

Abstract
Acute hepatic injury causes high morbidity and mortality world-wide. Management of severe acute hepatic failure continues to be one of the most challenging problems in clinical medicine. In present study, carbon tetrachloride (CCl4) was used to induce acute liver damage in mice and the protective effects of ethanol extract of Portulaca Oleracea L. (PO) were examined. The aminotransferase activities were biochemical estimated and the liver damage was tested by morphological histological analysis and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. The role of PO on the activity of NF-κB was determined by luciferase reporter gene assay and immunohistochemistry. The level of p-p65 was tested by western blot. Our results showed that PO administration on mice would decrease the serum aminotransferase level and reduced the liver histological damage. We also found that nuclear translocation of p65 was enhanced in liver tissues of mice treated with PO compared with control animals. In addition, in cultured hepatic cells, PO increased the NF-κB luciferase reporter gene activity and upregulated the level of phosphorylation of p65, but had no effects on mice liver SOD activity and MDA level. Collectively, PO attenuated CCl4 induced mice liver damage by enhancement of NF-κB activity.
Keywords: Carbon tetrachloride (CCl4), ethanol extract of Portulaca Oleracea L (PO), liver injury, NF-κB
Introduction
Because of the important role in detoxification, the liver is one of the most easily damaged organs [1]. Liver injury can be caused by ischemia, viral infection, autoimmune disorders, and several xenobiotics, including drugs, alcohol, or toxins. Management of severe liver injury continues to be one of the most challenging problems in clinical medicine [2].
Carbon tetrachloride (CCl4) has been widely used to induce chronic and acute liver damage in various animal models for decades [3]. Studies have demonstrated that reactive oxygen species (ROS) are heavily involved in the cause and progression of hepatic damage by CCl4. It is generally believed that CCl4 toxicity results from the bioactivation of the CCl4 molecule to the trichloromethyl free radical by cytochrome P450 2E1 of the endoplasmic reticulum [4]. Once the trichloromethyl radical is formed, it reacts with molecular oxygen to form the highly toxic trichloromethyl peroxy radical [5-7]. The free radicals then attack on polyunsaturated fatty acids of membrane lipids to propagate a chain reaction, leading to lipid peroxidation. These chains of events result in the breakdown of membrane structure, disrupting cell energy processes and protein synthesis and finally induced hepatic injury or necrosis [8-10]. Antioxidative therapy is therefore an effective means of preventing and attenuating oxidative stress related liver diseases.
In addition to the direct induction of hepatocyte damage by CCl4, its hepatotoxicity can also be mediated by the indirect activation of Kupffer cells/macrophages and neutrophils [11]. Activation of Kupffer cells with production of reactive oxygen species, up-regulation of proinflammatory cytokines, and neutrophil accumulation have been identified as contributing events to the inflammation-associated damage. Parenchymal and non-parenchymal cells, especially activated Kupffer cells, mediate the hepatic inflammation process by producing tumor necrosis factor-α (TNF-α) and other cytotoxic cytokines [12]. A previous study showed that the production of these inflammatory factors is associated with the nuclear factor-κB (NF-κB) pathway in liver after CCl4 treatment [13].
The Portulaca Oleracea L. (Portulacaceae) is a warm-climate annual and has a cosmopolitan distribution. It is an edible plant and is used as a vegetable in some place of the world including United Arab Emirates, Oman and some provinces of China. Portulacaceae is known as “vegetable for long life” in Chinese folklore and is widely used as a traditional Chinese herbal medicine [14,15]. A wide range of other pharmacological effects of Portulacaceae, such as antibacterial, analgesic, anti-inflammatory and wound-healing activities have been reported [15,16]. In our previous work, we demonstrated the anti-oxidative effects of the ethanol extract of Portulacaceae (PO) [17,18]. Based on all these information, we hypothesized that the PO may have the protective effects on acute liver injury induced by CCl4.
In present study, we studied the effects of the PO on mice liver administrated with CCl4. Our results showed that the PO protected the mice liver from damage induced by CCl4 may partially through regulating the activity of NF-κB.
Materials and methods
Preparation of the PO
The air dried aerial parts of Portulacaceae were purchased from the market in Yucheng, Henan province, China. The plant was identified and authenticated as Portulaca oleracea L. (Portulacaceae) by professor Han-chen Zheng (Department of Pharmacology, Second Military Medical University). A voucher specimen has been deposited in Department of Traditional Chinese Medicine (TCM), Second Military Medical University (20090829). The plant (40 kg) were ground and extracted with 8 times 80% ethanol for 2 times (1 h/time). The solvent was evaporated under vacuum to get rid of ethanol. The remained liquid were modified with 10% NaOH to the PH 6.5~7, then centrifuged (5000 rpm) to get the precipitation and oven dried to get the end extract (241.3 g) which appears black powder, lightly odorless taste. With GC-MS method, the chemical constituents of the end extract were investigated. Chromatographic conditions showed as followed. The column temperature started with 60°C for two minutes, rose to 300°C at the rate of 20°C/min and sustained for five minutes. The injection temperature was 250°C; carrier gas He flow rate was 1.0 ml/min; E1 was 70eV; detection temperature was 200°C. The results showed that 51 chromatographic peaks were separated and 44 chemical constituents were identified including palmitic acid 17.06%, ethyl linoleate 16.04%, palmitic acid, ethyl ester 14.47%, linolenic acid 12.72%, and linoleic acid 7.66% whose content was determined with the peak area normalization method. The other part includes alkaloids, tannins and flavonoids.
Animal and diet
Male C57 mice (8-10 weeks old) were purchased from the Shanghai-BK Ltd. Co. After adaptation for three days, the mice were divided into PBS/CCl4 group, PBS/oil group, PO/CCl4 group and PO/oil group. The PO administrated mice were subdivided into 100 mg/kg, 200 mg/kg and 300 mg/kg group. Each experimental group consisted of 10 mice. They were housed at 24 ± 1°C under a 12-h light/12-h dark cycle and had free access to standard pellet diet and tap water. PO administrated mice were orally administrated with 100 mg/kg, 200 mg/kg and 300 mg/kg of PO in 0.5 ml PBS and PBS administrated mice were orally administrated with 0.5 ml PBS each day for one week. The animals were deprived of food for 24 h before the experiment, with free access to drinking water, and received 30% CCl4 in Oil (v/v, 1 μl/g body weight). All animal treatments were strictly in accordance with international ethical guidelines and the National Institutes of Health Guide concerning the Care and Use of Laboratory Animals, and the experiments were carried out with the approval of the Committee of Experimental Animal Administration of the University.
Experimental protocols
The in vivo anti-hepatotoxic activity of the PO was carried out against CCl4-induced hepatotoxicity in C57 mice. Twenty-four hours after CCl4 injection, all the animals were anesthetized using pentobarbitone (35 mg/kg, i.p.). The abdominal artery was isolated and about 0.5 ml of blood was collected by using a 24 gauge hypodermic needle. The blood was allowed to clot for 30 minutes at room temperature and the serum was separated by centrifugation at 2500 rpm for 15 minutes and used for biochemical estimations.
After collection of the blood, the liver was removed from each mouse and washed several times with normal saline. Part of the liver was taken for biochemical examination and the remaining tissue was preserved in 10% v/v formal saline buffer for histopathological analysis. The remaining liver was frozen in liquid nitrogen and stored at -80°C for later use.
Histology and immunohistochemistry
Formalin fixed tissue samples were embedded in paraffin and 5 ìm sections were cut. Replicate sections were stained with hematoxylin-eosin (H-E) staining for evaluation of necrosis. For immunohistochemistry, sections were deparaffinized through xylene and rehydrated with graded alcohol. Endogenous peroxidase was then blocked with 3% H2O2 diluted in methanol for 20 min at room temperature. Antigen retrieval was performed by treating the slide in citrate buffer in a microwave for 10 min. The slides were blocked in 1% bovine serum albumin and incubated in a moist chamber with p65 monoclonal antibody (1:200) at 4°C overnight, respectively. After a brief wash in PBS, the slides were treated with goat anti-mouse or (EnVision Kits, DAKO) for 30 min at 37°C. After a brief wash in PBS, the slide was developed in 0.05% freshly prepared diaminobenzedine solution (DAB, Sigma, St. Louis, MO) for 8 min, and then counterstained with hematoxylin.
Immunofluorescence assay
Cultured L02 cells were washed twice with Tris-buffered saline (TBS) (10 mM Tris pH 7.5, 150 mM NaCl) and then fixed with 1% paraformaldehyde. 0.2% Triton in TBS was added for analysis of intracytoplasmic antigen. Controls included staining with isotype matched irrelevant antibodies. Secondary antibodies were Cy3 or fluorescein isothiocyanate linked. Olympus phase fluorescence microscopy system with an Olympus digital camera (Tokyo, Japan) was used to capture the images.
Western blot
Freshly isolated liver tissue was homogenized in lysis buffer for preparation of whole protein extracts. NEPER® (Pierce Biotechnology, Rockford, IL, USA) was used to extract nuclear proteins and cytosolic proteins according to the manufacturer’s instructions. Protein concentrations were determined using the BCA Protein Assay kit (Pierce Biotechnology). The equal quantity of protein samples were loaded and then separated by sodium dodecyl sulfate/polyacrylamide gel (10%~15%). After electrophoresis, transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) using the Semi-Dry Trans-Blot Cell (Bio-Rad Laboratories, Hercules, CA, USA). After transfer, the membranes were washed with 0.1% Tween-20 in 1×Tris-buffered saline (TBS/T) and blocked for 1 h at room temperature with 5% (w/v) skim milk powder in TBS/T. The blots were then incubated overnight at 4°C with primary antibodies, followed by incubation with an IRDye 800CW-conjugated secondary antibody and detection with LI-COR imaging system (LI-COR Biosciences). Primary antibodies against p65 (Cell Signaling Technology, Beverly, MA, USA; 1:1,000 dilution), p-p65 (Transduction Laboratories, San Jose, CA, USA; 1:1,000 dilution) and the signals were normalized to that of β-actin (Sigma, 1:2,500 dilution) .
TUNEL assays
Terminal deoxynucleotidyl transferase-mediated dUTP nickend labeling (TUNEL) assay was performed on 4% paraformaldehyde fixed and paraffin-embedded sections of liver and was performed using an ApopTag Peroxidase in Situ Apoptosis Detection kit (Chemicon International, Temecula, CA) according to the manufacturer’s instructions.
Cells transfection and luciferase assays
L02 cells were grown in DMEM with 10% fetal bovine serum supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were co-transfected with the mixture of NF-κB luciferase reporter plasmids, a renilla luciferase control reporter vector pRL-TK, and indicated amounts of constructs by PEI (Polyplus, AFAQ). After treatment, the cells were lysed and luciferase activity was detected using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s recommendations.
Assessment of hepatic reactive oxygen species (ROS) content
Hepatic ROS content was measured using 2’-7’-dichlorofluorescein. l of liver homogenates diluted 100-fold with phosphate-buffered? Briefly, 10 saline (PH 7.4), were loaded with 5 μmol/l 2’-7’-dichlorofluorescein and incubated at 37°C for 30 minutes. Fluorescence was measured at excitation wavelength of 485 nm and emission wave length of 530 nm as previously described [19].
Statistical analyses
For each set of experiments where two or more than two groups were compared, an analysis of variance (ANOVA) test was used to determine the significance of the differences. Differences between the PBS and the PO treated group were compared for significance using student’s t-test for non paired samples. All the values shown are the mean ± S.E.M.
Results
Protective effects of PO on CCl4 administrated mice liver
Aspartate transaminase (ALT) and aspartate transaminase (AST) activity were evaluated to estimate the extent of hepatic damage. Figure 1 showed the changes in content of both aminotransferase in serum of mice at the end of the experiment. Low levels of activity of both ALT and AST were seen in the groups without CCl4 treatment, whereas a marked increase in the levels of the two enzymes was observed in the serum of CCl4-treated mice. The plasma AST and ALT activities of the PBS/CCl4 group were significantly higher than those of the PBS/oil group and those of the PBS/CCl4 group were significantly higher than those of the PO/CCl4 group, indicating the protective effect of the PO against CCl4 induced liver injury. The results also showed that the protective effects of PO against CCl4 induced liver injury were in a dose-dependent manner. The 200 mg/kg and 300 mg/kg PO had the significantly protective effects.
Figure 1.
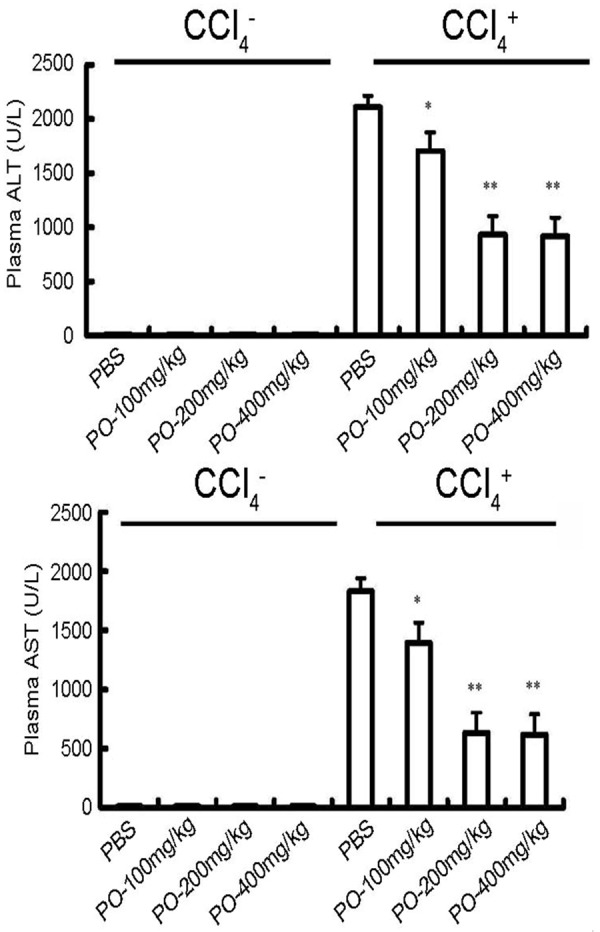
Effects of PO on mice plasma aminotransferase. Plasma ALT and AST activities were measured in mice 24 h after administration of oil or CCl4. PO-100, PO-200 and PO-400 group mice received 100 mg/kg, 200 mg/kg and 400 mg/kg PO in 0.5 ml PBS, respectively. PBS group mice received equal volume of PBS. All mice were orally administrated for seven days before CCl4 treatment. *P < 0.05 compared with PBS group; **P < 0.05 compared with PBS group. Data are expressed as the mean ± S.E.M. (n = 5~6 animals per group).
For further explored the role of PO, we further examined the liver damage by histological assays. As shown in Figure 2, the CCl4 treatment caused severe necrosis in hepatic tissues. The liver exposed to CCl4 showed multiple and extensive areas of portal inflammation and hepatocellular necrosis, randomly distributed throughout the parenchyma, as well as moderate increase in inflammatory cell infiltration. However, the PO did have obvious protective effect from hepatic damage induced by CCl4. The normal liver lobular architecture and cell structure were showed in the control group. The TUNEL assay also demonstrated the protective effects of the PO. These results were consistent with the plasma ALT and AST levels showed in Figure 1.
Figure 2.
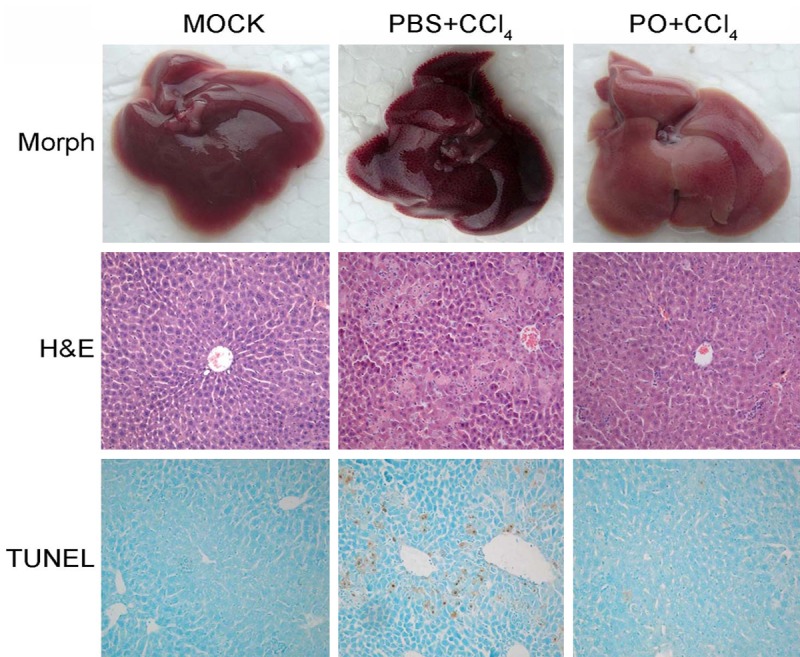
Effects of PO on morphological and histological change. Representative pictures of morphology, hematoxylin-eosin (H&E) staining, and DNA fragmentation (TUNEL assay) were showed (Original magnification ×100 for H&E and TUNEL panels).
Effects of the PO on expression of SOD and ROS content
Given that oxidative involves in the process of CCl4 induced mice liver injury, we tested the SOD activities and ROS level of the mice liver. Unexpectedly, the results showed that PO moderately increase the SOD activities (Figure 3A, 3B) and decrease ROS level (Figure 3C).
Figure 3.
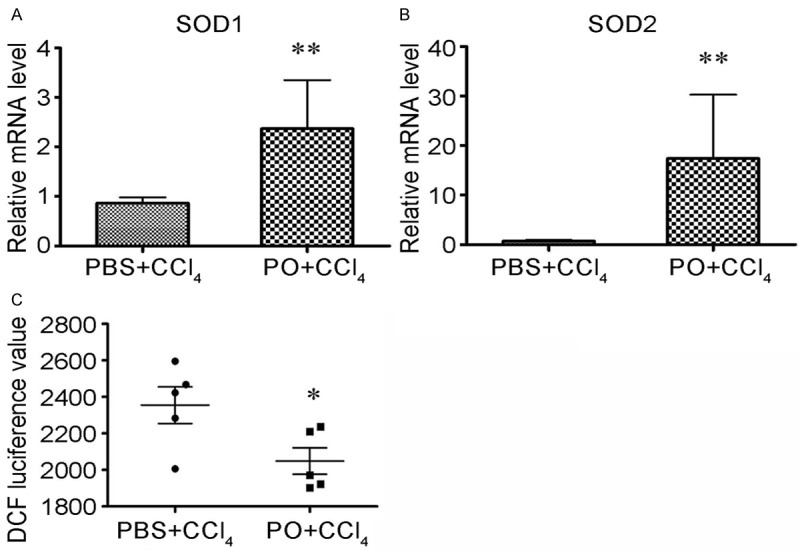
Effects of the PO on expression of SOD and ROS content. A, B. Mice liver tissues were lysed and RNA was extracted, qRT-PCR was performed to detected SOD1 and SOD2 levels. C. Liver homogenates diluted 100-fold with phosphate-buffered saline (PH 7.4), were loaded with 5 μmol/l 2’-7’-dichlorofluorescein and incubated at 37°C for 30 minutes. Fluorescence was measured at excitation wavelength of 485 nm and emission wave length of 530 nm.
Effects of the PO on activation of NF-κB
It has been reported that the inflammation play an important role during the process of liver injury induced by CCl4 and the NF-κB is an important inflammation mediating molecular [13]. So we tried to test whether PO influenced the activation of NF-κB. By immunohistochemistry we found that the PO could enhance the p-65 nuclear translocation (Figure 4A, 4B).
Figure 4.
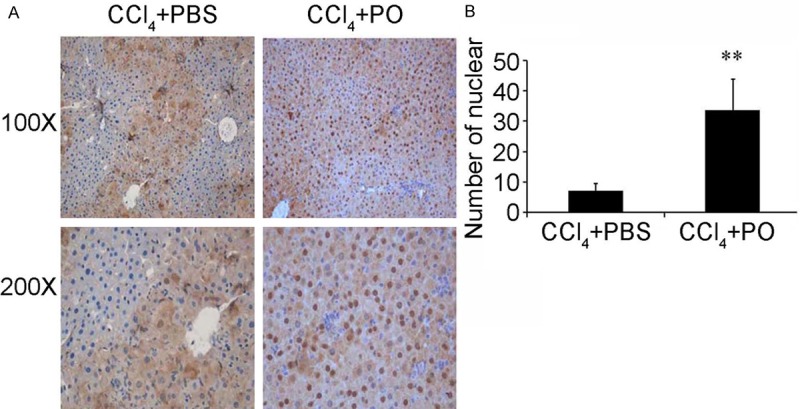
Effect of PO on the nucleic translocation of p65. The PO enhances the nucleic translocation of p65 in liver of mice administrated with CCl4.
To further confirm the effects of the PO on NF-κB activation, L02 cells were transfected with a luciferase reporter gene that contains putative binding sites for NF-κB, followed by the stimulation with or not hydrogen peroxide. The results showed that, PO treatment markedly enhanced the luciferase reporter gene activity compare with the control cells (Figure 5A). By western blot assay (Figure 5B), we also found that the PO promoted the phosphorylation of p65 of cells upon TNFα stimulation. The cell immunochemistry results also showed that PO pretreatment increased more p65 protein trans-located into cell nuclei compared with DMSO group after TNFα stimulation (Figure 5C).
Figure 5.
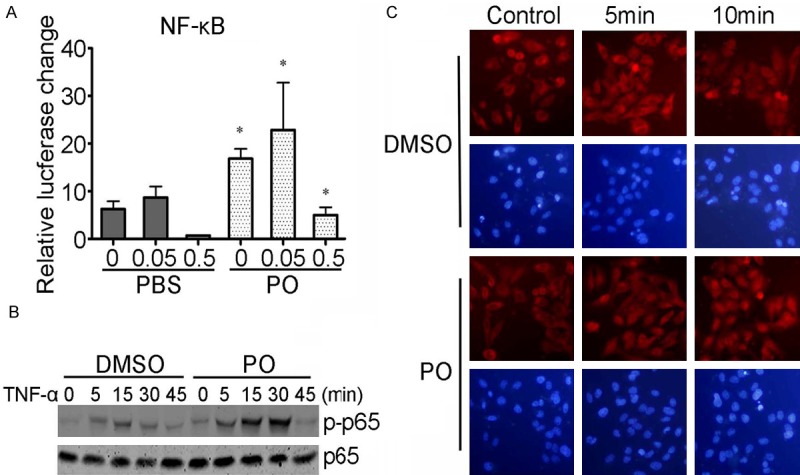
Effects of PO on NF-κB activity. (A) PO increased the luciferase reporter gene activity of transfected liver cell line L02 cells stimulated by hydrogen peroxide. *P < 0.05 compared with PBS group stimulated with equal concentration hydrogen peroxide (B) PO promoted the phosphorylation of p65 of L02 cells stimulated with TNF-α. (C) PO promoted the nucleic translocation of p65 in L02 cells stimulated by hydrogen peroxide.
Discussion
Present study mainly demonstrated that the protective effects of PO against mice liver damaged by CCl4. The PO could decrease the ALT and AST activity in blood plasma. The results also showed that the PO enhanced the nucleic translocation of NF-κB p-65 and the NF-κB luciferase reporter gene activity. All these results indicated that the mechanism of the hepatoprotective effects of the PO may be partly through modulating the activity of NF-κB.
The CCl4 induced liver damage have been studied in mice and rats and shown by significant elevation of serum aminotransferase levels [11,20,21]. In our previous experiments, we have used mice for the research and demonstrated the anti-oxidative effects of the PO [17,18]. The present results showed that the PO pretreatment significantly suppressed the CCl4 induced elevation of plasma aspartate aminotransferase and alanine aminotransferase activities. The CCl4 induced hepatic lesions were characterized by coagulation necrosis and/or vacuolation of hepatocytes mainly situated in central to middle part of hepatic lobules. By H-E staining and TUNEL assays, we demonstrated that PO pretreatment attenuated the liver damage of mice upon CCl4 administration. All these demonstrated the protective effects of the PO on the liver of mice towards CCl4.
It has been widely accepted that ROS are the main causes of CCl4 induced acute liver injury and anti-oxidative therapy is therefore an effective means of preventing and attenuating oxidative stress related liver diseases [3,10-12]. Our previous research demonstrated the anti-oxidative effects of the PO [17,18]. Based on all these information, we hypothesized that the PO could attenuate the liver damage induced by CCl4. The hepatotoxicity of CCl4 requires bioactivation by the cytochrome P450 phase I system in the liver and yields a reactive metabolic trichloromethyl radical (CCl3) and proxy trichloromethyl radical (OOCCl3) [3,22]. These free radicals can bind with polyunsaturated fatty acid and form alkoxy (R) and peroxy radicals (ROO) that can generate lipid peroxide, cause damage in the cell membrane, change enzyme activity and finally induce hepatic injury or necrosis [3,10,11,22]. So, we tested the activity of cytochrome P450 and found no obvious effects of PO (data not shown). Then we tested the SOD activities and ROS level of the mice liver. The results showed that PO increase the SOD activities and decrease ROS level. These results indicated the anti-oxidative action of the PO might partly involve in the protective effects against CCl4 induced acute injury.
The CCl4 can also indirectly activated the Kupffer cells, which mediate the hepatic inflammation process by producing tumor necrosis factor-α (TNF-α) and other cytotoxic cytokines [11,12]. It was found that baicalin protects hepatocytes from the oxidative damage caused by CCl4 and the protective effect of baicalin is likely due to the inhibition of the proinflammatory mediators [23]. NF-κB pathway is associated with the production of these inflammatory factors in liver after CCl4 treatment [13]. Furthermore, lots of studies have demonstrated that ROS are able to activate NF-κB pathway and antioxidants may inhibit NF-κB [23-26]. So, we further studied the effects of the PO on the NF-κB activity. NF-κB, which consists of the p50 and p65/RelA polypeptides, is a transcription factor expressed in a variety of cell types, including hepatocyte. When activated, it shifts to the nucleus and binds to promoters and enhancers of genes involved in inflammatory and proliferative responses [27-29]. Our results showed that the PO could promote the translocation of p65 (Figure 3), indicating that PO increased the activity of NF-κB. This was consistent with previous reports that NF-κB was increased in hepatic nuclear extracts at 2 and 24 hr after CCl4 administration, indicated the NF-κB levels was elevated in response to this toxicant [30] and that CCl4 decreased NF-κB translocation [31]. All these indicated that increase activity of NF-κB might participate in the protective effects of the PO on liver of mice administrated with CCl4. As all kown that, in the cytoplasm, NF-κB interacts with a specific inhibitor called IκBα [32,33]. IκBα can itself undergo rapid ubiquitin-dependent degradation by a variety of stimuli which activate the IκBα kinase (IKK) complex [34]. NF-κB activation requires sequential phosphorylation and degradation of IκBα, which finally disappears from the cytoplasm. Whether the PO activated the NF-κB by this canonical pathway or by other mechanism needs further research.
Although the research reported in the present study did not test individual components of the extract, this report did propose to test the whole extract for effects operative in vivo and in vitro. Since the resulting effect is a clinically important question. Furthermore, this might enlarge the using extent of the traditional herb and indicate the clue for researching the new medicine for treatment of liver diseases. Further studies regarding the isolation of major bioactive components responsible for the observed effect are the focus of ongoing and planned investigation.
In conclusion, it appeared that the PO reduced the acute liver injury induced by CCl4 involving the enhancement of NF-κB activity. We suggested that, the edible plant Portulaca Oleracea L., may be used to protect against toxic effects of CCl4 and other chemical agents in liver.
Acknowledgements
This work was supported by Medical Science and Technique projects for young talents of PLA (I3QNP035), National Natural Science Foundation of China (81270765) and Naval Medicine Research Project of Second Military Medical University (2009ZL01).
Disclosure of conflict of interest
None to declare.
References
- 1.Wang T, Shankar K, Ronis MJ, Mehendale HM. Mechanisms and outcomes of drug- and toxicant-induced liver toxicity in diabetes. Crit Rev Toxicol. 2007;37:413–459. doi: 10.1080/10408440701215100. [DOI] [PubMed] [Google Scholar]
- 2.Auzinger G, Wendon J. Intensive care management of acute liver failure. Curr Opin Crit Care. 2008;14:179–188. doi: 10.1097/MCC.0b013e3282f6a450. [DOI] [PubMed] [Google Scholar]
- 3.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 4.Wong FW, Chan WY, Lee SS. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol. 1998;153:109–118. doi: 10.1006/taap.1998.8547. [DOI] [PubMed] [Google Scholar]
- 5.Albano E, Tomasi A, Goria-Gatti L, Poli G, Vannini V, Dianzani MU. Free radical metabolism of alcohols by rat liver microsomes. Free Radic Res Commun. 1987;3:243–249. doi: 10.3109/10715768709069789. [DOI] [PubMed] [Google Scholar]
- 6.Connor HD, Lacagnin LB, Knecht KT, Thurman RG, Mason RP. Reaction of glutathione with a free radical metabolite of carbon tetrachloride. Mol Pharmacol. 1990;37:443–451. [PubMed] [Google Scholar]
- 7.McCay PB, Lai EK, Poyer JL, DuBose CM, Janzen EG. Oxygen- and carbon-centered free radical formation during carbon tetrachloride metabolism. Observation of lipid radicals in vivo and in vitro. J Biol Chem. 1984;259:2135–2143. [PubMed] [Google Scholar]
- 8.Packer JE, Slater TF, Willson RL. Reactions of the carbon tetrachloride-related peroxy free radical (CC13O. 2) with amino acids: pulse radiolysis evidence. Life Sci. 1978;23:2617–2620. doi: 10.1016/0024-3205(78)90378-8. [DOI] [PubMed] [Google Scholar]
- 9.Slater TF. Free-radical mechanisms in tissue injury. Biochem J. 1984;222:1–15. doi: 10.1042/bj2220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Recknagel RO, Glende EA Jr, Dolak JA, Waller RL. Mechanisms of carbon tetrachloride toxicity. Pharmacol Ther. 1989;43:139–154. doi: 10.1016/0163-7258(89)90050-8. [DOI] [PubMed] [Google Scholar]
- 11.Basu S. Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology. 2003;189:113–127. doi: 10.1016/s0300-483x(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 12.Akamatsu K, Yamasaki Y, Nishikawa M, Takakura Y, Hashida M. Synthesis and pharmacological activity of a novel water-soluble hepatocyte-specific polymeric prodrug of prostaglandin E(1) using lactosylated poly(L-glutamic hydrazide) as a carrier. Biochem Pharmacol. 2001;62:1531–1536. doi: 10.1016/s0006-2952(01)00799-7. [DOI] [PubMed] [Google Scholar]
- 13.Schmiedeberg P, Biempica L, Czaja MJ. Timing of protooncogene expression varies in toxin-induced liver regeneration. J Cell Physiol. 1993;154:294–300. doi: 10.1002/jcp.1041540212. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Q, Xu X, Huang Y, Xu L, Chen G. Field enhancement sample stacking for analysis of organic acids in traditional Chinese medicine by capillary electrophoresis. J Chromatogr A. 2012;1246:35–39. doi: 10.1016/j.chroma.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Xiang L, Xing D, Wang W, Wang R, Ding Y, Du L. Alkaloids from Portulaca oleracea L. Phytochemistry. 2005;66:2595–2601. doi: 10.1016/j.phytochem.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Rashed AN, Afifi FU, Disi AM. Simple evaluation of the wound healing activity of a crude extract of Portulaca oleracea L. (growing in Jordan) in Mus musculus JVI-1. J Ethnopharmacol. 2003;88:131–136. doi: 10.1016/s0378-8741(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 17.Wanyin W, Liwei D, Lin J, Hailiang X, Changquan L, Min L. Ethanol extract of Portulaca oleracea L. protects against hypoxia-induced neuro damage through modulating endogenous erythropoietin expression. J Nutr Biochem. 2012;23:385–391. doi: 10.1016/j.jnutbio.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 18.Chen CJ, Wang WY, Wang XL, Dong LW, Yue YT, Xin HL, Ling CQ, Li M. Anti-hypoxic activity of the ethanol extract from Portulaca oleracea in mice. J Ethnopharmacol. 2009;124:246–250. doi: 10.1016/j.jep.2009.04.028. [DOI] [PubMed] [Google Scholar]
- 19.Llacuna L, Mari M, Lluis JM, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Reactive oxygen species mediate liver injury through parenchymal nuclear factor-kappaB inactivation in prolonged ischemia/reperfusion. Am J Pathol. 2009;174:1776–1785. doi: 10.2353/ajpath.2009.080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu SJ, Tam KW, Tsai YH, Chang CC, Chao JC. Curcumin and saikosaponin a inhibit chemical-induced liver inflammation and fibrosis in rats. Am J Chin Med. 2010;38:99–111. doi: 10.1142/S0192415X10007695. [DOI] [PubMed] [Google Scholar]
- 21.Dharancy S, Body-Malapel M, Louvet A, Berrebi D, Gantier E, Gosset P, Viala J, Hollebecque A, Moreno C, Philpott DJ, Girardin SE, Sansonetti PJ, Desreumaux P, Mathurin P, Dubuquoy L. Neutrophil migration during liver injury is under nucleotide-binding oligomerization domain 1 control. Gastroenterology. 2010;138:1546–1556. doi: 10.1053/j.gastro.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Rechnagel RO, Glende EA Jr. Carbon tetrachloride hepatotoxicity: an example of lethal cleavage. CRC Crit Rev Toxicol. 1973;2:263–297. doi: 10.3109/10408447309082019. [DOI] [PubMed] [Google Scholar]
- 23.Park SW, Lee CH, Kim YS, Kang SS, Jeon SJ, Son KH, Lee SM. Protective effect of baicalin against carbon tetrachloride-induced acute hepatic injury in mice. J Pharmacol Sci. 2008;106:136–143. doi: 10.1254/jphs.fp0071392. [DOI] [PubMed] [Google Scholar]
- 24.Orfila C, Lepert JC, Alric L, Carrera G, Beraud M, Pipy B. Immunohistochemical distribution of activated nuclear factor kappaB and peroxisome proliferator-activated receptors in carbon tetrachloride-induced chronic liver injury in rats. Histochem Cell Biol. 2005;123:585–593. doi: 10.1007/s00418-005-0785-2. [DOI] [PubMed] [Google Scholar]
- 25.Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 26.Lee SM, Cho TS. Effect of Trolox C on hypoxia/reoxygenation-induced injury in isolated perfused rat liver. Arch Pharm Res. 1997;20:471–475. doi: 10.1007/BF02973942. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 28.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 30.Gruebele A, Zawaski K, Kaplan D, Novak RF. Cytochrome P4502E1- and cytochrome P4502B1/2B2-catalyzed carbon tetrachloride metabolism: effects on signal transduction as demonstrated by altered immediate-early (c-Fos and c-Jun) gene expression and nuclear AP-1 and NF-kappa B transcription factor levels. Drug Metab Dispos. 1996;24:15–22. [PubMed] [Google Scholar]
- 31.Campo GM, Avenoso A, Campo S, Nastasi G, Traina P, D’Ascola A, Rugolo CA, Calatroni A. The antioxidant activity of chondroitin-4-sulphate, in carbon tetrachloride-induced acute hepatitis in mice, involves NF-kappaB and caspase activation. Br J Pharmacol. 2008;155:945–956. doi: 10.1038/bjp.2008.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 33.Hayden MS, West AP, Ghosh S. SnapShot: NF-kappaB signaling pathways. Cell. 2006;127:1286–1287. doi: 10.1016/j.cell.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]