

Abstract
Osteoarthritis (OA) is the most common joint disease affecting close to 27 million Americans. The pathological change of OA joint is characterized by cartilage degradation and osteophyte formation that have been associated with OA initiation and progression respectively. Upon OA progression, articular chondrocytes undergo hypertrophic differentiation, a process usually occurs only in growth plate chondrocytes during endochondral ossification, suggesting a role of chondrocyte hypertrophy in OA pathogenesis. However, how altered chondrocyte hypertrophy, i.e. accelerated or delayed chondrocyte hypertrophy, influences OA development has not been fully elucidated. We have previously generated transgenic (TG) mice over-expressing Runx2, an essential transcription factor for chondrocyte hypertrophy, using hypertrophic chondrocyte-specific mouse type X collagen gene (Col10a1) control elements. These Col10a1-Runx2 TG mice show delayed chondrocyte hypertrophy and apoptosis in long bone sections of embryonic and new-born mice compared to their wild-type (WT) littermates. Here, we report further analysis of the skeletal phenotypes of these mice at postnatal stages. We have performed histological analysis of 1-month old TG and WT mice. Delayed chondrocyte hypertrophy was also observed in growth plate of TG mice. In addition, μCT analysis showed that the femur length was significantly shorter in TG mice (p = 0.033). Thinner cortical bone and markedly decreased BV/TV were also detected in TG mice compared to their WT littermates (p = 0.027), suggesting that delayed chondrocyte hypertrophy affects postnatal long bone development. Interestingly, histological analysis detected less articular cartilage absorption, while immunohistochemistry assay detected upregulated Sox9 expression in TG mouse joints compared to WT controls, implying that delayed chondrocyte hypertrophy may be OA protective. Indeed, we have performed Tgf-β1 injection and enforced uphill treadmill running (TTR model) to induce OA in TG and WT littermates. The results showed that WT littermates displayed characteristic pathology of fibrotic remodeling at the joint margins and focal cartilage erosion, while the joints in TG mice were essentially protected from remodeling responses, demonstrating that mice with delayed chondrocyte hypertrophy are not susceptible to developing OA. Further translational studies characterizing the role of chondrocyte hypertrophy during OA progression will facilitate identification of therapeutic targets to stop or slow down this degenerative and progressive human joint disease.
Keywords: Col10a1, Runx2, transgenic mice, Tgf-β1 injection, treadmill running, chondrocyte hypertrophy or maturation, osteoarthritis
Introduction
Osteoarthritis (OA) is a common degenerative joint disease that mostly affects the middle-aged or the elder people [1]. OA is also a progressive disorder that causes severe joint pain and dysfunction as its prominent symptoms. Current treatment of OA focuses on relieving these symptoms, but no therapeutic targets can prevent, slow down, or reverse OA progression [2]. Notably, articular chondrocyte hypertrophy, a remarkable pathological change of OA, has recently drawn extensive attention from scientists in OA field, as the hypertrophic differentiation of articular chondrocytes mimics the behavior of growth plate chondrocytes during endochondral ossification [3,4]. This makes it possible to inhibit chondrocyte hypertrophy so as to slow down OA progression [5]. However, chondrocyte hypertrophy is a complicated process including terminal chondrocyte differentiation, vascular invasion, and chondrocyte apoptosis that are regulated by many transcription factors and signaling pathways [6]. Currently, mouse models with distinctive accelerated or delayed chondrocyte hypertrophy are still lacking. Therefore, the specific correlation of chondrocyte hypertrophy with OA development remains to be determined.
To target chondrocyte hypertrophy, the type X collagen gene (Col10a1), a specific marker of hypertrophic chondrocytes is always of special interest. Previous studies have indicated that physiological distribution of type X collagen during chondrocyte hypertrophy is essential for endochondral ossification. Meanwhile, abnormal Col10a1 expression is often accompanied with abnormal chondrocyte hypertrophy that has been seen in multiple skeletal disorders, including OA [7-12]. These findings suggest that regulators that direct cell-specific Col10a1 expression are expected to play a role in chondrocyte hypertrophy. We have recently shown that Runx2 is an indispensible Col10a1 transactivator [13-15], whereas Runx2 has been implicated as a master transcription factor both for osteoblast differentiation and for chondrocyte hypertrophy [16-19]. We have also performed Runx2 gain-of-function studies by over-expressing Runx2 in hypertrophic chondrocytes using the cell-specific Col10a1 control elements. Interestingly, these Col10a1-Runx2 TG mice show delayed chondrocyte hypertrophy and apoptosis at embryonic and early postnatal stages compared to the WT littermate controls [20]. In this manuscript, we further analyzed the skeletal phenotypes and confirmed that delayed chondrocyte hypertrophy was also observed in postnatal stage (1 month) of TG mice compared to WT littermates. This provides us an opportunity to examine the correlation of delayed chondrocyte hypertrophy with OA progression in TG mice and WT controls using Tgf-β1 injection and enforced uphill treadmill running (TTR) approach [21].
Materials and methods
Mouse model, breeding, and PCR genotyping
The Col10a1-Runx2 (Tg-Runx2) TG mice have recently been described [20]. Briefly, flag-tagged Runx2 cDNA was driven by hypertrophic chondrocyte-specific Col10a1 promoter and enhancer elements that we previously defined [14]. These TG mice are on a FVB/N genetic background and exhibit delayed ossification, chondrocyte hypertrophy and reduced chondrocyte apoptosis at embryonic and early postnatal stages compared to WT littermates [20]. To generate TG and WT littermates at designated postnatal stages for relevant phenotypic analysis, sex-matured Col10a1-Runx2 TG mice (8-10 weeks’ age) were crossed with wild-type FVB/N mice. The offspring of multiple breeding pairs of mice were weaned at the age of 3-4 weeks and mouse tail tissues (~0.5 cm long) were prepared for genomic DNA extraction. TG and WT Mice were identified by PCR genotyping using Runx2 and flag-specific primers (5’-CTTCCCAAAGCCAGAGTGGAC-3’ and 5’-TGTCGTCATCGTCTTTGTAGC-3’). The animal studies were approved by the IACUC (Institutional Animal Care and Use Committee) committee at Rush University Medical Center.
Histological analysis
For histological analysis, mouse hind limbs at the age of 1 month were collected and fixed in 10% formalin for two days. The limbs were then decalcified in 0.5 M EDTA for 14 days and subjected to dehydration, paraffin embedding, and sectioning. Whole-joint saggital sections (5 µm) were obtained from different locations of the lateral and medial compartments. Comparable slides from both TG and WT littermates were selected for standard H&E (Hematoxylin & Eosin) and Safranin O/Fast Green staining to identify cartilage cells and matrices. For Safranin O/Fast Green staining, after de-paraffin with xylene and gradient ethanol treatment, slides were stained in Fast Green solution (0.1%) for 2 minutes followed by Safranin O (0.1%) staining for 4 minutes. At least 10 sagittal sections of the joint from both TG and WT littermates were observed under microscope (Nikon Eclipse 80i, Nikon Instruments Inc., Melville, NY USA) and analyzed using the Qcapture Suite software (version, 2.95.0, Quantitative Imaging Corp., USA).
Micro computed tomography (µCT) analysis
Six right femurs from each of the age/sex matched TG and WT littermate controls were analyzed at the 1 month and 2 month stages using micro-CT approach. The femur samples were placed vertically within custom holders that fit within the manufacturer’s 12 mm specimen holder and scanned in saline (Scanco Model 40, Wayne, PA) using 55 kVp, 145 µA, 300 ms integration with 12 µm isotropic resolution. The trabecular bone region of interest were identified as the volume extending from ~40 µm proximal to the primary spongiosa of the distal growth plate to 30% of the length of the bone measured from the distal condyles. This region was segmented from the surrounding cortical bone, at threshold 320 and the manufacturer’s software was used to calculate bone volume per tissue volume (BV/TV), as well as the trabecular thickness (Tb.Th), trabecular spacing (Tb.Sp), and connectivity density. Femur length was also measured by above µCT scanner. Two-way Analysis of Variance (ANOVA) was used for statistical analysis followed by Bonferroni t-Test using SPSS software (version 14.0, Chicago, IL). p < 0.05 was considered significant.
Immunohistochemistry (IHC) analysis
For immunohistochemical (IHC) analysis, comparable pretreated mouse hind limb/knee joint sections from TG and WT mice at 1 month’s age were incubated with anti-Runx2 (M-70, sc-10758, Santa Cruz, CA) and anti-Sox9 (H-90, sc-20095, Santa Cruz, CA) antibodies as previously described [20]. The concentration for primary anti-Runx2 antibody was at 1:50 dilution and anti-Sox9 antibody at 1:300. Non-immune mouse IgG was used as a negative control. The biotinylated anti-rabbit IgG (Santa Cruz, CA) was used as a secondary antibody. The ABC kit (Elite PK-6200 Universal, VECTOR laboratories, Burlingame, CA) was used for detection. Slides were counterstained with nuclear fast red (Poly Scientific R&D Corp., NY) before microscopic analysis as described above.
TGFβ1 injection and treadmill running (TTR) approach
After PCR genotyping, 12 male mice of TG and WT littermates (6 each) at around 12 weeks were selected from the Col10a1-Runx2 transgenic mouse lines that have been described previously [20]. These mice were subjected to joint injury by repeated intra-articular injection of TGFβ1 into the right knee joints using literature reported protocols with modifications [21,22]. Briefly, 200 ng recombinant human TGFβ1 (hr TGFβ1, active form, PrepoTech Inc) in 6 µl of sterile saline (0.9% NaCl) containing 0.1% ultrapure BSA (Sigma) was injected into the right knee of 3 TG and 3 WT mice using a Kendall Monoject U-100 Insulin syringe and a 29G needle on days 1, 3 and 10. 6 µl saline containing BSA was injected into the right knee of other 3 TG and 3 WT mice as controls. Mice were then subjected to enforced running on a flat or an uphill (15°) gradient Stoelting/Panlab treadmill (Barcelona, Spain) at 24-32 cm/seconds for 20 minutes/day for 14 days.
Macroscopic evaluation of cartilage using India ink
The use of Indian ink on the cartilage surface was carried out essentially as previously described [21,23]. Briefly, mice knee joints were carefully opened and rinsed with PBS. The cartilage surface was painted with India ink for 15 seconds using a paint brush. After rinsing, the menisci and articular surfaces were photographed and subjected to microscopic image analysis using the Nikon dissecting microscope (SMZ1000, X6). Cartilage erosion was scored on a scale ranging from 0-8. Four areas (the anterior and posterior aspects of the lateral and medial compartments) were recorded based on a visual estimate of affected surface lesions of the fraction (0/8, 1/8, 2/8, 7/8, 1 etc). The healthy complete femoral or tibial surface reaches a highest score of 32.
Statistical analysis
Values are presented as means ± SEM. One-way ANOVA was used to assess the significance across experimental groups. Student t-tests were used to compare mean differences between groups. p < 0.05 was considered statistically significant. SSPS v16.0 (Chicago, IL) and GraphPad Prism 5.0 (La Jolla, CA) were used for graph and statistics analysis.
Results
Delayed chondrocyte maturation in Col10a1-Runx2 mice
We have recently generated multiple Runx2 transgenic mouse lines that over-express Runx2 in hypertrophic chondrocytes using the hypertrophic chondrocyte-specific Col10a1 gene controlling elements [20]. These Col10a1-Runx2 mice show upregulated Runx2 and Col10a1 expression but delayed chondrocyte hypertrophy compared to WT littermates. The growth plate of TG mice displays more layers and disorganized hypertrophic chondrocytes at late embryonic (E17.5) and postnatal day 1 (P1) stages (Figure 1A, 1C) [20]. The hypertrophic chondrocytes in TG mice undergo less apoptosis compared to WT controls as demonstrated previously [20]. Here, we have performed histological analysis (Safranin O staining) of tibia growth plates of both TG and WT mice at 1 month’s age (~4 weeks). As illustrated in Figure 1B (WT) and 1D (TG), the TG mice also show more layers of hypertrophic chondrocytes, suggesting delayed chondrocyte hypertrophy and apoptosis as seen in late embryonic and P1 stages [20].
Figure 1.
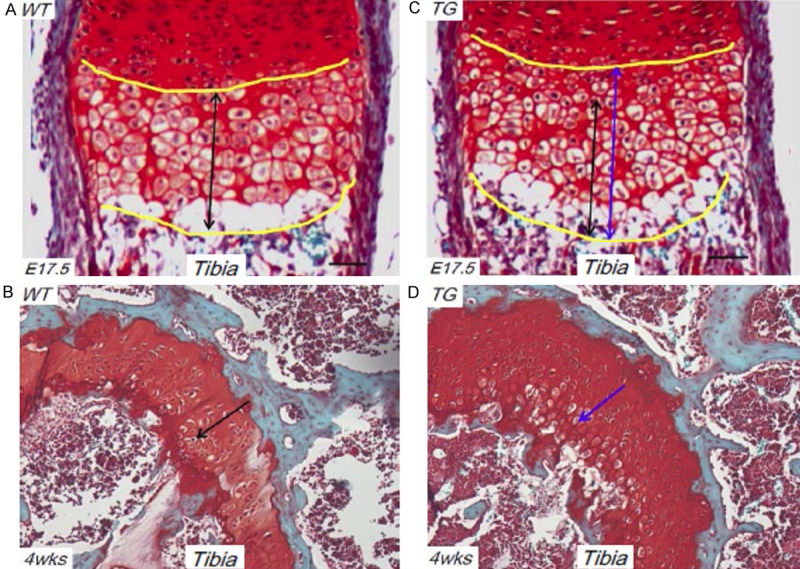
Delayed chondrocyte maturation in Col10a1-Runx2 mice. (A) Safranin O staining of sagittal section of tibia from E17.5 WT mouse embryo show organized hypertrophic chondrocytes as indicated by double black arrow. (B) Hypertrophic chondrocytes can be appreciated in tibia section of 4 wks WT mouse as shown by Safranin O staining (black arrow). (C) Compared to tibia section of E17.5 WT mouse (A), TG mouse displays much longer hypertrophic zone (more layers and disorganized hypertrophic chondrocytes) as indicated by double blue arrow. (D) Compared to tibia section of 4 wks WT mouse (B), 4 wks TG mouse also displays more layers and disorganized hypertrophic chondrocytes as indicated by blue arrow. WT: wild-type; TG: transgenic.
Impaired bone development in Col10a1-Runx2 mice
To examine how delayed chondrocyte maturation affects postnatal skeletal development, we performed skeletal phenotypic analysis using a micro-CT scanner (Scanco Model 40, Wayne, PA). The results showed that femur length was significantly shorter in TG mice compared to their WT littermates at the age of 1-month (p < 0.05), but not at 2-months (Figure 2A and data not shown). 3D imaging of microCT scanning detected thinner cortical bone in TG mice as measurement showing the bone and total area values (Figure 2B). The bone volume per tissue volume (BV/TV) was calculated and was markedly decreased in TG mice (p = 0.027, Figure 2C). No statistical significant difference was detected as to the trabecular thickness (Tb.Th), trabecular spacing (Tb.Sp), and connectivity density (data not shown).
Figure 2.
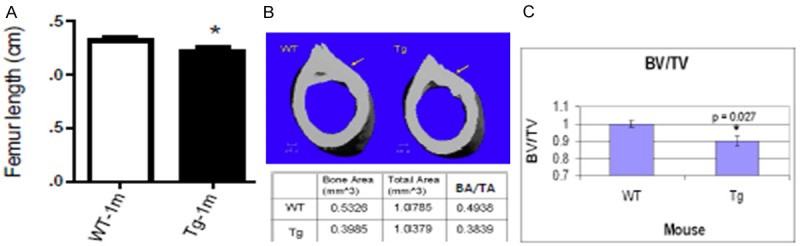
Impaired bone development in Col10a1-Runx2 mice. A. Femur length was significantly shorter in TG mice as compared to their WT littermates (Wt vs. Tg: 1.317 ± 0.03 vs. 1.218 ± 0.03, n = 6, t = 2.47, df = 10, p = 0.033) at 1-momth-age. B. 3D imaging of μCT scanning showed that TG mice have thinner cortical bone as compared to the WT controls. The bone and total area values were recorded in bottom panel. C. The bone volume per tissue volume (BV/TV) was markedly decreased in TG mice (Wt vs. Tg: 1.00 ± 0.02 Vs. 0.90 ± 0.03, n = 6, p = 0.027).
Less cartilage resorption and increased Sox9 expression in Col10a1-Runx2 mice
We have performed Safranin O/fast green staining of knee joint sections of 4-week old WT and TG mice. Much decreased cartilage resorption was observed at the femoral condyles of TG mice compared to the WT littermates (Figure 3A-D). We have also performed IHC analysis of these joint sections with anti-Runx2 and anti-Sox9 antibodies. Runx2 expression can be clearly appreciated in articular chondrocytes of WT mice, but diminished completely in the TG mouse joints (Figure 4A, 4B). Meanwhile, Sox9 was clearly expressed in articular chondrocytes of WT mice as expected. Notably, Sox9 was significantly upregulated in TG mouse joints compared to that of WT controls (Figure 4C, 4D), suggesting an anabonic effect in TG mice.
Figure 3.
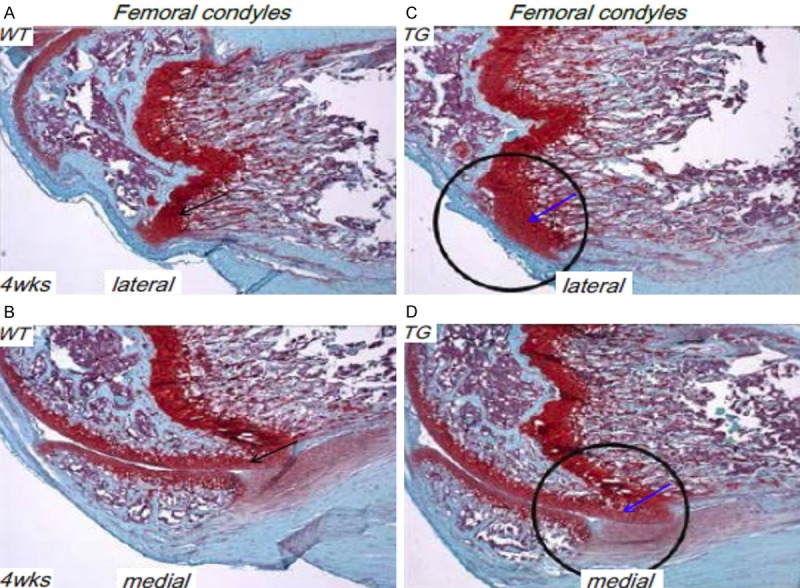
Less cartilage resorption in Col10a1-Runx2 mice. (A, B) Safranin O/fast green staining of sagittal section of femoral condyle from 4 wks WT mouse show clear cartilage resorption at the frontal margins of the proximal lateral (A) and medial (B) femoral condyles (black arrows). (C, D) Compared to the WT littermate controls, TG mice showed a much decreased cartilage resorption in corresponding lateral and medial femoral condyles (blue arrows).
Figure 4.
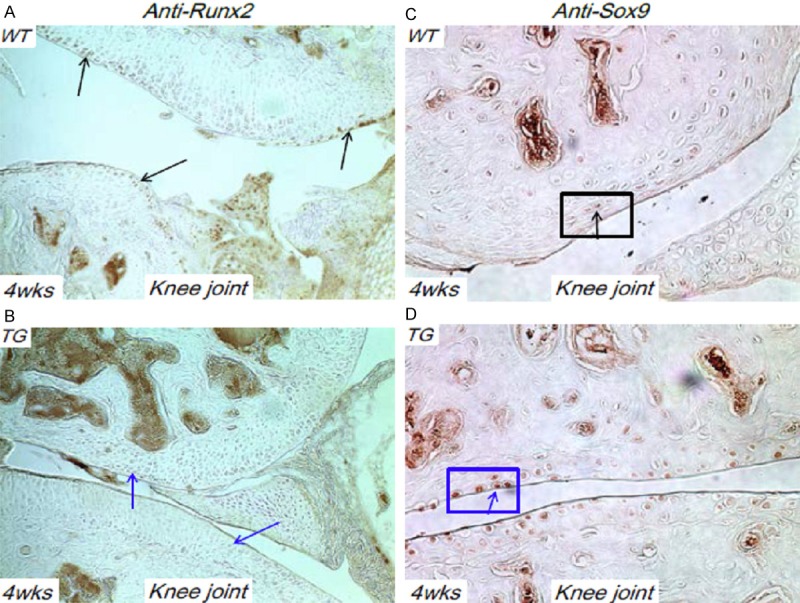
Runx2 and Sox9 expression in Col10a1-Runx2 mice. A. sagittal section of knee joint from 4 wks WT mouse was subjected to IHC analysis using anti-Runx2 antibody. Runx2 was clearly expressed in articular chondrocytes of WT mice (black arrow). B. No Runx2 expression was detected in comparable section of TG mouse (blue arrows). C. IHC analysis using sagittal section of knee joint from 4 wks WT mouse and anti-Sox9 antibody detect obvious Sox9 expression in articular chondrocytes (black arrow). D. Sox9 was significantly upregulated in articular chondrocytes of comparable TG mouse joint section compared to that of WT controls (blue arrow).
Protective effect in Col10a1-Runx2 mice upon OA induction
To determine how delayed chondrocyte hypertrophy influences cartilage integrity, we created joint injury by repeated intra-articular injection of TGFβ1 followed by treadmill overrunning to induce knee-joint OA in 12 weeks TG and WT controls [21,22]. As illustrated by India ink staining, the surface of tibial plateau in WT mice is rougher and displays shallow grooves compared to the smooth and shiny appearance in the joint surface of TG mice. Signs of synovitis were also shown in WT mice, but not in TG mice, suggesting different response of mice subjected to TGFβ1 injection and mechanic overuse (Figure 5A, 5B). Detailed histological analysis (H&E staining) of mouse knee joints showed that fibrotic remodeling (majorly cartilages erosions and fibrosis) at the margins of WT surfaces was seen but not in corresponding regions of the TG mice (Figure 5C, 5D). These results suggest a protection of delayed chondrocyte hypertrophy upon OA pathogenesis.
Figure 5.
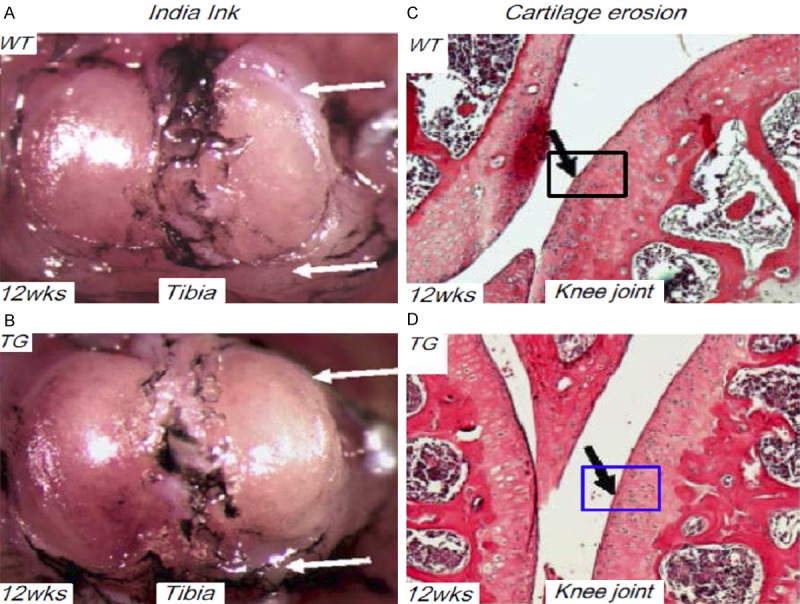
Protective effect in Col10a1-Runx2 mice upon OA induction. A. After repeated intra-articular TGFβ1 injection and treadmill overrunning, the representative mouse knee-joint of WT mouse at 12 weeks’ age was subjected to India ink staining. Clear rough joint surface/shallow grooves (top, white arrow) and signs of synovitis (bottom, white arrow) were observed. B. Compared to the WT mice, the corresponding joint surface of TG mice show smooth and shiny appearance and no signs of synovitis were shown (white arrows). C. H&E staining of WT mouse knee joints subjected to TTR treatment showed fibrotic remodeling (majorly cartilages erosions and fibrosis) at the margins of joint surfaces (black arrow). D. Compared to WT mice, the corresponding joint surface of TG mice did not show fibrotic remodeling as shown in WT mouse joints (black arrow).
Discussion
Runx2 impacts chondrocyte maturation during skeletal development and disorders
Runx2 is a well-known Runt domain containing transcription factor regulating both osteoblast differentiation and chondrocyte maturation [16-19]. Haploinsufficiency of RUNX2 in humans causes cleidocranial dysplasia (CCD) which is characterized by delayed closure of the fontanel, hypoplastic clavicles as well as short stature and cone epiphyses [9]. Interestingly, Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. This suggests that Runx2 fulfills antagonistic functions during chondrogenesis [24]. Runx2 was previously shown to be a candidate gene that promotes osteoarthritis progression [25]. In surgically induced OA mouse models, Runx heterozygotes showed decreased cartilage destruction and osteophyte formation, along with reduced type X collagen and MMP-13 expression compared with wild-type mice. This suggested that Runx2 contributes to OA pathogenesis through chondrocyte hypertrophy [4,25].
Mechanisms of impaired bone development and OA protection in Col10a1-Runx2 mice
Generally, Runx2 is a transactivator for Col10a1 that is known to upregulate cell-specific Col10a1 gene expression and promote chondrocyte hypertrophy [12-15,26-28]. Intriguingly, our Col10a1 -Runx2mice showed enhanced Runx2 and Col10a1 expression but delayed chondrocyte hypertrophy and less ossification at early skeletal developmental stages [20]. Consistently, delayed chondrocyte hypertrophy and apoptosis were also seen in TG mice at 1 month’s age, while micro-CT analysis detected significantly shorter femur length, less cortical bone, and markedly decreased BV/TV. These results demonstrate that delayed chondrocyte hypertrophy impaired endochondral ossification and skeletal development in Col10a1-Runx2 mice at both embryonic and postnatal stages (Figure 2) [20]. As to the potential mechanisms, we have shown that targeting Runx2 in hypertrophic chondrocytes upregulates Col10a1 as well as other marker genes, esp. Sox9, which dominantly over Runx2’s function [29]. This will change the local matrix environment, leading to delayed chondrocyte maturation, reduced apoptosis and matrix mineralization, and eventually, impaired endochondral ossification. It may also explain the chondroprotection in Col10a1-Runx2 mice upon OA induction, as less cartilage resorption and increased Sox9 expression was detected in articular chondrocytes of TG mice (Figures 3C, 3D, 4D). As mentioned above, articular chondrocyte hypertrophy is a remarkable pathological change of OA mimicking the endochondral pathway [3,4]. It has also been implicated that chondrocyte apoptosis contributes to OA [30,31], whereas Sox9 is a well-known promoter for early chondrogenesis but a major negative regulator for chondrocyte maturation and apoptosis during endochondral ossification [32,33].
Questions and future directions
It should be taken into consideration that many biological, genetics, and environmental factors have been shown to contribute to OA. Until now, there are no therapeutic targets that are able to cure or reverse OA progression. Given its specific correlation with OA, articular chondrocyte hypertrophy has recently drawn extensive attention in the field of cartilage biology. However, how altered chondrocyte hypertrophy and apoptosis affect OA development has not been fully elucidated. This is because articular chondrocyte hypertrophy is unlikely the single player for OA, while animal/mouse models that show definitive accelerated or delayed chondrocyte hypertrophy are still lacking, hindering characterization of the relationship between chondrocyte hypertrophy and OA. Our Col10a1-Runx2 mice show delayed chondrocyte hypertrophy. These mice are not susceptible to developing OA upon Tgf-β1 injection and enforced uphill treadmill running. However, due to limited resources, we only analyzed these mice using the single TTR model. Further characterization of these mice by surgically inducing OA using the DMM (destabilization of the medial meniscus) approach are needed to draw more reliable conclusions regarding the contribution (or protection) of altered chondrocyte hypertrophy upon OA development [21,34].
Acknowledgements
We are grateful to Drs. Rick Sumner, Meghan, Moran, and Mr. David Karwo for histological help and μCT analysis on transgenic mice at the Rush Histology Core. Dr. Anna Plaas from the Department of Biochemistry has helped to set up and analyze the TTR model. This work was supported by the National Cancer Institute (NCI/NIH) (R21CA161461, Q.Z.), the 2008 Rush pilot project (Q.Z.), the 2013 Innovation Program of Jiangsu Province (Q.Z.) and the NSFC (Nos. 31271399, 31440058, 81272034, 81472130, and 81472047, Q.Z., G.L., Y.L.).
Disclosure of conflict of interest
All authors have no conflict of interest.
References
- 1.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH National Arthritis Data Workgroup. Estimates of the Prevalence of Arthritis and Other Rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 2.Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, Malfait AM. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A. 2012;109:20602–20607. doi: 10.1073/pnas.1209294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drissi H, Zuscik M, Rosier R, O’Keefe R. Transcriptional regulation of chondrocyte maturation: potential involvement of transcription factors in OA pathogenesis. Mol Aspects Med. 2005;26:169–179. doi: 10.1016/j.mam.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Kawaguchi H. Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol Cells. 2008;25:1–6. [PubMed] [Google Scholar]
- 5.van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthriits Cartilage. 2012;20:223–232. doi: 10.1016/j.joca.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 7.Warman ML, Abbott M, Apte SS, Hefferon T, McIntosh I, Cohn DH, Hecht JT, Olsen BR, Francomano CA. A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nat Genet. 1993;5:79–82. doi: 10.1038/ng0993-79. [DOI] [PubMed] [Google Scholar]
- 8.Ikegawa S, Nishimura G, Nagai T, Hasegawa T, Ohashi H, Nakamura Y. Mutation of the type X collagen gene (COL10A1) causes spondylometaphyseal dysplasia. Am J Hum Genet. 1998;63:1659–1662. doi: 10.1086/302158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Q, Sebald E, Zhou G, Chen Y, Wilcox W, Lee B, Krakow D. Dysregulation of chondrogenesis in human cleidocranial dysplasia. Am J Hum Genet. 2005;77:305–312. doi: 10.1086/432261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamas JR, Rodríguez-Rodríguez L, Vigo AG, Alvarez-Lafuente R, López-Romero P, Marco F, Camafeita E, Dopazo A, Callejas S, Villafuertes E, Hoyas JA, Tornero-Esteban MP, Urcelay E, Fernández-Gutiérrez B. Large-scale gene expression in bone marrow mesenchymal stem cells: a putative role for COL10A1 in osteoarthritis. Ann Rheum Dis. 2010;69:1880–1885. doi: 10.1136/ard.2009.122564. [DOI] [PubMed] [Google Scholar]
- 11.Petit A, Wang HT, Girard-Lauriault PL, Wertheimer MR, Antoniou J, Mwale F. Novel insights into the mechanism of decreased expression of type X collagen in human mesenchymal stem cells from patients with osteoarthritis cultured on nitrogen-rich plasma polymers: implication of cyclooxygenase-1. J Biomed Mater Res A. 2010;94:744–750. doi: 10.1002/jbm.a.32739. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Qiao L, Lei G, Mira R, Gu J, Zheng Q. Col10a1 gene expression and chondrocyte hypertrophy during skeletal development and disease. Front Biol. 2014;9:195–204. [Google Scholar]
- 13.Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. 2003;162:833–842. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Q, Keller B, Zhou G, Napierala D, Chen Y, Zabel B, Parker AE, Lee B. Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J Bone Miner Res. 2009;24:1022–1032. doi: 10.1359/JBMR.081249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F, Lu Y, Ding M, Napierala D, Abbassi S, Chen Y, Duan X, Wang S, Lee B, Zheng Q. Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J Bone Miner Res. 2011;26:2899–2910. doi: 10.1002/jbmr.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 17.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 18.Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T, Komori T. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 19.Kim IS, Otto F, Zabel B, Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- 20.Ding M, Lu Y, Abbassi S, Li F, Li X, Song Y, Geoffroy V, Im HJ, Zheng Q. Targeting Runx2 expression in hypertrophic chondrocytes impairs endochondral ossification during early skeletal development. J Cell Physiol. 2012;227:3446–3456. doi: 10.1002/jcp.24045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Anemaet W, Diaz MA, Buchanan S, Tortorella M, Malfait AM, Mikecz K, Sandy JD, Plaas A. Knockout of ADAMTS5 does not eliminate cartilage aggrecanase activity but abrogates joint fibrosis and promotes cartilage aggrecan deposition in murine osteoarthritis models. J Orthop Res. 2011;29:516–522. doi: 10.1002/jor.21215. [DOI] [PubMed] [Google Scholar]
- 22.van Beuningen HM, Glansbeek HL, van der Kraan PM, van den Berg WB. Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-beta injections. Osteoarthritis Cartilage. 2000;8:25–33. doi: 10.1053/joca.1999.0267. [DOI] [PubMed] [Google Scholar]
- 23.Schmitz N, Laverty S, Kraus VB, Aigner T. Basic methods in histopathology of joint tissues. Osteoarthritis Cartilage. 2010;18(Supp 3):S113–116. doi: 10.1016/j.joca.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Hinoi E, Bialek P, Chen YT, Rached MT, Groner Y, Behringer RR, Ornitz DM, Karsenty G. Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev. 2006;20:2937–2942. doi: 10.1101/gad.1482906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G, Nakamura K, Chung UI, Kawaguchi H. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54:2462–2470. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- 26.Dong Y, Drissi H, Chen M, Chen D, Zuscik MJ, Schwarz EM, O’keefe RJ. Wnt-mediated regulation of chondrocyte maturation: Modulation by TGF-beta. J Cell Biochem. 2005;95:1057–1068. doi: 10.1002/jcb.20466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simões B, Conceição N, Viegas CS, Pinto JP, Gavaia PJ, Hurst LD, Kelsh RN, Cancela ML. Identification of a promoter element within the zebrafish colXalpha1 gene responsive to runx2 isoforms Osf2/Cbfa1 and til-1 but not to pebp2alphaA2. Calcif Tissue Int. 2006;79:230–244. doi: 10.1007/s00223-006-0111-6. [DOI] [PubMed] [Google Scholar]
- 28.Higashikawa A, Saito T, Ikeda T, Kamekura S, Kawamura N, Kan A, Oshima Y, Ohba S, Ogata N, Takeshita K, Nakamura K, Chung UI, Kawaguchi H. Identification of the core element responsive to runt-related transcription factor 2 in the promoter of human type X collagen gene. Arthritis Rheum. 2009;60:166–178. doi: 10.1002/art.24243. [DOI] [PubMed] [Google Scholar]
- 29.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson EO, Charchandi A, Babis GC, Soucacos PN. Apoptosis in osteoarthritis: morphology, mechanisms, and potential means for therapeutic intervention. J Surg Orthop Adv. 2008;17:147–152. [PubMed] [Google Scholar]
- 31.Kim HA, Blanco FJ. Cell death and apoptosis in osteoarthritic cartilage. Curr Drug Targets. 2007;8:333–845. doi: 10.2174/138945007779940025. [DOI] [PubMed] [Google Scholar]
- 32.Hattori T, Müller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bösl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B, von der Mark K. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137:901–911. doi: 10.1242/dev.045203. [DOI] [PubMed] [Google Scholar]
- 33.Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, Yoshikawa H, Tsumaki N. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development. 2011;138:1507–1519. doi: 10.1242/dev.057802. [DOI] [PubMed] [Google Scholar]
- 34.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK, Morris EA. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–648. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]