

Abstract
Objective: To investigate the role of PDGFR-β/PI3K/AKT signaling pathway in the myocardial fibrosis. Methods: CFs were divided into following 4 groups: control group (CON), PDGF-BB group (P), PDGF-BB+IMA group (IMA), and PDGF-BB+LY294002 (LY). Results: Immunofluorescence staining showed about 90% of cells were positive for vimentin and 10% for α-SMA. After incubation for 7 days, fluorescence microscopy revealed more than 90% of cells were positive for α-SMA, which was significantly higher than that in CON group (P < 0.01), but markedly lower than that in IMA group and LY group (P < 0.01). The mRNA and protein expression of PDGFR-β, Col I, Col III, PI3K and Akt increased dramatically at 48 h after PDGF-BB treatment when compared with CON group (P < 0.01). However, IMA and LY294002 significantly inhibited the expression of PDGFR-β and p-PI3K (P < 0.05). In addition, the mRNA expression of PDGFR-β, PI3K and Akt in IMA group and LY group was also markedly lower than those in P group (P < 0.01), and the mRNA and protein expression of Col I and Col III reduced remarkably when compared with P group (P < 0.01). Of note, the mRNA expression of PDGFR-α was comparable among 4 groups, and PDGFR-β expression after PDGF-BB treatment increased significantly when compared with PDGFR-α expression (P < 0.01). Conclusion: PDGF-BB may induce CF proliferation and their transformation into myofibroblasts, which leads to increased synthesis of collagen, resulting in myocardial fibrosis. This is closely associated with PDGFR-β, but not PDGFR-α. PDGFR-β/PI3K/Akt signaling pathway is involved in the PDGF-BB induced myocardial fibrosis.
Keywords: Myocardial fibrosis, myofibroblasts, platelet-derived growth factor, signaling pathway
Introduction
Myocardial fibrosis is a pathological feature at the end stage of some cardiovascular diseases and characterized by the proliferation of cardiac fibroblasts in the myocardial interstitium, the transformation of these fibroblasts into myofibroblasts and the excessive deposition of extracellular matrix (ECM). Collagen accounts for about 80% of ECM and is a major component of ECM. Collagen I (Col I) and collagen III (Col III) are the dominant types of collagen in the myocardial fibrosis [1]. Fibroblasts and myofibroblasts are the major cells which synthesize Col I and Col II during the myocardial fibrosis. Platelet-derived growth factor-BB (PDGF-BB) is an important mitogenic factor and can promote the proliferation of fibroblasts and secretion of collagens, playing an important role in the myocardial fibrosis [2]. However, how the PDGF-BB acts to activate fibroblasts and mediate the myocardial fibrosis is still poorly understood. Our previous study showed DOCA induced fibrosis in rats had significantly increased expression of PDGF and its receptors (PDGFRs) which were mainly localized in fibroblasts and myofibroblasts. In addition, PDGFR-β plays a key role in the proliferation, transformation and collagen secretion of fibroblasts [3,4]. PI3K/Akt signaling pathway is an important regulatory pathway and its activation has been found to be involved in the regulation of cell proliferation, migration, differentiation and angiogenesis [5]. In the present study, cardiac fibroblasts (CFs) were separated and purified, and then treated with PDGF-BB. The proliferation, transformation, collagen secretion, PDGFR and factors in the PDGFR-β/PI3K/Akt signaling pathway were determined in these CFs, aiming to explore the role of PDGF-BB in the pathogenesis of myocardial fibrosis and its down-stream mechanisms.
Materials and methods
Separation, purification and identification of CFs
A total of 30 SD rat pups aged 1-3 days were purchased from the Experimental Animal Center of Anhui Medical University. Under an aseptic condition, the ventricles were collected and cut into blocks. After washing in PBS thrice, these heart blocks were digested in 0.08% trypsin at a volume ratio of 1:10. The resultant suspension was allowed to stay at room temperature for 2 min. This cell suspension was transferred into a 15-ml centrifuge tube, and high glucose DMEM (10% HG/DMEM) containing 10% FBS, 100 mg/L streptomycin and 100 mg/L penicillin was added to stop the digestion. The residual heart blocks were digested repeatedly with above procedures. Then, centrifugation was done at 1200 r/min for 4 min, and the cells were re-suspended in 10 ml of 10% HG/DMEM. The resultant cell suspension was transferred into a 25-mm flask, followed by incubation for 90 min. After addition of 3 ml of 10% HG/DMEM, incubation continued, and the resultant cells were cardiac fibroblasts (CFs). The medium was refreshed once every 2 days. When the cell confluence reached 80%, passaging was done at a ratio of 1:2. Cells of the 2nd to 5th generation were used in the following experiments. CFs of 2nd generation were seeded into coverslip containing 6-well plates at a density of 104 cells/ml. Double fluorescence staining was done 48 h later for identification of CFs. In brief, the coverslips were washed 5 times with PBS (5 min for each), fixed in 40 g/L paraformaldehyde for 20 min, washed in PBS 5 times (5 min for each), blocked in 0.2% Triton X-100 at room temperature for 30 min, washed in PBS 5 times (5 min for each), blocked in 100 µl of normal goat serum for 30 min, treated with 100 µl of rabbit anti-rat vimentin (1:100; Abcam) at 4°C over night, washed in PBS 5 times (5 min for each), and incubated with 100 µl of mixture containing FITC conjugated goat anti-rabbit antibody (1:200) and DAPI (1:500) at room temperature in dark for 30 min. After washing in PBS thrice (5 min for each), cells were observed under a fluorescence microscope, and the positive cells were counted. Above procedures were also used to detect α-SMA (Abcam) positive cells.
Grouping
After separation and purification, CFs with stable status were divided into following groups: (1) blank control group (CON): cells were not treated; (2) PDGF-BB group (P): cells were treated with PDGF-BB (PeproThech); (3) PDGF-BB+IMA group (IMA): cells were treated with 20 ng/ml PDGF-BB and 100 µmol/l imatinib (IMA; a PDGFR phosphorylase inhibitor; Sigma); (4) PDGF-BB+LY294002 group (LY): cells were treated with 20 ng/ml PDGF-BB and 13 umol/l LY294002 (an inhibitor of PI3K). Cells were seeded at the same density and cultured in HG/DMEM containing 10% FBS.
Proliferation and transformation of CFs
MTT assay was used to detect the CF proliferation. In brief, cells were divided into 5 groups: medium control group (medium alone); cell control group (CON; cells and medium; P group, IMA group and LY group. CFs were seeded into a 96-well plate (5 wells/group). After culture for 48 h, cells were incubated with 5 mg/ml MTT (20 µl/well) for 4 h. The medium was removed, and DMSO (150 µl/well) was added, followed by incubation for 10 min. Optical density (OD) was measured at 490 nm with an automatic microreader. Experiment was done thrice, and the means were used to delineate a curve. Detection of CF transformation by immunofluorescence staining: Cells in CON group, P group and LY group were seeded into coverslip containing 6-well plates at a density of 104 cells/ml, followed by incubation for 7 d. Double immunofluorescence staining was done to detect α-SMA expression. Under a fluorescence microscope, α-SMA positive cells were counted.
mRNA expression of PDGFR-α, PDGFR-β, PI3K, Akt, Col I and Col III
Cells were seeded into 6-well plates (1 ml/well; 1 × 106 cells/well), and incubation was done in an environment with 5% CO2 for 24 h at 37°C. Cells of different groups were further cultured in conditioned medium for 48 h. Total RNA was extracted with TRIzol reagent, and the concentration and purification of total RNA were determined by UV spectrophotometry. The ratio of absorbance at 260 nm to that at 280 nm ranged from 1.8 to 2.0. Then, 5 µg of RNA was used for reverse transcription into cDNA with PrimeScriptTM RT reagent Kit (TaKaRa). PCR was done with a real time fluorescence quantification kit (SYBR Green, Qiagen) under following conditions: pre-denaturation at 95°C for 35 s, and 40 cycles of denaturation at 95°C for 5 s and annealing at 60°C for 34 s. Data were analyzed with ABI7500 analysis software, and Ct value was obtained. The 2-ΔΔCt method was used to calculate the relative expression of PDGFR-α, PDGFR-β, PI3K, Akt, Col I and Col III. The fold change of each gene was calculated as follow: ΔΔCt = Ct (target gene)-Ct (reference gene). Three samples were included in each group and experiment was repeated thrice. Primers were synthesized in Shanghai Sangon and are shown in Table 1.
Table 1.
Primers for different target genes used in this study
| Gene | Primers |
|---|---|
| PDGFR-α | Sense: CCTGGCATGATGGTTGATTCTACTT |
| Anti-sense: GGTCTCTTTTCGGGTTCACTGTTC | |
| PDGFR-β | Sense: GCACCGAAACAAACACACCTT |
| Anti-sense: ATGTAACCACCGTCGCTCTC | |
| PI3K | Sense: CTTGCCTCCATTCACCACCTCT |
| Anti-sense: GCCTCTAATCTTCTCCCTCTCCTTC | |
| Akt | Sense: CCGTTATCTTGATGTGCCCGTC |
| Anti-sense: TGTCTCGTGAGCGCGTGTTTT | |
| Col I | Sense: ACGCATGAGCCGAAGCTAAC |
| Anti-sense: AGGGACCCTTAGGCCATTGT | |
| Col III | Sense: ATAGACCTCAAGGCCCCAAG |
| Anti-sense: CCACCCATTCCTCCGACT |
Detection of protein expression of PDGFR-β, p-PDGFR-β, PI3K, p-PI3K, Col I and Col III
Cells in logarithmic growth phase were seeded into 6-well plates (1 ml/well; 1 × 106/well) and incubated at 37°C in an environment with 5% CO2 for 24 h. After culture in conditioned medium for 48 h, cells were digested and centrifuged, and CFs were harvested. Western blot assay was employed to detect the protein expression of PDGFR-β, p-PDGFR-β, PI3K, p-PI3K, Col I and Col III, and β-actin served as an internal reference. Cells were lysed with RIPA lysis buffer (1 ml) containing 1 mM PMSF, and total protein was extracted. BCA method was used to quantify the protein concentration. Then, proteins of equal amount were mixed with 2X loading buffer of equal volume, following by boiling for 10 min for denaturation. Then, these proteins were subjected to SDS-PAGE and transferred onto PVDF membrane. The membrane was incubated with primary antibody at 4°C over night and then treated with HRP conjugated secondary antibody (1:10000) at room temperature for 2 h. HRP-ECL substrate was used for visualization, and protein bands were captured, followed by detection of OD of each band. Image software was used to measure the OD of each band which was then normalized to that of actin. The OD in CON was defined as 1. Mouse anti-rat PDGFR-β antibody was purchased from Abcam, mouse anti-rat p-PDGFR-β, rabbit anti rat PI3K, rabbit anti-rat p-PI3K, rabbit anti-rat Col I, rabbit anti-rat Col III antibodies (Bioworld), goat anti-mouse secondary antibody and anti-goat/anti-rabbit secondary antibody (Beijing Zhongshan Goldenbridge) were used in the detection.
Statistical analysis
Statistical analysis was performed with SPSS version 13.0. Data were expressed as mean ± standard deviation (mean ± SD). Comparisons among groups were done with Dunnett-t test. A value of P < 0.05 was considered statistically significant.
Results
Identification of CFs
Under an inverted microscope, cells were observed. Cells immediately separated were ball-like and had different sizes. After culture for 90 min, cells became to be adherent (Figure 1A). CFs were firstly adherent to the wall, and had irregular shape or oval shape, and later became long-spindled. After culture for 2-3 days, cells began to proliferate and cell merge was observed (Figure 1B). These cells arranged closely and there was overlapping growth among these cells. There was no spontaneous beating. When the cell confluence reached 80%, cells were passaged. Cells of 5th generation showed obviously aging. Vimentin is the most important intermediate filaments among interstitial cells. Results showed CFs expressed vimentin which could be used to identify CFs. In addition, cardiomyocytes, smooth muscle cells and myofibroblast are positive α-SMA, and negativity for α-SMA may exclude these cells and be used for further identification of CFs. Under a fluorescence microscope, vimentin or α-SMA positive cells had green fluorescence and both proteins were mainly found in the cytoplasm. Blue fluorescence represented nucleus (DAPI). CFs were strong positive for vimentin, and the positive rate was about 90%. Immunofluorescence staining of α-SMA also revealed that about 90% of cells were negative for α-SMA (Figure 2).
Figure 1.
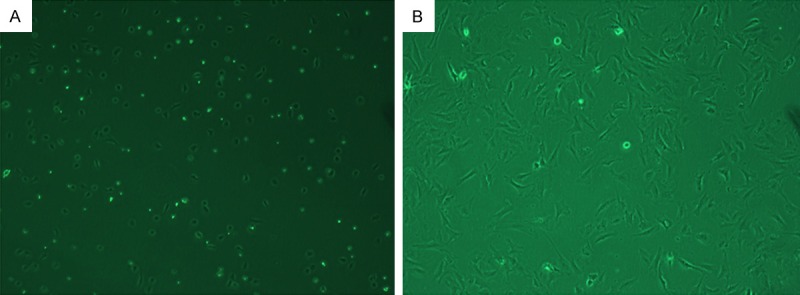
CFs under a light microscope. A: Primary cells (90 min; ×100), cells became to be adherent; B: After culture for 3 days (×100), cells began to proliferate and cell merge was observed.
Figure 2.
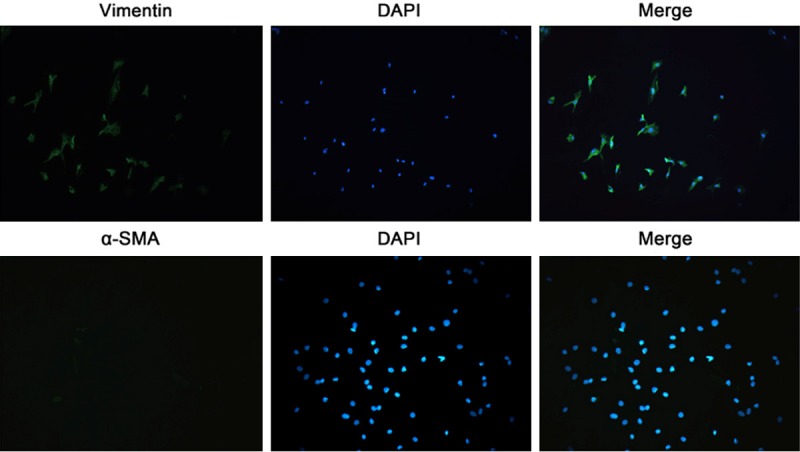
Vimentin and α-SMA expression in CFs by immunofluorescence. Expressed vimentin and negativity for α-SMA be used for further identification of CFs. The positive rate of Vimentin was about 90%, and about 90% of cells were negative for α-SMA (immunofluorescence staining; ×200).
Proliferation and transformation of CFs
After culture for 48 h, OD of each well was measured. OD represents the density absorbed by the detected substance. The higher the OD, the more the cells are; the lower the OD, the lesser the cells are. Thus, OD value can be used to reflect the number of cells. In P group, the OD was 0.4991 ± 0.009, which was significantly higher than that in CON group (0.2072 ± 0.0028; P < 0.05). In IMA group, the OD was 0.2783 ± 0.0193, which was markedly lower than that in P group (P < 0.05). In LY group, the OD was 0.2323 ± 0.0059, which was significantly lower than that in P group (P < 0.05). As shown in Figure 3, fluorescence microscopy showed more than 90% of cells were positive for α-SMA, but nearly none were positive for α-SMA in CON group, showing significant difference (P < 0.01). However, about 10% of cells were positive for α-SMA in LY group, which was markedly lower than that in P group (P < 0.01) (Figure 4). Thus, we speculate that the PDGFR-β/PI3K/AKT signaling pathway might be involved in the PDGF-BB induced proliferation of fibroblasts and their transformation into CFs.
Figure 3.
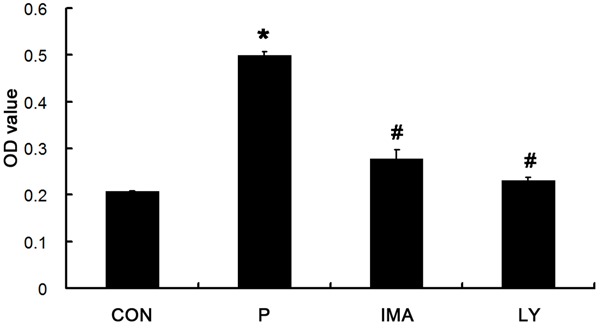
Effect of PDGF-BB, IMA and LY294002 on the proliferation of CFs. The number of cells in P group was significantly higher than CON group (*P < 0.05 vs. CON), and it was significantly lower in IMA and LY group than that P group, (#P < 0.05 vs. P group).
Figure 4.
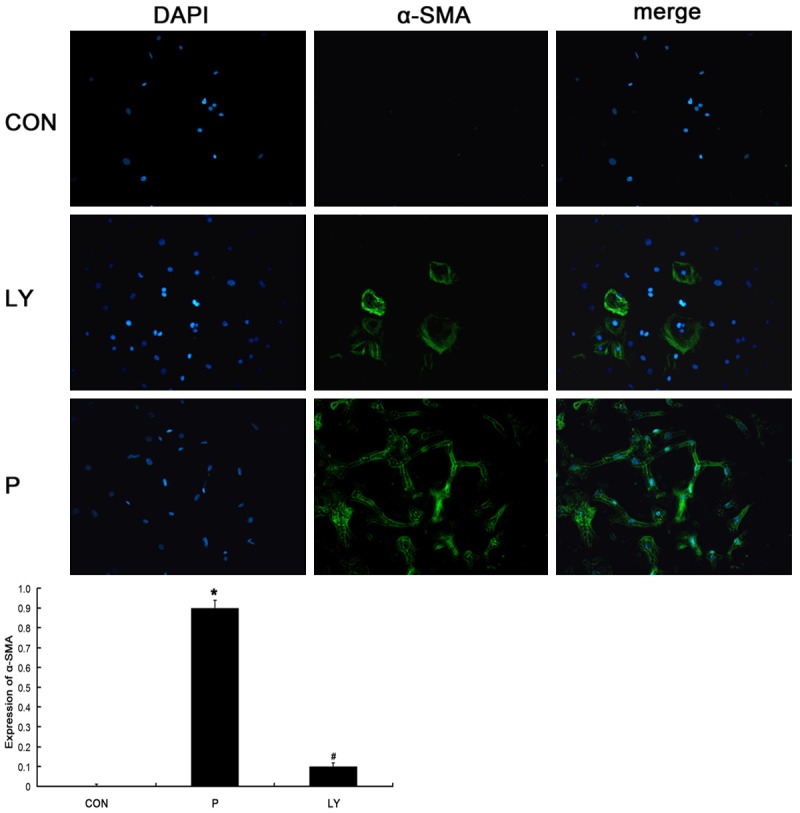
Effect of PDGF-BB, IMA and LY294002 on the α-SMA expression in CFs. Nearly none were positive for α-SMA in CON group and more than 90% of cells were positive for α-SMA in P group, however, about 10% of cells were positive for α-SMA in LY group. *P < 0.05 vs. CON; #P < 0.05 vs. P group.
mRNA expression of PDGFR-α, PDGFR-β, PI3K, Akt, Col I and Col III
In P group, the relative mRNA expression of PDGFR-β was 1.257 ± 0.3263, which was significantly higher than that in CON group (0.2024 ± 0.1995; P < 0.01). In IMA group, the relative mRNA expression of PDGFR-β was 0.4914 ± 0.0875, which was markedly lower than that in P group (P < 0.05). In LY group, the relative mRNA expression of PDGFR-β was 0.5517 ± 0.1473, which was significantly lower than that in P group (P < 0.05). The mRNA expression of PI3K in CON group was 0.0159 ± 0.0124, which was significantly lower than that in P group (0.0595 ± 0.0101; P < 0.05). In IMA group and LY group, the PI3K mRNA expression was 0.0178 ± 0.0062 and 0.0249 ± 0.0013, which were markedly lower than that in P group (P < 0.05). The mRNA expression of AKT in P group was 0.1212 ± 0.0356, which was significantly higher than that in CON group (P < 0.01). In IMA group and LY group, the AKT mRNA expression was 0.0477 ± 0.0121 and 0.0587 ± 0.0049, respectively, which was significantly lower than that in P group (P < 0.01). In IMA group, the mRNA expression of Col I and Col III was 1.0246 ± 0.3173 and 1.1173 ± 0.4278, respectively, which were significantly lower than those in P group (9.8151 ± 0.5897 and 12.1257 ± 0.199, respectively; P < 0.01). In IMA group, the mRNA expression of Col I and Col III was 2.9383 ± 0.032 and 1.8025 ± 0.055, respectively, which were markedly lower than those in P group (P < 0.01). In LY group, the mRNA expression of Col I and Col III was 2.3867 ± 0.0568 and 1.7351 ± 0.009, respectively, which were significantly lower than those in P group (P < 0.01). The mRNA expression of PDGFR-α was 0.1363 ± 0.0436 in CON group, 0.156 ± 0.123 in P group, 0.1046 ± 0.0248 in IMA group and 0.2076 ± 0.0442 in LY group, and no significant difference was observed between P group and CON group (P > 0.05). In addition, the mRNA expression of PDGFR-α in P group was also comparable to that in IMA group and LY group (P > 0.05) (Figure 5).
Figure 5.
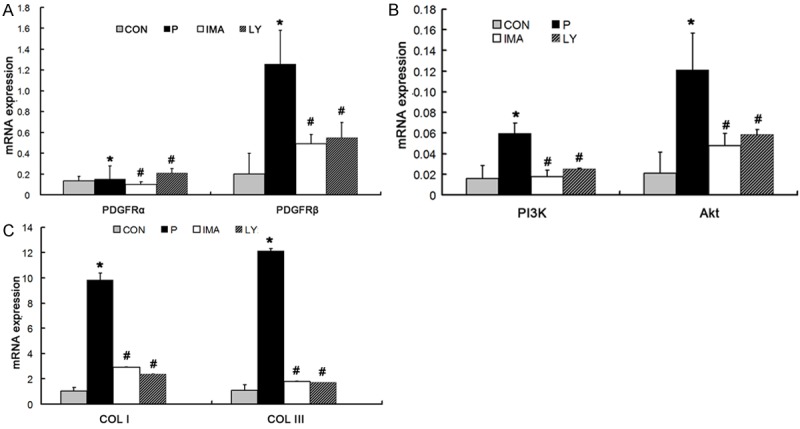
mRNA expression of different factors in PI3K/AKT signaling pathway in Con group, P group, IMA group and LY294002 group. A: No significant difference was observed between four group of PDGFRα, and the mRNA expression of PDGFRβin P group was significantly higher than other three groups. B: The mRNA expression of PI3K and Akt in P group was significantly higher than other three groups. C: The mRNA expression of Col I and Col III in P group was significantly higher than other three groups. *P < 0.05 vs. CON; #P < 0.05 vs. P group.
Protein expression of PDGFR-β, p-PDGFRβ, PI3K, p-PI3K, Col I and Col III
Western blot assay showed the OD of p-PDGFRβ and p-PI3K in P group was 2.3158 ± 0.554 and 2.2895 ± 0.788, respectively, which were significantly higher than those in CON group (0.5145 ± 0.147 and 0.9983 ± 0.431, respectively; P < 0.05). In IMA group, the OD of p-PDGFRβ and p-PI3K was 1.1257 ± 0.231 and 1.4132 ± 0.255, respectively, which were markedly lower than those in P group (P < 0.05). In LY group, the OD of p-PDGFRβ and p-PI3K was 0.5051 ± 0.135 and 0.9912 ± 0.238, which were markedly lower than those in P group (P < 0.05). In P group, the OD of PDGFR-β and PI3K was 1.7521 ± 0.734 and 1.4243 ± 0.633, respectively, which were markedly higher than those in CON group (0.3266 ± 0.301 and 0.5578 ± 0.304, respectively; P < 0.05). In IMA group, the OD of PDGFR-β and PI3K was 0.7094 ± 0.2 and 0.6675 ± 0.34, respectively, which were markedly lower than those in P group (P < 0.05). In LY group, the OD of PDGFR-β and PI3K was 0.3185 ± 0.085 and 0.2754 ± 0.186, which were markedly lower than those in P group (P < 0.05). The protein expression of Col I and Col III in P group was 1.2331 ± 0.527 and 1.5527 ± 0.758, respectively, which were significantly higher than that in CON group (0.3098 ± 0.181and 0.5591 ± 0.154, respectively; P < 0.05). In IMA group, the OD of Col I and Col III was 0.696 ± 0.154 and 0.9479 ± 0.245, respectively, which were markedly lower than those in P group (P < 0.05). In LY group, the OD of Col I and Col III was 0.4874 ± 0.118 and 0.6037 ± 0.225, respectively, which were markedly lower than those in P group (P < 0.05; Figure 6).
Figure 6.
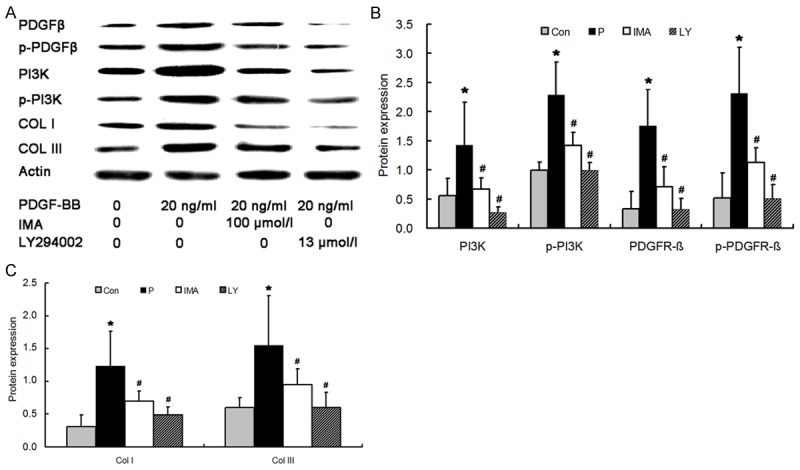
Protein expression of PDGFR-β, p-PDGFR-β, PI3K, p-PI3K (A, B), Col I and Col III (A, C) in Con group, P group, IMA group and LY294002 group. The protein expression of PDGFR-β, p-PDGFR-β, PI3K, p-PI3K, Col I and Col III in P group were significantly higher than other three groups. *P < 0.05 vs CON group; #P < 0.05 vs. P group.
Discussion
Myocardial fibrosis is as a result of excessive deposition of ECM in the myocardium. Collagen is a major component of ECM. In the myocardium, there are collagens I, III, IV, V and VI, and collagen and collagen III account for more than 90% of ECM. Moreover, these collagens are secreted by cardiac fibroblasts and myofibroblasts transformed from these fibroblasts [6]. Under normal conditions, there is a small amount of collagens in the myocardium which is essential for the maintenance of normal structure and function of the heart. In addition, the degradation of collagens in the interstitium is regulated by MMP and TIMP [7]. Under normal conditions, the synthesis and degradation of collagens are dynamically balanced, which avoids the fibrosis. However, under pathological conditions (such as denaturation or necrosis of cardiomyocytes), some inflammatory cells (such as monocytes and macrophages) aggregate and secret a lot of growth factors (such as PDGF), resulting in the proliferation of CFs, which induces the transformation of CFs into myofibroblasts. The consequence is excessive synthesis of collagens overwhelming their degradation, causing myocardial fibrosis [8]. Currently, the mechanisms underlying the activation of fibroblasts by PDGF and its upstream factors are still unclear.
PDGF is a potent growth factor that can promote the division of fibroblasts. In recent years, a series of studies demonstrate that members of PDGF family are involved in the pathogenesis of myocardial fibrosis [9]. In the fibrosis of several organs, there are interstitial inflammation and excessive secretion of PDGF by inflammatory cells such as monocytes and macrophages, which is also accompanied by increased expression of PDGFR on the fibroblasts. A variety of studies have revealed that the myocardial fibrosis is closely associated with the activation of PDGF signaling pathway and excessive division and proliferation of fibroblasts [10,11]. PDGF family includes PDGF-AA, PDGF-BB, PDGF-CC, PDGF-DD and PDGF-AB. PDGFR belongs to tyrosine kinase receptor and includes PDGFR-α and PDGFR-β. Thus, the PDGFR signaling pathway can be classified as PDGFR-α signaling pathway and PDGFR-β signaling pathway [12]. PDGFR without binding to PDGF exist in the form of inactivated monomers. When PDGF binds to the extracellular domain of PDGFR, PDGFR forms homodimer, resulting in phosphorylation of tyrosine residues. The activated tyrosine may further activate down-stream factors, mediating different cell activities, such as cell growth and differentiation. PDGF-AA and PDGF-CC are the major ligands of PDGFR-α and may act in the PDGFR-α signaling pathway. PDGF-BB is mainly involved in the PDGFR-β mediated myocardial fibrosis [13]. There is evidence showing that PDGFR-α signaling pathway exerts effects at the early stage of myocardial fibrosis and PDGFR-β signaling pathway acts in the whole process of myocardium fibrosis [3,4]. Thus, PDGFR-β signaling pathway is more important in the myocardial fibrosis when compared with PDGFR-α signaling pathway. Thus, the present study focused on the PDGFR-β signaling pathway.
The downstream pathway of PDGFR-β is very complex and has cross talk with a lot of pathways. Studies have revealed that PI3K/Akt signaling pathway plays important roles in the cell proliferation, migration, differentiation and angiogenesis [14]. Studies on CFs treated with aldosterone, angiotensin II and statins showed PI3K/Akt signaling pathway was involved in the pathological processes of fibrosis [15-19]. Studies on cancer prevention reveal PDGF may act on PI3K signaling pathway to regulate the expression of proteins related to cell metabolism, apoptosis, proliferation and differentiation, and to inhibit the PI3K signaling pathway may kill cancer cells [20]. In lung fibrosis and liver fibrosis animal models, results also demonstrate that PDGFR-β and its downstream PI3K/Akt are involved in the proliferation, transformation and collagen synthesis of fiber cells [21,22]. In studies on the vascular repair and angiogenesis, PDGFR-β was found to function via PI3K/Akt signaling pathway in the endothelial progenitor cells with PDGFR-β over-expression [23]. To date, few studies have been conducted to investigate the role of PDGFR-β and PI3K/Ak signaling pathway in the myocardium fibrosis.
In our previous study on mineralocorticoid induced myocardial fibrosis in rats, the expression of PDGFs and PDGFRs increased significantly, PDGFRs were mainly localized in fibroblasts and myofibroblasts and the number of fibroblasts and myofibroblasts and collagen secretion increased dramatically, suggesting that PDGFs/PDGFRs are involved in the pathogenesis of myocardial fibrosis [4]. However, the role of PDGFR-β/PI3K/Akt signaling pathway in the myocardial fibrosis is still poorly understood. In the present study, differential adherent method was employed to separate fibroblasts, and the purified CFs were treated with PDGF-BB. Results showed the PDGFR-β/PI3K/Akt signaling pathway was significantly activated, and collagen secretion increased obviously. After addition of IMA (an inhibitor of PDFGR phosphorylase) or LY294002 (an inhibitor of PI3K signaling pathway), the PDGFR-β/PI3K/Akt signaling pathway was markedly inhibited, accompanied by reduction in collagen secretion. Our study for the first time confirmed that PDGFR-β/PI3K/Akt signaling pathway is involved in the pathogenesis of fibroblast induced myocardial fibrosis.
Studies have shown that PDGF-BB treatment may markedly increase the number of fibroblasts. Immunofluorescence staining showed the proportion of myofibroblasts was higher than 90%, suggesting that fibroblasts transform into myofibroblasts. However, after addition of PDGFR phosphorylase inhibitor IMA, the myofibroblasts reduced dramatically. However, the PDGFR-α expression remained unchanged after PDGF-BB treatment, suggesting that PDGFR-α is not involved in the PDGF-BB induced myocardial fibrosis. In addition, after addition of LYA294002 (an inhibitor of PI3K/Akt signaling pathway), the fibroblasts and myofibroblasts reduced significantly accompanied by decreased collagen synthesis. Thus, we speculate that, in the fibroblast induced myocardial fibrosis, PDGF-BB binds to and activates PDGFR-β on fibroblasts. The phosphorylated PDGFR-β further activates PI3K/Akt signaling pathway, resulting in proliferation of CFs and their transformation into myofibroblasts, which significantly increases collagen synthesis and finally cause myocardial fibrosis.
Taken together, CFs were separated and cultured in vitro in the present study, and PDGF-BB was used to prepare the CF induced myocardial fibrosis in vitro. Our results showed PDGF-BB could induce the proliferation of CFs and their transformation into myofibroblasts. PDGFR phosphorylase inhibitor IMA and PI3K/Akt signaling pathway inhibitor LY294002 could markedly attenuate myocardial fibrosis. These findings suggest that PDGFRβ/PI3K/AKT signaling pathway is involved in the CF induced myocardial fibrosis after PDGF-BB treatment, but the specific mechanism is required to be further studied.
Disclosure of conflict of interest
None.
References
- 1.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–2323. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 3.Fan B, Ma L, Li Q, Wang L, Zhou J, Wu J. Role of PDGFs/PDGFRs signaling pathway in myocardial fibrosis of DOCA/salt hypertensive rats. Int J Clin Exp Pathol. 2014;7:16–27. [PMC free article] [PubMed] [Google Scholar]
- 4.Fan B, Ma L, Li Q, Wang L, Zhou J, Wu J. Correlation between platelet-derived growth factor signaling pathway and inflammation in desoxycorticosterone-induced salt-sensitive hypertensive rats with myocardial fibrosis. Int J Clin Exp Pathol. 2013;6:2468–2475. [PMC free article] [PubMed] [Google Scholar]
- 5.Sui JQ, Xie KP, Zou W, Xie MJ. Emodin Inhibits Breast Cancer Cell Proliferation through the ERalpha-MAPK/Akt-Cyclin D1/Bcl-2 Signaling Pathway. Asian Pac J Cancer Prev. 2014;15:6247–6251. doi: 10.7314/apjcp.2014.15.15.6247. [DOI] [PubMed] [Google Scholar]
- 6.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 7.Yari K, Rahimi Z, Moradi MT. The MMP-2 -735 C Allele is a Risk Factor for Susceptibility to Breast Cancer. Asian Pac J Cancer Prev. 2014;15:6199–6203. doi: 10.7314/apjcp.2014.15.15.6199. [DOI] [PubMed] [Google Scholar]
- 8.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leipner C, Grun K, Muller A, Buchdunger E, Borsi L, Kosmehl H, Berndt A, Janik T, Uecker A, Kiehntopf M, Bohmer FD. Imatinib mesylate attenuates fibrosis in coxsackievirus b3-induced chronic myocarditis. Cardiovasc Res. 2008;79:118–126. doi: 10.1093/cvr/cvn063. [DOI] [PubMed] [Google Scholar]
- 11.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 12.Wu E, Palmer N, Tian Z, Moseman AP, Galdzicki M, Wang X, Berger B, Zhang H, Kohane IS. Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS One. 2008;3:e3794. doi: 10.1371/journal.pone.0003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao CH, Akazawa H, Tamagawa M, Ito K, Yasuda N, Kudo Y, Yamamoto R, Ozasa Y, Fujimoto M, Wang P, Nakauchi H, Nakaya H, Komuro I. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J Clin Invest. 2010;120:242–253. doi: 10.1172/JCI39942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirton JP, Xu Q. Endothelial precursors in vascular repair. Microvasc Res. 2010;79:193–199. doi: 10.1016/j.mvr.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Huang PH, Chen YH, Tsai HY, Chen JS, Wu TC, Lin FY, Sata M, Chen JW, Lin SJ. Intake of red wine increases the number and functional capacity of circulating endothelial progenitor cells by enhancing nitric oxide bioavailability. Arterioscler Thromb Vasc Biol. 2010;30:869–877. doi: 10.1161/ATVBAHA.109.200618. [DOI] [PubMed] [Google Scholar]
- 17.Antonio N, Fernandes R, Rodriguez-Losada N, Jimenez-Navarro MF, Paiva A, de Teresa Galvan E, Goncalves L, Ribeiro CF, Providencia LA. Stimulation of endothelial progenitor cells: a new putative effect of several cardiovascular drugs. Eur J Clin Pharmacol. 2010;66:219–230. doi: 10.1007/s00228-009-0764-y. [DOI] [PubMed] [Google Scholar]
- 18.Chung CC, Hsu RC, Kao YH, Liou JP, Lu YY, Chen YJ. Androgen attenuates cardiac fibroblasts activations through modulations of transforming growth factor-beta and angiotensin II signaling. Int J Cardiol. 2014;176:386–93. doi: 10.1016/j.ijcard.2014.07.077. [DOI] [PubMed] [Google Scholar]
- 19.Tibaldi E, Zonta F, Bordin L, Magrin E, Gringeri E, Cillo U, Idotta G, Pagano MA, Brunati AM. The tyrosine phosphatase SHP-1 inhibits proliferation of activated hepatic stellate cells by impairing PDGF receptor signaling. Biochim Biophys Acta. 2014;1843:288–298. doi: 10.1016/j.bbamcr.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Ding J, Li XM, Liu SL, Zhang Y, Li T. Overexpression of platelet-derived growth factor-D as a poor prognosticator in endometrial cancer. Asian Pac J Cancer Prev. 2014;15:3741–3745. doi: 10.7314/apjcp.2014.15.8.3741. [DOI] [PubMed] [Google Scholar]
- 21.Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, Schirmacher P, Biesterfeld S, Galle PR, Lohse AW, Kanzler S. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45:419–428. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Li Y, Liang C, Yang W. CCN5 overexpression inhibits profibrotic phenotypes via the PI3K/Akt signaling pathway in lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis and in an in vivo model of lung fibrosis. Int J Mol Med. 2014;33:478–486. doi: 10.3892/ijmm.2013.1565. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Wang YC, Hu XB, Zhang BF, Dou GR, He F, Gao F, Feng F, Liang YM, Dou KF, Han H. Notch-RBP-J signaling regulates the mobilization and function of endothelial progenitor cells by dynamic modulation of CXCR4 expression in mice. PLoS One. 2009;4:e7572. doi: 10.1371/journal.pone.0007572. [DOI] [PMC free article] [PubMed] [Google Scholar]