virgin HWt. Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J Virol. 2005;79:14149–14160. doi: 10.1128/JVI.79.22.14149-14160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007;128:445–458. doi: 10.1016/j.cell.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–191. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canny SP, Reese TA, Johnson LS, Zhang X, Kambal A, Duan E, Liu CY, Virgin HW. Pervasive transcription of a herpesvirus genome generates functionally important RNAs. MBio. 2014;5:e01033–01013. doi: 10.1128/mBio.01033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong KT, Gresser I, Mims CA. Interferon as a defence mechanism in mouse cytomegalovirus infection. J Gen Virol. 1983;64(Pt 2):461–464. doi: 10.1099/0022-1317-64-2-461. [DOI] [PubMed] [Google Scholar]
- Cliffe LJ, Humphreys NE, Lane TE, Potten CS, Booth C, Grencis RK. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science. 2005;308:1463–1465. doi: 10.1126/science.1108661. [DOI] [PubMed] [Google Scholar]
- Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, Woude GF, Sher A, Taylor GA. Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med. 2001;194:181–188. doi: 10.1084/jem.194.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creamer B. The turnover of the epithelium of the small intestine. Br Med Bull. 1967;23:226–230. doi: 10.1093/oxfordjournals.bmb.a070561. [DOI] [PubMed] [Google Scholar]
- Cross M, Mangelsdorf I, Wedel A, Renkawitz R. Mouse lysozyme M gene: isolation, characterization, and expression studies. Proc Natl Acad Sci U S A. 1988;85:6232–6236. doi: 10.1073/pnas.85.17.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238:91–98. doi: 10.1111/j.1749-6632.2011.06220.x. [DOI] [PubMed] [Google Scholar]
- De Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- De Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282:20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- Di Domizio J, Cao W. Fueling autoimmunity: type I interferon in autoimmune diseases. Expert Rev Clin Immunol. 2013;9:201–210. doi: 10.1586/eci.12.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchateau PN, Movsesyan I, Yamashita S, Sakai N, Hirano K, Schoenhaus SA, O'Connor-Kearns PM, Spencer SJ, Jaffe RB, Redberg RF, et al. Plasma apolipoprotein L concentrations correlate with plasma triglycerides and cholesterol levels in normolipidemic, hyperlipidemic, and diabetic subjects. J Lipid Res. 2000;41:1231–1236. [PubMed] [Google Scholar]
- Dutia BM, Allen DJ, Dyson H, Nash AA. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology. 1999;261:173–179. doi: 10.1006/viro.1999.9834. [DOI] [PubMed] [Google Scholar]
- El Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Farache J, Zigmond E, Shakhar G, Jung S. Contributions of dendritic cells and macrophages to intestinal homeostasis and immune defense. Immunol Cell Biol. 2013;91:232–239. doi: 10.1038/icb.2012.79. [DOI] [PubMed] [Google Scholar]
- Farin HF, Van Es JH, Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology. 2012;143:1518–1529. e1517. doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- Feng CG, Collazo-Custodio CM, Eckhaus M, Hieny S, Belkaid Y, Elkins K, Jankovic D, Taylor GA, Sher A. Mice deficient in LRG-47 display increased susceptibility to mycobacterial infection associated with the induction of lymphopenia. J Immunol. 2004;172:1163–1168. doi: 10.4049/jimmunol.172.2.1163. [DOI] [PubMed] [Google Scholar]
- Haegebarth A, Clevers H. Wnt signaling, lgr5, and stem cells in the intestine and skin. Am J Pathol. 2009;174:715–721. doi: 10.2353/ajpath.2009.080758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HH, Lee J, Kim JH, Ryu KJ, Kang SA, Park C, Sung K, Nam DH, Kang WK, Park K, et al. STAT1 and Nmi are downstream targets of Ets-1 transcription factor in MCF-7 human breast cancer cell. FEBS Lett. 2005;579:3941–3946. doi: 10.1016/j.febslet.2005.06.011. [DOI] [PubMed] [Google Scholar]
- King KY, Baldridge MT, Weksberg DC, Chambers SM, Lukov GL, Wu S, Boles NC, Jung SY, Qin J, Liu D, et al. Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood. 2011;118:1525–1533. doi: 10.1182/blood-2011-01-328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreit M, Paul S, Knoops L, De Cock A, Sorgeloos F, Michiels T. Inefficient type I interferon-mediated antiviral protection of primary mouse neurons is associated with the lack of apolipoprotein l9 expression. J Virol. 2014;88:3874–3884. doi: 10.1128/JVI.03018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F, Davis CR, Wang HT, Chu P, Lee M, Yuan J, Nusse R, Kuo CJ. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci U S A. 2004;101:266–271. doi: 10.1073/pnas.2536800100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, White LS, Hurov KE, Stappenbeck TS, Piwnica-Worms H. Response of small intestinal epithelial cells to acute disruption of cell division through CDC25 deletion. Proc Natl Acad Sci U S A. 2009;106:4701–4706. doi: 10.1073/pnas.0900751106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K, et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;343:1246980. doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, Grossniklaus E, Schoenborn AA, Sartor RB, Taylor GA. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2013;305:G573–584. doi: 10.1152/ajpgi.00071.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 2001;3:333–340. doi: 10.1016/s1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- Mahoney ZX, Stappenbeck TS, Miner JH. Laminin alpha 5 influences the architecture of the mouse small intestine mucosa. J Cell Sci. 2008;121:2493–2502. doi: 10.1242/jcs.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol. 2006;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- Newby CM, Sabin L, Pekosz A. The RNA binding domain of influenza A virus NS1 protein affects secretion of tumor necrosis factor alpha, interleukin-6, and interferon in primary murine tracheal epithelial cells. J Virol. 2007;81:9469–9480. doi: 10.1128/JVI.00989-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Strong DW, McCune BT, Pohl CS, Virgin HW. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J Virol. 2013;87:327–334. doi: 10.1128/JVI.01864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packey CD, Ciorba MA. Microbial influences on the small intestinal response to radiation injury. Curr Opin Gastroenterol. 2010;26:88–94. doi: 10.1097/MOG.0b013e3283361927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellettieri J, Sanchez Alvarado A. Cell turnover and adult tissue homeostasis: from humans to planarians. Annu Rev Genet. 2007;41:83–105. doi: 10.1146/annurev.genet.41.110306.130244. [DOI] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Pfefferle PI, Renz H. The mucosal microbiome in shaping health and disease. F1000Prime Rep. 2014;6:11. doi: 10.12703/P6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten CS. Stem cells in gastrointestinal epithelium: numbers, characteristics and death. Philos Trans R Soc Lond B Biol Sci. 1998;353:821–830. doi: 10.1098/rstb.1998.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, Merkler D, Detje C, Gutcher I, Mages J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28:675–686. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Ramirez-Alcantara V, LoGuidice A, Boelsterli UA. Protection from diclofenac-induced small intestinal injury by the JNK inhibitor SP600125 in a mouse model of NSAID-associated enteropathy. Am J Physiol Gastrointest Liver Physiol. 2009;297:G990–998. doi: 10.1152/ajpgi.00219.2009. [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijke RP, Plaisier H, Hoogeveen AT, Lamerton LF, Galjaard H. The effect of continuous irradiation on cell proliferation and maturation in small intestinal epithelium. Cell Tissue Kinet. 1975;8:441–453. doi: 10.1111/j.1365-2184.1975.tb01231.x. [DOI] [PubMed] [Google Scholar]
- Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41:D1040–1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales BS, Huffnagle GB. The microbiome in wound repair and tissue fibrosis. J Pathol. 2013;229:323–331. doi: 10.1002/path.4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, Stappenbeck TS. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci U S A. 2009;106:256–261. doi: 10.1073/pnas.0803343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19:850–858. doi: 10.1101/gr.085647.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumaria N, van Dommelen SL, Andoniou CE, Smyth MJ, Scalzo AA, Degli-Esposti MA. The roles of interferon-gamma and perforin in antiviral immunity in mice that differ in genetically determined NK-cell-mediated antiviral activity. Immunol Cell Biol. 2009;87:559–566. doi: 10.1038/icb.2009.41. [DOI] [PubMed] [Google Scholar]
- Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2011;146:621–632. doi: 10.1016/j.cell.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, Kapadia SB, Speck SH, Virgin HWt. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol. 2003;77:7696–7701. doi: 10.1128/JVI.77.13.7696-7701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhollebeke B, Pays E. The function of apolipoproteins L. Cell Mol Life Sci. 2006;63:1937–1944. doi: 10.1007/s00018-006-6091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW. The virome in Mammalian physiology and disease. Cell. 2014;157:142–150. doi: 10.1016/j.cell.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.