

Graphical abstract

Keywords: Magnolol, Honokiol, Cross-coupling, GABAA, Subtype selectivity
Abstract
We present the synthesis of new derivatives of natural products magnolol (1) and honokiol (2) and their evaluation as allosteric ligands for modulation of GABAA receptor activity. New derivatives were prepared via metal assisted cross-coupling reactions in two consecutive steps. Compounds were tested by means of two-electrode voltage clamp electrophysiology at the α1β2γ2 receptor subtype at low GABA concentrations. We have identified several compounds enhancing GABA induced current (IGABA) in the range similar or even higher than the lead structures. At 3 μM, compound 8g enhanced IGABA by factor of 443, compared to 162 and 338 of honokiol and magnolol, respectively. Furthermore, 8g at EC10–20 features a much bigger window of separation between the α1β2γ2 and the α1β1γ2 subtypes compared to honokiol, and thus improved subtype selectivity.
Magnolol (1) and honokiol (2) (Scheme 1) are natural products belonging to the class of neolignans isolated from Magnolia officinalis. They have been widely used in Eastern traditional medicine for treatment of gastric and psychiatric disorders. Therapeutic applications of Magnolia constituents were summarized comprehensively in the recent literature.1 Previously, several studies have revealed additional pharmaceutical effects of magnolol or honokiol as anti-angiogenetic,2 antiepileptic,3 neuroprotective,4 or antimicrobial and antiproliferative activity.5 Honokiol has been shown to have somnogenic effects in mice.6 Furthermore, interaction with the PPARγ receptor, involved in treatment of type 2 diabetes, metabolic syndrome, and potential anti-inflammatory target, was shown as well.7 In the central nervous system, neolignans interact with GABAA receptors and modulate their GABA-induced activity leading to the assumption that their antiepileptic and somnogenic in vivo effects are mediated by these receptors.8,9
Scheme 1.
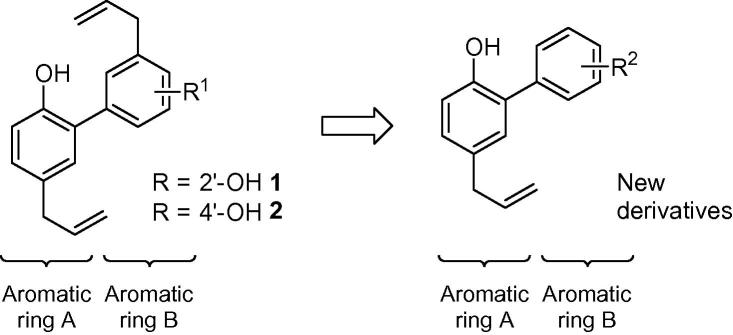
Structure of lead compounds and of new derivatives.
The family of GABAA receptors comprises at least 26 distinct subtypes.10 These pentameric GABA-gated chloride channels assemble as homo- or heteropentamers in mammals from a repertoire of 19 distinct subunits that belong to eight classes. There are six α, three β, γ and ρ as well as one δ, π, ε and θ subunits. The most abundant receptor consists of two α1, two β2 and one γ2 subunits. The GABA binding site in this type of receptor is located at the extracellular interface between the beta and alpha subunits.11 The properties and the pharmacology of the receptor subtypes depend on the subunit composition.12 In addition to the site for the endogenous agonist GABA, at which other agonists or antagonists such as muscimol, bicuculline or gaboxadol also bind, there are multiple allosteric binding sites, such as the one for benzodiazepines, sites for barbiturates, neurosteroids, and several more.11
Magnolol and analogues have been shown to be allosteric modulators of GABAA receptors, thus their binding site does not overlap with the one for GABA, muscimol, bicuculline or gaboxadol.8,9 The antiepileptic and somnogenic effects of magnolol and honokiol, respectively, have been connected with the benzodiazepine binding site because they can be diminished in vivo by the benzodiazepine antagonist flumazenil.4,6 However, neolignans and several synthetic analogs have been shown to modulate both GABAA receptors with and without a benzodiazepine binding site.8,9 The benzodiazepine binding site requires the presence of an α and a γ2 subunit.12 Recently, it was shown that flumazenil also antagonizes modulation by aryl pyrazoles in receptors lacking a γ2 subunit and, thus lacking the high affinity benzodiazepine binding site.13 Consequently, flumazenil sensitive effects in vivo do not necessarily indicate that the benzodiazepine binding site is eliciting them.
Certain subunits have been connected with specific pharmacological effects: α1 containing receptors are thought to mediate sedative and addictive effects of benzodiazepines, while activity of ligands at receptors containing a β1 subunit have been proposed to mediate ataxic effects.14 With respect to GABAA receptor subtypes, the natural neolignans have been demonstrated as relatively unselective compounds, and safety concerns have been raised. Subtype selective synthetic analogs have been proposed to be potentially useful.9,13,15
Here, we present a facile synthetic strategy towards new magnolol/honokiol derivatives and their evaluation on GABAA receptors. The strength of our approach consists in a simplification of the final molecules, allowing their easy synthetic access (compared to the lead structures), while increasing the biological effect at the same time.
Magnolol and honokiol are small biaryl isomeric molecules (Scheme 1). Aromatic ring A is identical for both molecules, while ring B differs in the position of the hydroxyl group. Our approach was based on leaving the ring A untouched and carrying out simplification of the ring B. Instead of allyl and hydroxyl groups, single substituents with diverse properties were introduced in all possible positions of ring B.
Very efficiently, magnolol can be accessed by metal or enzyme mediated oxidative coupling of two 4-allylanisol units and demethylation.16–18 Such strategy, nevertheless, leads inevitably to the symmetric product. Unsymmetrical honokiol can be prepared by bromination of 4-allylanisole and subsequent Suzuki coupling with 4-hydroxyphenylboronic acid. Further modifications (including O-allylation, Claisen rearrangement and demethylation) lead to the desired structure.19 4-Allyl-2-bromoanisole is the key intermediate which could be used within the synthesis of compounds of our interest. A drawback of this pathway is the bromination step, where overbromination of the double bond takes place and debromination with zinc is necessary, conflicting with aspects of atom efficiency. Alternatively, directed ortho-lithiation and introduction of the boronic acid into 4-allylanisole followed by subsequent coupling with aryl bromide was reported.20,21 However, we encountered problems with a reproduction of the protocol.
To circumvent the above outlined limitations, we have developed a simple two steps protocol, relying on palladium catalyzed transformations. In the first step, the allyl moiety is regioselectively introduced into the more reactive 4-position of 4-bromo-2-chlorophenol 3 using allylborate. In the second step, Suzuki coupling of 4-allyl-2-chlorophenol 6 with corresponding boronic acids enabled access to the products of interest. First, we focused our attention on optimization of the allylation reaction of 3 and 4 (Scheme 2). Initial experiments utilizing Pd2dba3 as a catalyst, KF as a base, dioxane/H2O (9:1) mixture as solvent at 150 °C (30 min) under microwave irradiation led to partial success. Three different ligands were tested (Table 1, entries 1–3) of which dppf provided most promising results. Full consumption of the starting material was not observed, however, formation of the desired product was accompanied by debromination of the starting material as side reaction, leading to 2-chlorophenol. Changing the palladium source to heterogenous PdEn40®, which can be easily separated by filtration, led to a decrease of the side reaction; however, full consumption of 3 could not be achieved (entry 4). Further investigation revealed that both conversion and side reaction can be controlled by the base used. Several bases were investigated (entries 5–9): using triethylamine or sodium acetate led to full consumption, however debromination was observed (entries 5 and 6). Slight improvement could be achieved by exploiting sodium carbonate or sodium hydroxide. However, in all cases, side product was still formed. Full conversion and complete suppression of the side reaction could be achieved finally by utilizing potassium carbonate (entry 9). Reaction time could be successfully shortened to 7 min and no sign of debromination was observed (entry 10) facilitating isolation of compound 6 in 77% of yield. Changing pinacol allylboronate 4 to potassium allytrifluoroborate 5 turned out to be beneficial for purification of the product, since pinacolester fragments often contaminated the reaction product complicating isolation. After optimization of reaction conditions target compound 6 was finally isolated in 80% (entry 11).
Scheme 2.
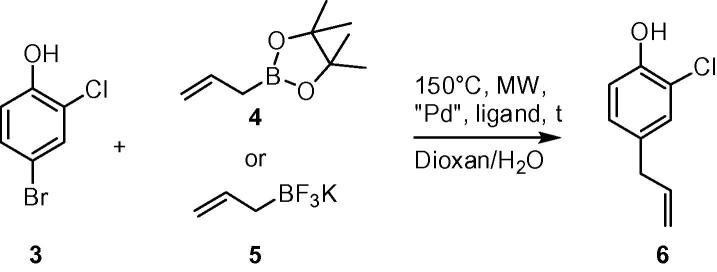
Introduction of allyl moiety.
Table 1.
Optimization of the reaction conditions for introduction of the allyl moiety
| Entry | Pd source | Ligand | Borate | Base | Time (min) | Remaining SM | Debromination | Isolated yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Pd2dba3 | SPhos | 4 | KF | 30 | Yes | Yes | n.d. |
| 2 | Pd2dba3 | ±BINAP | 4 | KF | 30 | Yes | Yes | n.d. |
| 3 | Pd2dba3 | dppf | 4 | KF | 30 | Yes | Yes | n.d. |
| 4 | PdEn40 | dppf | 4 | KF | 30 | Yes | Yes | n.d. |
| 5 | PdEn40 | dppf | 4 | TEA | 30 | No | Yes | n.d. |
| 6 | PdEn40 | dppf | 4 | NaOAc | 30 | No | Yes | n.d. |
| 7 | PdEn40 | dppf | 4 | NaOH | 30 | No | Traces | n.d. |
| 8 | PdEn40 | dppf | 4 | Na2CO3 | 30 | No | Yes | n.d. |
| 9 | PdEn40 | dppf | 4 | K2CO3 | 30 | No | No | 65 |
| 10 | PdEn40 | dppf | 4 | K2CO3 | 7 | No | No | 77 |
| 11 | PdEn40 | dppf | 5 | K2CO3 | 7 | No | No | 80 |
For the second step, we adopted a modified protocol by Denton et al.21 for the coupling of phenyboronic acid with chloro compound 6 under microwave irradiation; 4-allyl-2-phenylphenol 8a was obtained in 58% of yield (Table 2, entry 1), giving an overall yield of 46%. For comparison, 8a was also synthesized according to the above discussed pathway (demethylation/bromination of 4-allylanisole and Suzuki coupling), giving a 15% overall yield after three steps in our hands.22
Table 2.
Synthesized derivatives and their effect at α1β2γ2 GABAA receptor
| Compound | R | Yield | % IGABA at 3 μM |
|---|---|---|---|
| 8a | H | 58 | Inactive |
| 8b | o-Me | 34 | 207 ± 33 |
| 8c | m-Me | 39 | Inactive |
| 8d | p-Me | 46 | 84 ± 11 |
| 8e | o-OMe | 54 | Inactive |
| 8f | m-OMe | 48 | 136 ± 32 |
| 8g | p-OMe | 34 | 443 ± 60 |
| 8h | Lactone | 31 | Inactive |
| 8i | m-COOMe | 34 | 406 ± 70 |
| 8j | p-COOMe | 36 | Inactive |
| 8k | o-NO2 | 42 | Inactive |
| 8l | m-NO2 | 86 | Inactive |
| 8m | p-NO2 | 60 | Inactive |
| 2 | — | — | 162 ± 31 |
| 1 | — | — | 338 ± 93 |
This facile two-step protocol was subsequently employed within the preparation of a small library of compounds to obtain some preliminary structure activity relationship. Indicative substitution patterns contained weakly electron donating methyl groups in positions ortho, meta and para, strongly electron donating methoxy groups and methoxycarbonyl as well as nitro groups in all possible positions as representative electron withdrawing substituents. Only in a single case we have encountered a problem with this synthetic route: ortho-methoxycarbonyl substitution resulted in spontaneous lactonization, since a stable six-membered ring was formed (Scheme 3, 8h). All compounds were obtained in moderate to good yields employing the outlined reaction sequence (Table 2).
Scheme 3.

Introduction of the aryl moiety.
The ability of the synthesized honokiol derivatives to modulate recombinantly expressed α1β2γ2 GABAA receptors from rat was determined in Xenopus laevis oocytes (Table 2). Cells were clamped at a holding potential of −80 mV. A GABA concentration titrated to trigger 0.5% of the respective maximum GABA-elicited current of the individual oocyte (=GABA EC0.5) was applied together with a final concentration of 3 μM of the compound in the measurement buffer. Use of low GABA concentration allows detecting compounds exerting low efficacy modulation. Compounds were applied for 20 s. None of the compounds could open the channel without GABA, so they all are only modulators of the channel. For details on the electrophysiological recordings see Lüscher et al.23
Data was obtained from 5 oocytes from at least 4 different oocyte batches for the active compounds; inactives were measured with n = 2. The response to 3 μM honokiol (2) was 162 ± 31% of the response to an EC0.5 GABA concentration and two of the tested compounds (8g and 8i) possess higher efficacy than the natural product.
A methoxy group in meta (8f) or para (8g) position is beneficial; similar behavior was encountered in case of a m-methoxycarbonyl group (8i).
Surprisingly, also high efficacy was achieved with the o-methyl group (8b). Compounds with a nitro group did not show any effect.
Since both electron donating (methyl, methoxy) as well as electron withdrawing (methoxycarbonyl) substituents can increase the efficacy, a hydrogen bond formation might be involved.
To investigate selectivity, the concentration dependent effect of 8g, the best performing compound at 3 μM (443 ± 60% stimulation), was characterized in α1β2γ2 and α1β1γ2 receptors at a higher GABA concentration (at GABA EC10–20) for a better comparison with published literature (EC10–20),8 and compared with honokiol. In α1β2γ2 the EC50 value was at approximately 7 μM, (20 μM for honokiol) and the efficacy at 100 μM was 539 ± 98%. The EC50 in the β1 containing subtype is 30 μM (20 μM for honokiol), the efficacy only 206 ± 60%. The effect of 8g in a mutant α1β2N265Sγ2 receptor was investigated at EC0.5 (see Supplementary Fig. 1). This mutation in the β2 subunit is known to reduce the efficacy of several compounds including loreclezole and etomidate. With 8g we observed a pronounced drop in response, confirming the key role of β2N265 for the beta subtype selectivity. The stimulation in the wild type receptor at the same compound concentration is for comparison 2462%.
Compounds not acting on β1 receptors are thought to be non-sedative—a desired property for compounds used as anxiolytic or anticonvulsive medication (Fig. 1).14
Figure 1.

Dose–response curves for IGABA potentiation by 8g and honokiol in α1β1γ2 and α1β2γ2 using GABA EC10–20.
In conclusion, we present a rapid and facile synthetic approach to magnolol and honokiol analogs with unsymmetrical aryl substitution employing consecutive cross coupling reactions. The protocol turned out versatile towards highly diverse aryl coupling partners.
Biological assessment of a first small product library revealed compound 8g to possess a highly interesting combination of increased potency compared to the natural analogue in the most abundant α1β2γ2 receptor subtype, while showing strongly reduced responses in the β1 containing subtype that has been connected to ataxic effects.14 Such an action profile has not been reported for synthetic neolignan derivatives or the natural products, so far, underscoring the relevance of this report.8 It has been suggested that compounds with antiepileptic or anxiolytic properties would have fewer side effects if they are inactive in β1 containing receptor subtypes14 The activity difference we report here for our lead compound is much more pronounced compared to the natural products or other synthetic analogs for which this has been studied.8,9
Based on these preliminary findings, exploitation of the synthetic strategy towards investigation of further substitution effects in an elaborated compound library is presently underway in our laboratories.
Acknowledgements
This project was in part supported by the Austrian Science Fund (FWF): S10710 (NFN Drugs from Nature Targeting Inflammation). Financial support by the graduate school program Molecular Drug Targets MolTag (FWF, grant no. W1232) to L.R. and R.P. is acknowledged. The authors gratefully acknowledge Prof. Erwin Sigel (University of Bern) for hosting R.P. during an internship to collect additional data for this publication.
Supplementary data
Experimental procedures.
References and notes
- 1.Lee Y.-J., Lee Y.M., Lee C.-K., Jung J.K., Han S.B., Hong J.T. Pharmacol. Ther. 2011;130(2):157. doi: 10.1016/j.pharmthera.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Ma L., Chen J., Wang X., Liang X., Luo Y., Zhu W., Wang T., Peng M., Li S., Jie S., Peng A., Wei Y., Chen L. J. Med. Chem. 2011;54(19):6469. doi: 10.1021/jm200830u. [DOI] [PubMed] [Google Scholar]
- 3.Chen C.R., Tan R., Qu W.M., Wu Z., Wang Y., Urade Y., Huang Z.L. Br. J. Pharmacol. 2011;164(5):1534. doi: 10.1111/j.1476-5381.2011.01456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tripathi S., Chan M.-H., Chen C. Bioorg. Med. Chem. Lett. 2012;22(1):216. doi: 10.1016/j.bmcl.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 5.Jada S., Reddy Doma M., Singh P.P., Kumar S., Malik F., Sharma A., Khan I.A., Qazi G.N., Kumar H.M.S. Eur. J. Med. Chem. 2012;51:35. doi: 10.1016/j.ejmech.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 6.Qu W.-M., Yue X.-F., Sun Y., Fan K., Chen C.-R., Hou Y.-P., Urade Y., Huang Z.-L. Br. J. Pharmacol. 2012;167(3):587. doi: 10.1111/j.1476-5381.2012.02010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fakhrudin N., Ladurner A., Atanasov A.G., Heiss E.H., Baumgartner L., Markt P., Schuster D., Ellmerer E.P., Wolber G., Rollinger J.M., Stuppner H., Dirsch V.M. Mol. Pharmacol. 2010;77(4):559. doi: 10.1124/mol.109.062141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taferner B., Schuehly W., Huefner A., Baburin I., Wiesner K., Ecker G.F., Hering S. J. Med. Chem. 2011;54(15):5349. doi: 10.1021/jm200186n. [DOI] [PubMed] [Google Scholar]
- 9.Alexeev M., Grosenbaugh D.K., Mott D.D., Fisher J.L. Neuropharmacology. 2012;62(8):2507. doi: 10.1016/j.neuropharm.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen R.W., Sieghart W. Neuropharmacology. 2008;56(1):141. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sieghart W. Pharmacol. Rev. 1995;47(2):181. [PubMed] [Google Scholar]
- 12.Sieghart W., Ernst M. Curr. Med. Chem.: Cent. Nerv. Syst. Agents. 2005;5(3):217. [Google Scholar]
- 13.Mascia M.P., Ledda G., Orru A., Marongiu A., Loriga G., Maciocco E., Biggio G., Ruiu S. Eur. J. Pharmacol. 2014;733:1. doi: 10.1016/j.ejphar.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 14.Gee K.W., Tran M.B., Hogenkamp D.J., Johnstone T.B., Bagnera R.E., Yoshimura R.F., Huang J.-C., Belluzzi J.D., Whittemore E.R. J. Pharmacol. Exp. Ther. 2010;332(3):1040. doi: 10.1124/jpet.109.161885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paola Mascia M., Fabbri D., Antonietta Dettori M., Ledda G., Delogu G., Biggio G. Eur. J. Pharmacol. 2012;693(1–3):45. doi: 10.1016/j.ejphar.2012.07.048. [DOI] [PubMed] [Google Scholar]
- 16.Tzeng S.-C., Liu Y.-C. J. Mol. Catal. B: Enzym. 2004;32(1–2):7. [Google Scholar]
- 17.Kong Z.-L., Tzeng S.-C., Liu Y.-C. Bioorg. Med. Chem. Lett. 2005;15(1):163. doi: 10.1016/j.bmcl.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Tamura H. Jpn. Kokai Tokkyo Koho, Chem. Abstr. 2007:1442479. [Google Scholar]
- 19.Chen C.-M., Liu Y.-C. Tetrahedron Lett. 2009;50(10):1151. [Google Scholar]
- 20.Denton R.M., Scragg J.T. Synlett. 2010:633. [Google Scholar]
- 21.Denton R.M., Scragg J.T., Galofre A.M., Gui X., Lewis W. Tetrahedron. 2010;66(40):8029. [Google Scholar]
- 22.General procedures: 4-Bromo-2-chlorophenol (1.5 g, 7.25 mmol) was charged into a microwave vial, together with potassium allyltriflouroborate (534.2 mg, 3.61 mmol), Pd(En)30TM (300 mg, 0.05 equiv), dppf (134 mg, 0.1 equiv) and K2CO3 (667 mg, 14.5 mmol). A mixture of dioxane/water (9:1) was added (20 mL), the vial was flushed with argon and sealed. The reaction solution was heated to 150 °C for 7 min in a microwave oven (Biotage Initiator 60). After complete reaction, the mixture was filtered through a pad of Celite, the solvent was evaporated under reduced pressure and compound 6 was purified via Kugelrohr distillation to give 80% of a yellowish oil. This material (80 mg, 0.47 mmol) was charged into a microwave vial, together with potassium fluoride (67.2 mg, 1.17 mmol), Pd2dba3 (21.5 mg, 0.05 equiv), SPhos (19.3 mg, 0.1 equiv) and the corresponding boronic acid (0.71 mmol, 1.5 equiv). A dioxane/water mixture (9:1) was added (5 mL), the vial was flushed with argon and sealed. The reaction solution was heated to 150 °C for 10 min in a microwave oven (Biotage Initiator 60). After the reaction was finished, the mixture was filtered through a pad of Celite and the solvent was evaporated under reduced pressure; the crude material was absorbed onto silicagel and purified via column chromatography using PE/EtOAc mixture (0–5% gradient if not stated differently—see electronic Supporting material).
- 23.Lüscher B.P., Baur R., Goeldner M., Sigel E. PLoS ONE. 2012;7(7):e42101. doi: 10.1371/journal.pone.0042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures.