

Abstract
Objective
Cathepsin G (CatG) is a serine protease that mediates angiotensin-I (Ang-I) to angiotensin-II (Ang-II) conversion and is highly expressed in human abdominal aortic aneurysms (AAAs). However, it remains untested whether this protease participates in the pathogenesis of AAA.
Methods and Results
Immunofluorescent double staining demonstrated the expression of CatG in smooth-muscle cells (SMCs), macrophages, and endothelial cells (ECs) in human AAA lesions (n=12), but not in AAA-free aortas (n=10). While inflammatory cytokines induced CatG expression, high glucose increased CatG activity in producing Ang-II and angiotensin-converting enzyme (ACE) in SMCs, which could be fully blocked by a CatG-selective inhibitor or its siRNA. To test whether CatG contributes to AAA development, we generated CatG and low-density lipoprotein receptor (LDLr) double deficient (Ldlr−/−Ctsg−/−) mice and their littermate controls (Ldlr−/−Ctsg+/+). Absence of CatG did not affect Ang-II infusion-induced AAAs. In contrast, in Ang-II-independent AAAs induced by peri-aortic CaCl2 injury (n=12 per group), CatG deficiency significantly reduced aortic diameter increase (58.33%±6.83% vs. 31.67%±5.75%, P=0.007), aortic lesion area (0.35±0.04 mm2 vs. 0.21±0.02 mm2, P=0.005), and aortic wall elastin fragmentation grade (2.75±0.18 vs. 1.58±0.17, P=0.002) along with reduced lesion collagen content grade (2.80±0.17 vs. 2.12±0.17, P=0.009) without affecting indices of lesion inflammation, angiogenesis, cell proliferation, or apoptosis. In vitro elastin degradation assays demonstrated that CaCl2-induced AAA lesions from Ldlr−/−Ctsg−/− mice contained much lower elastinolytic activity than in those from littermate control mice. Gelatin gel zymogram assay suggested that absence of CatG in CaCl2-induced AAA lesions also reduced the activity of elastinolytic matrix metalloproteinase (MMP)-2 and MMP-9.
Conclusion
CatG may contribute to CaCl2-induced experimental AAAs directly via its elastinolytic activity and indirectly by regulating lesion MMP-2 and MMP-9 activities. Increased expression of CatG in vascular and inflammatory cells of human AAAs, and its increased activity in producing Ang-II and ACE by SMCs suggest additional mechanism by which CatG contributes to AAA lesion progression.
INTRODUCTION
Mast cells and neutrophils express the serine protease cathepsin G (CatG).1,2 This enzyme can generate angiotensin-II (Ang-II) from its precursor Ang-I or even from angiotensinogen.3 CatG also proteolytically activate matrix metalloproteinases (MMP)-1, -2, -3, and -9,4–7 that participate in the pathogenesis of cardiovascular diseases, including abdominal aortic aneurysms (AAAs) and atherosclerosis.8,9 By having multiple enzymatic activities, i.e. by being a collagenase activator,6,10 a collagenase,11 and an elastase,12 and by being expressed by mast cells and neutrophils which are found in the luminal layer of the intraluminal thrombus and in the adventitia of most human AAA lesions,13 CatG may contribute to collagen and elastin degradation in the aneurysmal aortic wall.14 Compared with healthy aortas, human AAA lesions with or without thrombus had significantly higher CatG mRNA, protein, and activity, associating CatG with AAAs.14
In AAA patients, the plasma levels of a 10-amino acid peptide, hemorphin 7, which is derived from hemoglobin proteolyzed by CatG proteolysis, was found to be several fold increased and to correlate positively with thrombus volume and aortic diameters.15 Increased CatG expression and activity in AAA lesions suggest its direct participation in the pathogenesis of this disease. In this study we used CatG-deficient (Ctsg−/−) mice and established a role for CatG elastinolytic activity in arterial wall remodeling and experimental AAA pathogenesis.
METHODS
Human AAA lesion immunohistology
Discarded human AAA specimens were obtained from patients (n=12) who underwent invasive repair. AAA-free arteries were obtained from transplant donors (n=10), according to protocols pre-approved by the Human Investigative Review Committee of Brigham and Women’s Hospital. Human AAA serial cryostat sections (6 μm) were prepared and stained for CatG (1:50 immunofluorescent, Calbiochem, San Diego, CA), CD68 (macrophages, 1:400 immunofluorescent, Dako, Carpinteria, CA), CD31 (endothelial cells [ECs], 1:30 immunofluorescent, Dako), and α-actin (SMCs, 1:30 immunofluorescent, Enzo Diagnostics Inc., Farmingdale, NY).
Human aortic SMC culture, ELISA, and immunoblot analysis
Human aortic SMCs were isolated by an explant outgrowth method from minced human abdominal aorta, as described previously,16 and subcultured at passages 2~5 in Dulbecco’s Modified Eagle Medium (DMEM). To study CatG expression in human SMCs activated with inflammatory cytokines, we cultured human SMCs in low-glucose DMEM containing interferon (IFN)-γ (20 ng/mL), tumor necrosis factor (TNF)-α (10 ng/mL), interleukin 6 (IL-6, 20 ng/mL), and fibroblast growth factor-2 (b-FGF, 10 ng/mL) (all from R&D Systems) for 48 hours, followed by immunoblot analysis. An equal amount of protein from each cell preparation was separated by SDS-PAGE, blotted, and detected with antibodies against CatG (1:1,000, Calbiochem), β-actin, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:1000, Santa Cruz Biotechnology Inc.).
To test the role for CatG in high-glucose–induced production of Ang-II and possibly angiotensin-converting enzyme (ACE), we pre-treated human aortic SMCs with the cell permeable CatG-selective inhibitor Ac-Phe-Val-Thr-(4-guanidine)Phg(P)-(OPh4-SMe)2 (10 μg/ml), previously validated with a Kobs/I of 256000 M−1s−1,17 in low-glucose DMEM (1 g/L) for 30 minutes and then replaced with CatG inhibitor-containing high-glucose DMEM (4.5 g/L) for 24 hours, followed by ELISA determination of Ang-II (Assaypro, St. Charles, MO) and ACE (R&D Systems, Minneapolis, MN) in cell lysate and those secreted to the culture media. To confirm CatG activity in Ang-II and ACE production, we also performed a CatG knockdown experiment by transfecting CatG siRNA or control siRNA (Santa Cruz Biotechnology Inc., Santa Cruz, CA) into human aortic SMCs. In brief, human SMCs on a 6-well plate were transiently transfected with 100 nmol/L control siRNA or CatG siRNA per well with Lipofectamine 2000 reagent in an OptiMEM medium (Invitrogen, Grand Island, NY). Two days later, cells were cultured in low or high glucose DMEM containing 10% fetal bovine serum for another 48 hours. Cell culture medium and cell lysates were prepared for ELISA to determine Ang-II and ACE levels.
Mouse experimental AAA
We crossbred Ctsg−/− mice (C57BL/6/129/SvJ)18 with Ldlr−/− mice (C57BL/6, N11, The Jackson Laboratory, Bar Harbor, ME) to generate Ldlr+/−Ctsg+/− breeding pairs, which then produced Ldlr−/−Ctsg−/− mice and their littermate Ldlr−/−Ctsg+/+ control mice. All mice used in this study were males. To generate experimental AAAs, we used 10-week-old Ldlr−/−Ctsg−/− mice and their littermate Ldlr−/−Ctsg+/+ control mice. AAA was induced by chronic infusion of 1000 ng.kg−1.min−1 Ang-II (Sigma, St. Louis, MO) delivered subcutaneously by Alzet model 2004 osmotic minipumps (DURECT Corp, Cupertino, CA) for 28 days while on a Western diet (C12108; Research Diets, Inc., New Brunswick, NJ). We also generated experimental AAA in 10-week-old Ldlr−/−Ctsg−/− mice and Ldlr−/−Ctsg+/+ littermate control mice by peri-aortic application of 0.25 mM CaCl2, as described previously.19,20 NaCl (0.9%) was substituted for CaCl2 in sham control mice. We measured blood pressures and aortic diameters before aneurysm induction and at sacrifice, 4 weeks after the surgery. Aortic diameters were measured in situ before CaCl2 injury and 4 weeks after injury before harvest, under a surgical microscope with a microruler-installed eyepiece. Each AAA lesion was harvested, cut at the middle of the largest expansion, after which one half was embedded vertically in OCT compound for frozen section preparation and the other half was used for tissue extract preparation. Aortic cross-sections were used for AAA lesion area measurements and AAA lesion characterizations. Lesion characterizations of mouse AAAs, including macrophages (Mac-3, 1:900, BD Biosciences, San Jose, CA), T cells (CD4, 1:90, BD Biosciences), MHC class II–positive cells (MHC class-II, 1:250, BD Biosciences), SMCs (α-actin, 1:750, Sigma), microvessels (CD31, 1:1500, BD Biosciences), cell proliferation (Ki67, 1:750, Vector Laboratories, Burlingame, CA), apoptosis (TUNEL, EMD Millipore, Billerica, MA), collagen (picrosirius red birefringence), and elastin (Verhoeff–van Gieson) were performed as described previously.20 We captured images with a digital system; the staining area was measured using computer-assisted image quantification (Image-Pro Plus software, Media Cybernetics), and immunopositive cells were counted manually. Elastin fragmentation was graded using our previous grading keys,20 whereas new grading keys for collagen degradation in mouse AAA lesions were generated based on our prior experience.21 All mouse experiments were performed, and data were analyzed in a blinded fashion, by at least three observers. All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health, and were approved by the Harvard Medical School Standing Committee on Animals (protocol # 03759).
Blood pressure and plasma Ang-II and ACE level measurements
Blood pressure was measured before and after peri-aortic CaCl2 injury-induced AAA. To measure blood pressure from live mice, we used the CODA standard non-invasive blood pressure system — the tail-cuff method, according to the manufacturer’s instructions (Kent Scientific Corporation, Torrington, CT). Briefly, mice were trained for 3–5 times before the experiments to ascertain that they became accustomed to the tail-cuff procedure. A single investigator recorded blood pressure in a quiet environment without disturbance. At least 30 measurements were obtained from each mouse to determine the mean values of systolic and diastolic blood pressure.
Blood samples were collected by retro-orbital venous plexus puncture. Plasma Ang-II and ACE levels were determined using the Ang-II (USCN Life Science Inc., Houston, TX) and ACE (R&D Systems, Minneapolis, MN) ELISA kits, respectively, according to the manufacturers’ instructions.
In vitro elastin and type I collagen degradation and gelatin gel zymogram assay with human neutrophil CatG and aortic tissue extract
In 100 μl of reaction buffer containing 90 mM HEPES, pH 7.5, 450 mM NaCl, and 1.28 M DMSO,22 100 μl fluorogenic DQ™ bovine neck ligament elastin (Invitrogen) or 100 μl fluorescein conjugated bovine Achilles tendon type I collagen (Calbiochem) with and without different amounts of human neutrophil CatG (EMD Millipore) from 1 mU to 3.9 μU, were added, and the reaction was incubated at 37 °C for 2 days. Optical density was read at an excitation of 450 nm and emission of 510 nm for elastin degradation and excitation of 485 nm and emission of 530 nm for collagen degradation. To measure AAA lesion CatG elastase activity, AAA tissue fragment was pulverized and lysed in a buffer containing 90 mM HEPES, pH 7.5, 450 mM NaCl, 1% Triton X-100, and 1.28 M DMSO. In 100 μl of the same buffer, 20 μg AAA lesion tissue extract and 100 μl fluorogenic DQ™ bovine neck ligament elastin (Invitrogen) were incubated with and without a CatG inhibitor (10 μg/ml) at 37 °C for 2 days, followed by reading the optical density. The same volume buffer and fluorogenic DQ™ elastin or fluorogenic were used as baseline control. AAA lesion (20 μg per sample) MMP activity was determined using gelatin gel zymogram assay as previously reported and gel density was measured using ImageJ software (National Institute of Health).20
Statistical analysis
All mouse data were expressed as mean ± SEM. Due to our small sample sizes and often skewed data distributions, we performed a pairwise non-parametric Mann-Whitney test followed by Bonferroni corrections to examine the statistical significances. SPSS 16.0 was used for analysis.
RESULTS
CatG expression in human AAA lesions
Prior studies demonstrated CatG expression in mast cells and neutrophils in human AAA lesions.13,14 Immunofluorescent double staining demonstrated negligible CatG expression in AAA-free abdominal aortas (Fig 1). However, α-actin-positive SMCs, CD68-positive macrophages, and even CD31-positive ECs in human AAA lesions were also immunoreactive to anti-CatG polyclonal antibody. (Fig 1), suggesting that the majority CatG in human AAA lesions comes from these dominant lesion cell types.
Figure 1.

CatG expression in human AAA lesion and AAA-free aorta. Immunofluorescent double staining with anti-α-actin and anti-human CatG antibodies showed negligible CatG expression in AAA-free aorta, but localized CatG expression in SMCs (α-actin, from the media), macrophages (CD68, from the intima), and ECs (CD31, from the media) in human AAA lesions. Inserts with higher magnifications are shown in the bottom row. Magnifications are indicated.
Expression and activity of CatG in human aortic SMCs
AAA is a chronic inflammatory disease. Besides producing proteases, inflammatory infiltrates are also important sources of inflammatory cytokines. SMCs are the most dominant vascular cells in healthy aortas and these cells otherwise do not express CatG (Fig 1). However, in AAA lesions, SMCs encounter a reservoir of inflammatory cytokines that may stimulate the expression of CatG and other proteases23. SMC localization between elastin layers in the tunica media make them essential to aortic wall remodeling and AAA growth. To test this hypothesis, we treated primary cultured human aortic SMCs with several common inflammatory cytokines, including IFN-γ, TNF-α, IL-6, and b-FGF, and found that these cytokines greatly enhanced CatG expression in human SMCs, as determined by immunoblot analysis (Fig 2, A).
Figure 2.

Inflammatory cytokine and high glucose induced CatG expression and activity in Ang-II and ACE production from human aortic SMCs (huSMCs). A. Immunoblot determined CatG expression in huSMCs treated with different inflammatory cytokines for 48 hours. B. ELISA determined ACE and Ang-II levels in lysates and culture medium from huSMCs cultured in low-glucose or high-glucose DMEM with and without CatG inhibitor for 24 hours. C. Immunoblot determined CatG expression in huSMCs from low-glucose and high-glucose medium with and without CatG inhibitor for 24 hours. D. CatG immunoblot in huSMCs transfected with CatG siRNA or control siRNA and treated with low or high glucose (glu). E. ELISA determined Ang-II and ACE levels in culture medium and cell lysates from huSMCs from panel D. GAPDH or β-actin blots were used to ensure equal protein loading for immunoblots. Bar figures are mean ± SEM from three to six independent experiments.
Hyperglycemia associates with the risk of coronary heart disease.24 Both human and mouse AAA lesions show enhanced glucose uptake (glycolysis) and expression of glucose transporters, which correlate with increased infiltration of macrophages and T cells and elevated MMP-2 and MMP-9 activities.25,26 Interruption of glycolysis reduces CaCl2 peri-aortic injury-induced AAAs.26 Although detailed mechanisms by which glucose contributes to AAA formation remain incompletely understood, high glucose may act like other inflammatory cytokines in inducing CatG expression from SMCs. This hypothesis is supported by observations from rat vascular SMCs, which express serine protease chymase after exposure to high glucose (4.5 g/L).27 As in rat SMCs, human vascular SMCs produced high levels of Ang-II and its processing enzyme, ACE in the cell lysates and secreted to the cell culture media after treatment with high glucose, as determined by ELISA (Fig 2, B). From these human SMCs, however, we were unable to detect chymase by reverse transcription and polymerase chain reaction, followed by cDNA cloning (data not shown). In SMCs treated with the CatG-selective inhibitor Ac-Phe-Val-Thr-(4-guanidine)Phg(P)-(OPh4-SMe)2 (10 μg/ml),17 the high glucose-increased production of Ang-II and ACE was fully suppressed (Fig 2, B). This finding suggests that human SMCs produce Ang-II and ACE depending on CatG activity, although a mechanism by which suppressed CatG activity reduced ACE production remains to be investigated. CatG inhibitor not only affected CatG activity, but also reduced CatG protein levels, whereas high glucose only increased CatG activity but not its protein levels (Fig 2, A, B and C). To explore a specific function of CatG in Ang-II and ACE production, we performed an RNA interference experiment to suppress CatG expression in human SMCs using CatG siRNA. As anticipated, CatG siRNA efficiently reduced CatG protein levels in human SMCs exposed to low or high glucose as determined by CatG immunoblot analysis (Fig 2, D). High glucose-increased production of Ang-II and ACE in cell lysate preparation and secretion in culture medium fell substantially in cells transfected with CatG siRNA, but not in those transfected with control siRNA (Fig 2, E).
CatG contributes to peri-aortic CaCl2 injury-induced AAAs by degrading arterial wall elastin and type I collagen
Suppression of Ang-II and ACE production by CatG-selective inhibitor or CatG RNA interference in aortic SMCs, and likely in other CatG-positive cells in the vasculature, suggests that CatG contributes to AAA development by producing these vasoconstrictive molecules. To test a direct role of CatG in AAAs, we implanted Ang-II–containing minipumps (1000 ng.kg−1.min−1 Ang-II) into Ldlr−/−Ctsg−/− mice (n=15) and Ldlr−/−Ctsg+/+ mice (n=17). Mice were harvested after 28 days of Ang-II infusion. Ldlr−/− mice develop negligible atherosclerotic lesions after consuming 2 months of an atherogenic diet.28 We detected no noticeable atherosclerosis from the aortic arches or thoracic aortas from either Ldlr−/−Ctsg+/+ mice or Ldlr−/−Ctsg−/− mice after mice consuming 28 days of an atherogenic diet. Maximal aortic diameters also did not differ significantly between the two groups (1.69±0.23 mm vs. 1.57±0.33 mm, P=0.764). High dose of exogenous Ang-II from the minipump may have overridden the differences in CatG-derived endogenous Ang-II production, thereby obscuring the difference in AAA formation between the two groups of mice with deficient and sufficient CatG expression. To test further the role of CatG in AAA formation, we introduced Ldlr−/−Ctsg−/− mice (n=12) and Ldlr−/−Ctsg+/+ mice (n=12) into peri-aortic CaCl2 injury-induced AAAs, which do not require exogenous Ang-II or develop atherosclerosis. We chose to use the same mice as those used in Ang-II infusion experiment for the peri-aortic NaCl (sham operation) and CaCl2 injury experiment to be certain that any difference in CaCl2 injury-induced AAAs between the Ldlr−/− Ctsg+/+ mice and Ldlr−/−Ctsg−/− mice was not due to the variation of mouse strain. While sham (NaCl)-operated mice showed negligible increases in aortic diameter and had no differences between the two groups (3.19%±0.81% vs. 2.98%±0.15%, P=0.99), CaCl2 injury-induced abdominal aortic diameter increase (58.33%±6.83% vs. 31.67%±5.75%, P=0.007) and AAA lesion areas (0.35±0.04 mm2 vs. 0.21±0.02 mm2, P=0.005) were significantly higher in Ldlr−/−Ctsg+/+ mice than those in Ldlr−/−Ctsg−/− mice (Fig 3). Absence of CatG did not affect plasma Ang-II or ACE levels, and neither did systolic and diastolic blood pressure differ between Ldlr−/−Ctsg−/− mice and Ldlr−/−Ctsg+/+ mice before or after AAA production (Fig 4, A and B). Therefore, neither the Ang-II-induced AAAs nor the CaCl2 peri-aortic injury-induced AAAs tested a role of CatG in Ang-II production in AAAs. We did not detect any significant effect of the absence of CatG on AAA lesion content of macrophages, CD4+ T cells, or levels of MHC-class II (Fig 4, C). Nor did CatG deficiency affect AAA lesion SMC content, CD31+ microvessel number, cell proliferation (Ki67+ cell number), or apoptosis (TUNEL-positive cell number) (Fig 4, D). In contrast, AAA lesions from Ldlr−/−Ctsg−/− mice had not only significantly lower aortic wall elastin fragmentation grade (2.75±0.18 vs. 1.58±0.17, P=0.0002) but also lower collagen grade (2.80±0.17 vs. 2.12±0.17, P=0.009) than in those from Ldlr−/−Ctsg+/+ mice, as shown by Verhoeff–van Gieson staining (Fig 5, A)20 and picrosirius red birefringence (Fig 5, B),21 respectively. These observations suggest that CatG participates in peri-aortic CaCl2 injury-induced AAA production via its elastinolytic activities without affecting lesion inflammation, angiogenesis, cell proliferation, or apoptosis.12
Figure 3.

Aortic diameter and lesion areas from peri-aortic CaCl2 injury-induced AAAs in Ldlr−/−Ctsg−/− and Ldlr−/−Ctsg+/+ mice. Data are mean ± SE. Number of mice per experimental group is indicated in each bar.
Figure 4.

Characterization of peri-aortic CaCl2 injury-induced AAAs between Ldlr−/−Ctsg−/− and Ldlr−/−Ctsg+/+ mice. Plasma Ang-II and ACE levels (A), systolic and diastolic blood pressures before and after AAA production (B), lesion macrophages, CD4+ T cells, and MHC class-II (C), and lesion SMCs, CD31+ microvessels, Ki67+ proliferating cells, and TUNEL-positive apoptotic cells (D). Data are mean ± SE. Number of mice per experimental group is indicated in each bar.
Figure 5.

AAA lesion elastin fragmentation grades (A) and collagen content grades (B) in Ldlr−/−Ctsg−/− and Ldlr−/−Ctsg+/+ mice at 4 weeks after peri-aortic CaCl2 injury. Representative elastin fragmentation (with magnifications indicated) and collagen grading keys are illustrated to the right of each panel. Data are mean ± SE. Number of mice per experimental group is indicated in each bar.
To test the role of CatG in elastin fragmentation and collagen degradation in peri-aortic CaCl2 injury-induced AAAs, we first confirmed a role for CatG in degrading elastin and type I collagen, the dominant collagen type in human aortas and AAA lesions.29 Purified human neutrophil CatG (from Calbiochem) degraded elastin and type I collagen dose-dependently (Fig 6, A and B). When tissue extracts prepared from AAA lesions from Ldlr−/−Ctsg−/− mice and Ldlr−/−Ctsg+/+ mice were incubated with and without CatG-selective inhibitor under the same experimental condition as that of purified human CatG, we found that elastin degradation was significantly higher in AAA extracts from Ldlr−/−Ctsg+/+ mice than in those from Ldlr−/−Ctsg−/− mice, and also observed that the elevated elastase activity in Ldlr−/−Ctsg+/+ mice was effectively suppressed by a CatG inhibitor (Fig 6, C). CatG mediates MMP activation4–7 and expression.30 Reduced elastase activity (Fig 6, C) and elastin fragmentation (Fig 5, A) in AAA lesions from Ldlr−/−Ctsg−/− mice could also be due to reduced MMP activation or expression. Gelatin gel zymogram allowed us to measure AAA lesion tissue elastinolytic MMP-2 and MMP-9 activity.20 Active forms of MMP-2 and MMP-9 and pro-MMP-2 activity were all significantly reduced in AAA lesions from Ldlr−/−Ctsg−/− mice, compared with those from Ldlr−/−Ctsg+/+ mice (Fig 6, D).
Figure 6.
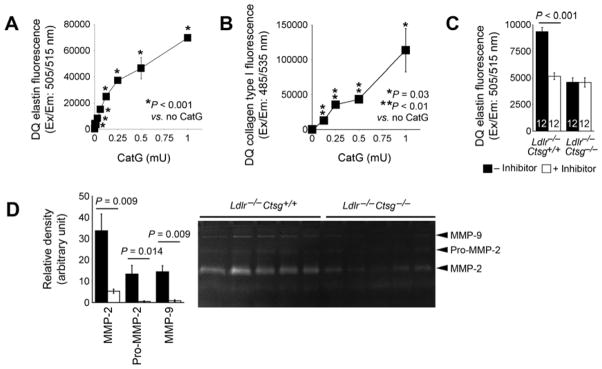
In vitro elastin and type I collagen degradation assays. A. DQ-elastin degradation by different amounts of purified human neutrophil CatG in mU. B. DQ-type I collagen degradation by different amounts of purified CatG in mU. Data are mean ± SE of four independent experiments. C. DQ-elastin degradation by AAA lesion tissue extracts from Ldlr−/−Ctsg−/− mice and Ldlr−/−Ctsg+/+ mice at 4 weeks after peri-aortic CaCl2 injury. D. Gelatin gel zymogram assay tested MMP-2, pro-MMP-2, and MMP-9 (indicated) activities in AAA lesions from Ldlr−/−Ctsg−/− mice and Ldlr−/−Ctsg+/+ mice. Representative zymograph is shown to the right panel. Data are mean ± SE of four experiments. Number of mice per experimental group is indicated in each bar.
DISCUSSION
This study establishes an important role for the elastinolytic activity of CatG in the pathogenesis of CaCl2 peri-aortic injury-induced AAAs. In this experimental model, CatG expression did not affect inflammation, angiogenesis, cell proliferation or apoptosis in the AAA lesions (Fig 4, C and D). We did not explore the mechanism of CatG in Ang-II-induced AAAs in this study due to insignificant differences in AAA sizes between the CatG-deficient and CatG-sufficient mice. The present observation that CatG has a role in arterial wall elastin degradation in AAAs agrees with prior observations using the same AAA model. Overexpression of the CatG endogenous inhibitor α1-anti-chymotrypsin significantly suppressed peri-aortic CaCl2 injury–induced AAA formation. Although detailed mechanisms were not fully executed in these experimental AAAs, α1-anti-chymotrypsin overexpression has been shown to preserve the aortic lesion elastin fibers,31 supporting the conclusion that CatG mediates elastin fragmentation during experimental AAA formation, a mechanism that has never been proposed. By accounting for about 75% of total collagen, type I collagen is the most abundant collagen type in AAA lesions.29 We demonstrated for the first time that CatG degrades type I collagen (Fig 6, B), but such activity may have minimal contribution to reduced AAAs in CatG-deficient mice, consistent with prior human studies that collagen contributes to AAAs via their architecture and network behavior, rather than collagen turnover.32 In addition to its direct role in elastin degradation, CatG is also known to activate MMPs, such as MMP-1, -2, -3, and -9 zymogens4, 5, 33 or to enhance the expression of these MMPs.30 Among these MMPs, both MMP-2 and MMP-9 have elastinolytic activity.34,35 These prior studies may explain the observations that both MMP-2 and MMP-9 activities and pro-MMP-2 activity were significantly lower in AAA lesions from Ldlr−/−Ctsg−/− mice than those from Ldlr−/−Ctsg+/+ mice (Fig 6, D). Therefore, CatG may contribute to arterial elastin fragmentation indirectly by regulating these elastinolytic MMPs.
CatG expression in human AAA lesions was suggested previously to contribute to AAA pathogenesis by its activity in producing Ang-II and the Ang-II–converting serine protease, ACE, thereby enhancing blood pressure and aortic expansion.1–3,13,14 This study demonstrated that, in addition to producing Ang-II from liver-derived angiotensin-I or even angiotensinogen,3,36 CatG also generates Ang-II and increases ACE production from aortic SMCs (Fig 2, B and E), suggesting an additional pathological role of increased CatG expression in aortic SMCs in human AAA lesions (Fig 1). Both inflammatory cytokines and high glucose appeared to regulate CatG expression and activity, but differently. Thus, while inflammatory cytokines increased directly CatG protein levels from human SMCs (Fig 2, A), high glucose did not significantly change CatG protein levels (Fig 2, C and D), but rather increased CatG activity in generating Ang-II and ACE (Fig 2, B and E). The two experimental AAA models used in this study, however, did not test a role for CatG in Ang-II and ACE production in AAAs. Although Ang-II infusion–induced AAAs in Apoe−/− mice or Ldlr−/− mice increased arterial pressure,37 excessive exogenous Ang-II release from the minipumps may have overridden any differences in endogenous Ang-II production between the Ldlr−/−Ctsg−/− and Ldlr−/−Ctsg+/+ mice — possibly contributing to the lack of significant differences in maximal aortic diameters between these mice. In AAAs induced by peri-aortic CaCl2, we detect no changes in blood Ang-II and ACE between Ldlr−/−Ctsg−/− and Ldlr−/−Ctsg+/+ mice (Fig 4, A), and we also found similar systolic or diastolic blood pressures in these two groups of mice before or after CaCl2 application (Fig 4, B). Therefore, a different experimental AAA model may be necessary to test the role for CatG in Ang-II production and blood pressure regulation in AAAs. Insignificant difference in Ang-II-induced AAAs between Ldlr−/−Ctsg−/− and Ldlr−/−Ctsg+/+ mice does not depreciate the impact of CatG in Ang-II production in AAAs. Instead, increased CatG expression in human AAA lesions (Fig 1) and CatG activity in Ang-II production from human vascular SMCs (Fig 2, B and E) suggest a participation of CatG in Ang-II production in human AAA pathogenesis. Therefore, CatG may contribute to human AAAs by both its activities in producing Ang-II and mediating aortic wall elastin degradation. Inhibition of CatG may have therapeutic potential among AAA patients, a hypothesis that merits further investigation.
The role of glucose and its molecular mechanism in AAA formation remain unclear. As discussed, human and mouse AAA lesions have increased expression of glucose transporters and enhanced glycolysis, which correlate with enhanced inflammatory cell infiltration and protease (MMP) activities.25,26 Reduced experimental AAA formation after glycolysis interruption suggests a detrimental role of glucose in AAAs. Our data support this hypothesis. In addition to statistical correlation analysis between protease (MMP) activity and AAA lesion cell glucose transporter expression and glycolysis from prior studies, we provided direct evidence that glucose upregulated vascular cell Ang-II and ACE production and CatG activity (Fig 2), which may also indirectly affect lesion MMP activities (Fig 6, D). In contrast to these proposed mechanisms, it is also known that patients with diabetes are protected from AAAs.38 Although we do not have explanation to these contradictory observations among different studies, it is possible that glucose activity in regulating Ang-II and ACE production and CatG activity in AAA lesions requires expression of functional glucose transporters,25,26 which are often deficient or genetically silenced among diabetic patients.39–44
In conclusion, the results of this study reveal that— in addition to mast cells and neutrophils —there are several cellular sources of CatG in AAAs, notably SMCs, ECs, and macrophages in the lesions. The results also establish an essential role for CatG in arterial wall elastin fragmentation in experimental AAAs. Neutrophils and mast cells produce CatG, and mast cells also produce chymase, for Ang-II and ACE production, and our study, by using molecular cloning techniques, excluded the possibility of human SMCs in producing chymase. Instead, expression of CatG in cultured human SMCs, and in SMCs, ECs, and macrophages from human AAA lesions suggests that these cells are at least partly responsible for glucose uptake and associated inflammatory cell infiltration in AAA lesions,25,26 for Ang-II and ACE production in AAA lesions, and for high blood pressure in humans and animals with AAA.45
Clinical Relevance.
Cathepsin G (CatG) is a serine protease from mast cells and neutrophils to produce angiotensin II (Ang-II), thereby promoting abdominal aortic aneurysms (AAAs). This study demonstrates CatG expression in macrophages, smooth muscle cells (SMCs), and endothelial cells from human AAA lesions and CatG from SMCs produces Ang-II. Inhibition of CatG also reduces elastin and type-I collagen degradation in vitro. Genetic deficiency of CatG reduces aortic wall elastin fragmentation and collagen degradation and protects mice from AAA formation, suggesting a therapeutic potential of CatG inhibition in human AAAs.
Acknowledgments
The authors thank Drs. Timothy J. Ley, M.D. and Christine T. Pham, M.D. from Washington University Medical School, St Louis, MO 63110, USA, for providing the cathepsin G-deficient mice, Wendy Yu and Eugenia Shvartz for technical assistance, and Sara Karwacki for editorial assistance.
SOURCES OF FUNDING
This study is supported by grants from the National Institutes of Health (HL60942, HL81090, HL88547, to G.P.S.; HL56985, to P.L.), and by an EIA award (0840118N) from the American Heart Association (to GPS). The Jenny and Antti Wihuri Foundation supported the Wihuri Research Institute.
Footnotes
DISCLOSURES
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helske S, Syväranta S, Kupari M, Lappalainen J, Laine M, Lommi J, et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–1504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- 2.Heck LW, Rostand KS, Hunter FA, Bhown A. Isolation, characterization, and amino-terminal amino acid sequence analysis of human neutrophil cathepsin G from normal donors. Anal Biochem. 1986;158:217–27. doi: 10.1016/0003-2697(86)90612-3. [DOI] [PubMed] [Google Scholar]
- 3.Owen CA, Campbell EJ. Angiotensin II generation at the cell surface of activated neutrophils: novel cathepsin G-mediated catalytic activity that is resistant to inhibition. J Immunol. 1998;160:1436–43. [PubMed] [Google Scholar]
- 4.Okada Y, Nakanishi I. Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 (‘gelatinase’) by human neutrophil elastase and cathepsin G. FEBS Lett. 1989;249:353–6. doi: 10.1016/0014-5793(89)80657-x. [DOI] [PubMed] [Google Scholar]
- 5.Saunders WB, Bayless KJ, Davis GE. MMP-1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D collagen matrices. J Cell Sci. 2005;118:2325–40. doi: 10.1242/jcs.02360. [DOI] [PubMed] [Google Scholar]
- 6.Wilson TJ, Nannuru KC, Singh RK. Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Mol Cancer Res. 2009;7:1224–33. doi: 10.1158/1541-7786.MCR-09-0028. [DOI] [PubMed] [Google Scholar]
- 7.Shamamian P, Schwartz JD, Pocock BJ, Monea S, Whiting D, Marcus SG, et al. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. 2001;189:197–206. doi: 10.1002/jcp.10014. [DOI] [PubMed] [Google Scholar]
- 8.Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–14. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 9.Keeling WB, Armstrong PA, Stone PA, Bandyk DF, Shames ML. An overview of matrix metalloproteinases in the pathogenesis and treatment of abdominal aortic aneurysms. Vasc Endovascular Surg. 2005;39:457–64. doi: 10.1177/153857440503900601. [DOI] [PubMed] [Google Scholar]
- 10.Capodici C, Muthukumaran G, Amoruso MA, Berg RA. Activation of neutrophil collagenase by cathepsin G. Inflammation. 1989;13:245–58. doi: 10.1007/BF00914392. [DOI] [PubMed] [Google Scholar]
- 11.Kielty CM, Lees M, Shuttleworth CA, Woolley D. Catabolism of intact type VI collagen microfibrils: susceptibility to degradation by serine proteinases. Biochem Biophys Res Commun. 1993;191:1230–6. doi: 10.1006/bbrc.1993.1349. [DOI] [PubMed] [Google Scholar]
- 12.Boudier C, Godeau G, Hornebeck W, Robert L, Bieth JG. The elastolytic activity of cathepsin G: an ex vivo study with dermal elastin. Am J Respir Cell Mol Biol. 1991;4:497–503. doi: 10.1165/ajrcmb/4.6.497. [DOI] [PubMed] [Google Scholar]
- 13.Mäyränpää MI, Trosien JA, Fontaine V, Folkesson M, Kazi M, Eriksson P, et al. Mast cells associate with neovessels in the media and adventitia of abdominal aortic aneurysms. J Vasc Surg. 2009;50:388–95. doi: 10.1016/j.jvs.2009.03.055. discussion 395–6. [DOI] [PubMed] [Google Scholar]
- 14.Gacko M, Chyczewski L. Activity and localization of cathepsin B, D and G in aortic aneurysm. Int Surg. 1997;82:398–402. [PubMed] [Google Scholar]
- 15.Dejouvencel T, Féron D, Rossignol P, Sapoval M, Kauffmann C, Piot JM, et al. Hemorphin 7 reflects hemoglobin proteolysis in abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2010;30:269–75. doi: 10.1161/ATVBAHA.109.198309. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Bai Y, Qin L, Zhang P, Yi T, Teesdale SA, et al. Interferon-gamma induces human vascular smooth muscle cell proliferation and intimal expansion by phosphatidylinositol 3-kinase dependent mammalian target of rapamycin raptor complex 1 activation. Circ Res. 2007;101:560–9. doi: 10.1161/CIRCRESAHA.107.151068. [DOI] [PubMed] [Google Scholar]
- 17.Sieñczyk M, Lesner A, Wysocka M, Legowska A, Pietrusewicz E, Rolka K, et al. New potent cathepsin G phosphonate inhibitors. Bioorg Med Chem. 2008;16:8863–7. doi: 10.1016/j.bmc.2008.08.069. [DOI] [PubMed] [Google Scholar]
- 18.MacIvor DM, Shapiro SD, Pham CT, Belaaouaj A, Abraham SN, Ley TJ. Normal neutrophil function in cathepsin G-deficient mice. Blood. 1999;94:4282–93. [PubMed] [Google Scholar]
- 19.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–32. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117:3359–68. doi: 10.1172/JCI31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitamoto S, Sukhova GK, Sun J, Yang M, Libby P, Love V, et al. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation. 2007;115:2065–75. doi: 10.1161/CIRCULATIONAHA.107.688523. [DOI] [PubMed] [Google Scholar]
- 22.Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–53. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–83. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanter JE, Johansson F, LeBoeuf RC, Bornfeldt KE. Do glucose and lipids exert independent effects on atherosclerotic lesion initiation or progression to advanced plaques? Circ Res. 2007;100:769–81. doi: 10.1161/01.RES.0000259589.34348.74. [DOI] [PubMed] [Google Scholar]
- 25.Reeps C, Essler M, Pelisek J, Seidl S, Eckstein HH, Krause BJ. Increased 18F-fluorodeoxyglucose uptake in abdominal aortic aneurysms in positron emission/computed tomography is associated with inflammation, aortic wall instability, and acute symptoms. J Vasc Surg. 2008;48:417–23. doi: 10.1016/j.jvs.2008.03.059. discussion 424. [DOI] [PubMed] [Google Scholar]
- 26.Tsuruda T, Hatakeyama K, Nagamachi S, Sekita Y, Sakamoto S, Endo GJ, et al. Inhibition of development of abdominal aortic aneurysm by glycolysis restriction. Arterioscler Thromb Vasc Biol. 2012;32:1410–7. doi: 10.1161/ATVBAHA.111.237065. [DOI] [PubMed] [Google Scholar]
- 27.Lavrentyev EN, Estes AM, Malik KU. Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. Circ Res. 2007;101:455–64. doi: 10.1161/CIRCRESAHA.107.151852. [DOI] [PubMed] [Google Scholar]
- 28.Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, et al. Deficiency of cathepsin S reduces atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;111:897–906. doi: 10.1172/JCI14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rizzo RJ, McCarthy WJ, Dixit SN, Lilly MP, Shively VP, Flinn WR, et al. Collagen types and matrix protein content in human abdominal aortic aneurysms. J Vasc Surg. 1989;10:365–73. doi: 10.1067/mva.1989.13151. [DOI] [PubMed] [Google Scholar]
- 30.Son ED, Kim H, Choi H, Lee SH, Lee JY, Kim S, et al. Cathepsin G increases MMP expression in normal human fibroblasts through fibronectin fragmentation, and induces the conversion of proMMP-1 to active MMP-1. J Dermatol Sci. 2009;53:150–2. doi: 10.1016/j.jdermsci.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Wågsäter D, Johansson D, Fontaine V, Vorkapic E, Bäcklund A, Razuvaev A, et al. Serine protease inhibitor A3 in atherosclerosis and aneurysm disease. Int J Mol Med. 2012;30:288–94. doi: 10.3892/ijmm.2012.994. [DOI] [PubMed] [Google Scholar]
- 32.Lindeman JH, Ashcroft BA, Beenakker JW, van Es M, Koekkoek NB, Prins FA, et al. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. Proc Natl Acad Sci U S A. 2010;107:862–5. doi: 10.1073/pnas.0910312107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okada Y, Gonoji Y, Naka K, Tomita K, Nakanishi I, Iwata K, Yamashita K, et al. Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. J Biol Chem. 1992;267:21712–9. [PubMed] [Google Scholar]
- 34.Mecham RP, Broekelmann TJ, Fliszar CJ, Shapiro SD, Welgus HG, Senior RM. Elastin degradation by matrix metalloproteinases. Cleavage site specificity and mechanisms of elastolysis. J Biol Chem. 1997;272:18071–6. doi: 10.1074/jbc.272.29.18071. [DOI] [PubMed] [Google Scholar]
- 35.Yasmin, McEniery CM, Wallace S, Dakham Z, Pulsalkar P, Maki-Petaja K, et al. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:372. doi: 10.1161/01.ATV.0000151373.33830.41. [DOI] [PubMed] [Google Scholar]
- 36.Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol. 2012;23:1181–9. doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, et al. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–5. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torsney E, Pirianov G, Cockerill GW. Diabetes as a negative risk factor for abdominal aortic aneurysm - does the disease aetiology or the treatment provide the mechanism of protection? Curr Vasc Pharmacol. 2013;11:293–8. doi: 10.2174/1570161111311030003. [DOI] [PubMed] [Google Scholar]
- 39.Kampmann U, Christensen B, Nielsen TS, Pedersen SB, Ørskov L, Lund S, et al. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS One. 2011;6:e27854. doi: 10.1371/journal.pone.0027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kouidhi S, Berrhouma R, Rouissi K, Jarboui S, Clerget-Froidevaux MS, Seugnet I, et al. Human subcutaneous adipose tissue Glut 4 mRNA expression in obesity and type 2 diabetes. Acta Diabetol. 2013;50:227–32. doi: 10.1007/s00592-011-0295-8. [DOI] [PubMed] [Google Scholar]
- 41.Kipmen-Korgun D, Bilmen-Sarikcioglu S, Altunbas H, Demir R, Korgun ET. Type-2 diabetes down-regulates glucose transporter proteins and genes of the human blood leukocytes. Scand J Clin Lab Invest. 2009;69:350–8. doi: 10.1080/00365510802632163. [DOI] [PubMed] [Google Scholar]
- 42.Gaster M, Staehr P, Beck-Nielsen H, Schrøder HD, Handberg A. GLUT4 is reduced in slow muscle fibers of type 2 diabetic patients: is insulin resistance in type 2 diabetes a slow, type 1 fiber disease? Diabetes. 2001;50:1324–9. doi: 10.2337/diabetes.50.6.1324. [DOI] [PubMed] [Google Scholar]
- 43.Giacchetti G, Faloia E, Taccaliti A, Morosini PP, Arnaldi G, Soletti F, et al. Decreased expression of insulin-sensitive glucose transporter mRNA (GLUT-4) in adipose tissue of non-insulin-dependent diabetic and obese patients: evaluation by a simplified quantitative PCR assay. J Endocrinol Invest. 1994;17:709–15. doi: 10.1007/BF03347765. [DOI] [PubMed] [Google Scholar]
- 44.Tao T, Tanizawa Y, Matsutani A, Matsubara A, Kaneko T, Kaku K. HepG2/erythrocyte glucose transporter (GLUT1) gene in NIDDM: a population association study and molecular scanning in Japanese subjects. Diabetologia. 1995;38:942–7. doi: 10.1007/BF00400583. [DOI] [PubMed] [Google Scholar]
- 45.Lu H, Rateri DL, Cassis LA, Daugherty A. The role of the renin-angiotensin system in aortic aneurysmal diseases. Curr Hypertens Rep. 2008;10:99–106. doi: 10.1007/s11906-008-0020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]