

Abstract
Animal stroke models suggest that valproate has multiple neuroprotective mechanisms against ischemic brain damage. This study investigated whether valproate improves functional recovery in patients with acute middle cerebral artery (MCA) infarction. This was an open-label controlled trial. Three to 24 hours after acute MCA infarction, patients were assigned to either the valproate group (n = 17) or the non-valproate group (n = 17). The valproate group received intravenous valproate (400 mg) at enrollment, and then every 12 hours for three days, followed by oral valproate (500 mg) every 12 hours for three months. Neurological function, laboratory data, and brain magnetic resonance imaging were examined at stroke onset, and at two-week and three-month follow-up. No significant differences were observed between the groups with regard to demographics or baseline characteristics. All patients were elderly, had a high pretreatment score on the NIH stroke scale (NIHSS), and slow stroke lesion growth with a final large infarct volume at two-week follow-up. At the three-month follow-up, functional outcome between pre- and post-treatment had improved significantly in the valproate group (NIHSS, p = 0.004; modified Rankin scale (mRS), p = 0.007; Barthel index (BI), p = 0.001). No such improvement was noted in the NIHSS or mRS for the non-valproate group, though mild improvement was seen on the BI (p = 0.022). This open-label trial is the first to demonstrate that valproate treatment markedly improves functional outcome in patients with acute MCA infarction.
Keywords: Ischemic stroke, valproate, stroke outcome, neuroprotection
Introduction
Ischemic stroke is the most common stroke incident. Increasing evidence suggests that expansion of the cerebral infarct size of no-reflow by secondary injury recruits the peripheral zones into the ischemic core; this is due to a triggered ischemic cascade that includes ionic imbalance, excitotoxicity, excessive neuroinflammation, and oxidative stress [1,2]. Current therapies for acute ischemic stroke are aimed at early recanalization of the occluded cerebral vessel to minimize brain damage and to modify the ischemic cascade pathway in order to rescue injured neurons. Thrombolysis with recombinant tissue plasminogen activator (rtPA) is presently the only approved therapy for acute ischemic stroke, and eligible patients treated with rtPA show better neurological performance [3,4]. However, rtPA thrombolysis is not an ideal therapy because of the narrow therapeutic window and the strict patient selection requirements. To date, no clinically neuroprotective drug for stroke has yet proven effective [5,6]. Thus, there is a clear clinical urgency to develop novel and effective neuroprotective drugs or new strategies to improve current treatment for patients experiencing acute ischemic stroke [7,8].
Sodium valproate is a simple eight-carbon branched-chain fatty acid. It is a widely used and effective drug for the treatment of seizures and bipolar mood disorder, as well as for migraine prophylaxis [9-12]. Although its underlying therapeutic mechanisms remain unclear, a growing body of evidence from animal stroke models suggests that valproate comprises multiple mechanisms that contribute both neuroprotective and neurotrophic effects against ischemic stroke (for a review, see [13-15]). Well-established data have demonstrated that post-insult treatment with valproate in rodent ischemic stroke models markedly reduces infarct volume and improves functional outcome [16-20]. The mechanisms underlying these beneficial effects in experimental stroke models likely involve inhibition of histone deacetylases (HDACs), which induce key neuroprotective molecules that suppress ischemia-induced neuroinflammation, protect against blood-brain barrier disruption, and promote angiogenesis, thus facilitating functional recovery. However, to date, no clinical trial has investigated the putative benefits of valproate in patients with acute ischemic stroke.
This open-label controlled study investigated patients with acute middle cerebral artery (MCA) infarction who received valproate as an add-on treatment between three and 24 hours after stroke onset, and for three months post-insult. The primary objective of the study was to examine whether the patients receiving post-insult treatment with valproate for three months showed reduced neurological deficits and decreased infarct volume.
Patients and methods
Study design
This prospective study was an open-label, controlled-group trial evaluating the use of valproate (Depakine® (Sanofi, Paris, France)) in patients with acute MCA infarction. It was carried out in the Department of Neurology, Tri-Service General Hospital (Taipei, Taiwan) between July 01, 2006 and August 31, 2009. The study was approved by the hospital’s Institutional Review Board for Human Studies (Clinical Trials Registration: TSGHIRB Approval Number: 095-05-002), and that it conforms to the provisions of the Declaration of Helsinki and all patients provided written informed consent before enrollment.
Selection of study patients and clinical characteristics (Figure 1)
Figure 1.
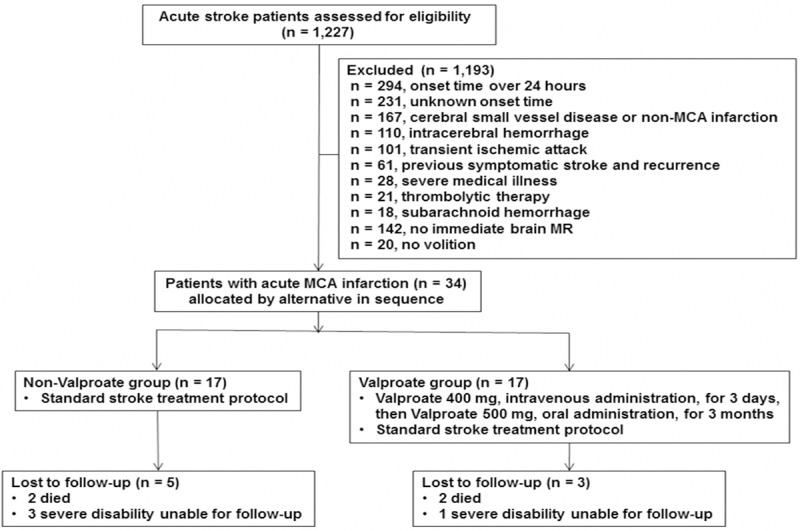
Study patient selection. MCA, middle cerebral artery. MR, magnetic resonance.
Eligible patients were experiencing the clinical symptoms of acute MCA infarction, and stroke onset time was between three and 24 hours of their arrival at the hospital. This was subsequently confirmed by brain magnetic resonance (MR) diffusion-weighted imaging (DWI).
Exclusion criteria included radiological evidence comparable to their previous clinical symptoms of disabling stroke, brain stroke precipitated during surgery or angiography, non-MCA infarction, receiving therapeutic anticoagulation, history of bleeding diathesis or illicit drug use, or other concomitant serious medical illness, including malignancy, uraemia, cirrhosis of the liver, sepsis, autoimmune disorders, evidence of cerebral vascular malformation, or a history of allergy to valproate.
An experienced neurologist explained the treatment protocol to eligible patients or their relatives, and obtained informed consents. At study enrollment, all patients were examined by an experienced neurologist. Pre-trial screening laboratory tests included systolic and diastolic blood pressure, body mass index, electrocardiogram, complete blood cell counts, serum electrolyte determinations, and blood biochemistry analyses. A clinical history was obtained for all patients based on a structured checklist that assessed major vascular risk factors, such as hypertension, diabetes, hypercholesterolemia, hypertriglyceridemia, coronary artery disease, atrial fibrillation, previous stroke, alcohol consumption, and smoking. A detailed neurological evaluation was also obtained using the following stroke outcome scales: the National Institutes of Health Stroke Scale (NIHSS), the modified Rankin scale (mRS), and the Barthel index (BI). Patients were subsequently re-evaluated two weeks and three months post-insult.
Cranial magnetic resonance imaging (MRI) was performed on a 1.5-Tesla scanner (GE, Signa HDx, Milwaukee, USA) using a conventional spin-echo T1-weighted, T2-weighted sequence with a rectangular field of view, T2-FLAIR (fluid attenuated inversion recovery) imaging, and other sequences including DWI and time-of-flight MR angiography. Brain MR volumetry for stroke lesion was conducted using DWI and T2-FLAIR data. Diffusion weighted imaging was performed using a single-shot echoplanar spin-echo sequence with two 180° radiofrequency pulses to minimize eddy current warping. Twenty images per slice were acquired at b = 0 s/mm2 followed by 20 at b =1000 s/mm2 in six directions (TR/TE 5000/80–110 ms, field of view 24 cm, matrix 128 × 128 zero-filled to 256 × 256, 6-mm slice thickness, 1.2-mm gap). T2-FLAIR imaging was performed using TR/TE 9000/120 to 140 ms, field of view 24 cm, matrix size 256 × 256, 5-mm thickness, 2-mm gap.
All patients met the inclusion criteria established by brain MRI including MCA moderate stenosis to occlusion (> 50%) and a corresponding DWI hyperintensity representing the core of the infarct at entry. Brain FLAIR imaging was used to assess final stroke lesion volume at two weeks and again at three months post-insult. Head MRI scans were performed not only to delineate the volume of the stroke lesion, but also to exclude intracranial hemorrhage, cerebral vascular malformation, meningoencephalitis, or brain tumor. Irregular infarct area on each axial slice was semi-manually delineated and segmented based on thresholding in combination with region-growing techniques within the AMIRA software (VSG Inc., Burlington, MA, USA). The final infarct volume was estimated and visualized by the volume rendering technique (see Figure 2). All MRI scans were read by two investigators blind to treatment assignment and clinical outcomes, but not to the time point of MRI examination.
Figure 2.
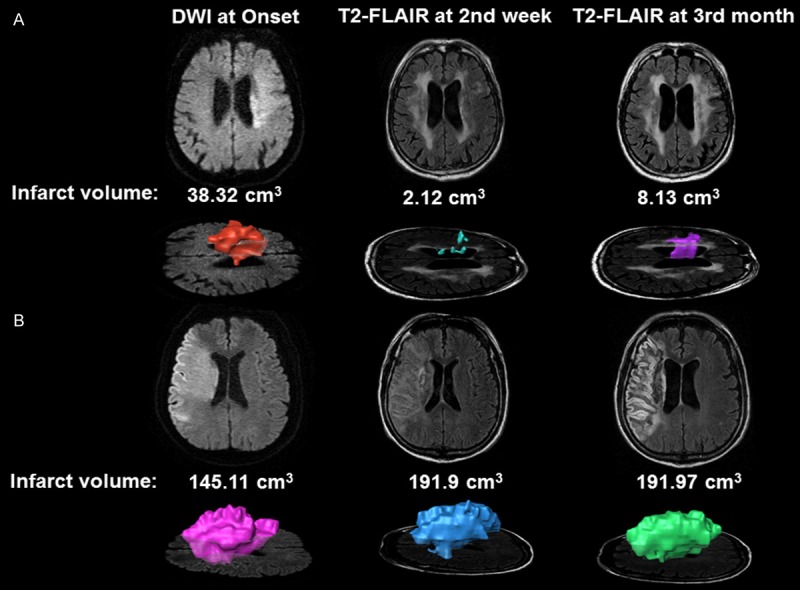
Infarct volume scans of patients with MCA infarction. A series of infarct volume scans were semi-manually measured using AMIRA software. A. Representative image from a patient who received valproate treatment and had a reduced infarct volume at the two-week and three-month follow-up, as measured by T2-fluid attenuated inversion recovery (FLAIR) imaging. B. Representative image from a patient who did not receive valproate treatment and demonstrated delayed stroke lesion growth at the two-week follow-up.
During the period of clinical trial, a total of 1227 acute stroke patients have been screened. A total of 1193 patients were excluded, as 294 were with onset time over 24 hours, 231 with unknown onset time, 167 with cerebral small vessel disease or non-MCA infarction, 110 with intracerebral hemorrhage, 101 with transient ischemic accident, 61 with previous symptomatic stroke and recurrence, 28 with severe medical illness, 21 with acute thrombolytic therapy, 18 with subarachnoid hemorrhage, 142 without immediate brain MR study, and 20 without volition. Thus, only thirty-four eligible patients were alternatively assigned to the valproate or non-valproate groups in sequence. These subjects received standard treatment for acute cerebral infarction. Seventeen patients receiving no treatment of valproate were used as the control. The remaining patients in the valproate group received intravenous (IV) administration of valproate (Depakine®, Sanofi; 400 mg) starting at the time of enrollment and then every 12 hours for three days, followed by oral valproate (500 mg) every 12 hours for three months. All patients in the valproate group were checked for liver function and blood valproate levels at two weeks after treatment began, and again at three months.
Outcome measures
For all patients, neurological function, laboratory data, and brain MRI were assessed at onset, at two weeks post-insult, and at three months post-insult. The efficacy criterion was defined as the results of functional outcomes measured by NIHSS, mRS, and BI, as well as the infarct volume obtained by brain MRI. All the evaluators have been certified for measuring the scores of functional outcomes. Primary end point for assessing efficacy was 90 days after the stroke event. Safety variables included vital signs and laboratory data of all subjects. Laboratory data were monitored and further evaluated. Adverse events were also recorded throughout the study.
Statistical analysis
Categorical variables were expressed as numbers and percentages and compared using a Fisher’s exact test. Continuous variables were calculated as mean ± standard deviation and compared using Mann-Whitney U tests between the valproate and non-valproate groups. Paired t-tests were used to compare treatment effects within the same group of patients by comparing pre-treatment and three months post-treatment data. In addition, the ordinal scales of NIHSS, mRS, and BI were also quoted as median and interquartile range. Statistical analyses were performed using SPSS version 19 (SPSS, Chicago, IL, USA).
Results
Table 1 shows the basic clinical characteristics for all 34 patients with acute MCA (20 M, 14 F), who were equally distributed into the valproate or non-valproate groups. No significant differences were observed between the groups for any demographic or baseline characteristics, including mean age of onset (71.47 ± 12.32 years vs 72.71 ± 9.12 years) or NIHSS scores at admission (17.65 ± 8.21 vs 17.35 ± 8.61). Functional outcomes and laboratory data are documented in Table 2. Four patients died during the study period, two from each treatment group. In addition, four patients withdrew from the study; one from the valproate group and three from the non-valproate group. The withdrawal was most likely due to severe disability (NIHSS > 20) that prevented participation in a subsequent follow-up study.
Table 1.
Demographic and baseline characteristics in patients with acute middle cerebral artery infarction in the valproate and non-valproate groups
| Variables | Valproate (n = 17) | Non-Valproate (n = 17) | P |
|---|---|---|---|
| Age, y (mean ± SD) | 71.5 ± 12.3 | 72.7 ± 9.1 | 0.74 |
| Sex, M:F | 9:8 | 11:6 | 0.72 |
| Infarction side, R:L | 8:9 | 8:9 | 1.00 |
| Body mass index, kg/m2 | 23.4 ± 6.6 | 23.6 ± 3.0 | 0.92 |
| SBP at ER, mmHg | 172 ± 28 | 165 ± 29 | 0.50 |
| DBP at ER, mmHg | 94 ± 36 | 86 ± 19 | 0.43 |
| Onset to treatment time, hr | 8.5 ± 4.4 | 11.2 ± 4.6 | 0.16 |
| Medical history, n (%) | |||
| Hypertension | 15 (88.2) | 11 (64.7) | 0.17 |
| Diabetes Mellitus | 4 (23.5) | 5 (29.4) | 1.00 |
| Hyperlipidemia | 5 (29.4) | 6 (35.3) | 1.00 |
| CAD | 2 (11.8) | 3 (17.6) | 1.00 |
| Atrial fibrillation | 11 (64.7) | 10 (58.8) | 1.00 |
| Hyperuricemia | 3 (17.6) | 4 (23.5) | 1.00 |
| Ever smoking | 7 (41.2) | 4 (23.5) | 0.46 |
| Previous stroke/TIA | 1 (5.9) | 4 (23.5) | 0.17 |
Patients received stroke treatment immediately after pretrial brain MR scans were completed. CAD, coronary artery disease; DBP, diastolic blood pressure; ER, emergency room; F, female; L, left; M, male; R, right; SBP, systolic blood pressure; TIA, transient ischemic attack; y, year. Statistically significant difference was evaluated using Fisher’s exact test for the categorical variables and Mann-Whitney U test for continuous variables between the tested groups.
Table 2.
Functional outcome and laboratory data in patients with acute middle cerebral artery infarction in the valproate and non-valproate groups
| Time to check/Variables | Valproate | Non-Valproate | P | ||
|---|---|---|---|---|---|
| At onset, n | 17 | 17 | |||
| Sex, M:F | 9:8 | 11:6 | |||
| Outcome scales | Mean ± SD | Median (IQR) | Mean ± SD | Median (IQR) | |
| NIHSS | 17.7 ± 8.2 | 16.0 (14.5) | 17.4 ± 8.6 | 16.0 (11.0) | 0.92 |
| mRS | 4.4 ± 1.0 | 5.0 (1.0) | 4.1 ± 1.3 | 5.0 (1.0) | 0.55 |
| BI | 10.6 ± 14.4 | 0.0 (22.5) | 8.8 ± 11.5 | 5.0 (20.0) | 0.70 |
| Infarct volume, cm3 | 55.0 ± 51.5 | 94.7 ± 86.8 | 0.12 | ||
| Laboratory data | |||||
| GOT, U/L | 34.0 ± 30.7 | 31.6 ± 12.7 | 0.77 | ||
| GPT, U/L | 27.5 ± 21.4 | 27.9 ± 15.0 | 0.95 | ||
| Two-week follow-up, n | 16† | 17 | |||
| Sex, M:F | 9:7 | 11:6 | |||
| Outcome scales | Mean ± SD | Median (IQR) | Mean ± SD | Median (IQR) | |
| NIHSS | 15.3 ± 10.1 | 14.5 (19.8) | 16.4 ± 11.2 | 13.0 (22.5) | 0.78 |
| mRS | 3.8 ± 1.8 | 4.5 (2.5) | 4.0 ± 1.3 | 4.0 (1.5) | 0.64 |
| BI | 24.7 ± 33.0 | 15.0 (30.0) | 31.2 ± 36.9 | 10.0 (55.0) | 0.60 |
| Infarct volume, cm3 | 110.7 ± 137.1 | 106.5 ± 84.5 | 0.92 | ||
| Laboratory data | |||||
| Valproate level, mcg/mL | 64.6 ± 25.2 | ||||
| GOT, U/L | 40.7 ± 37.9 | ||||
| GPT, U/L | 36.8 ± 42.9 | ||||
| Three-month follow-up, n | 14‡ | 12§ | |||
| Sex, M:F | 8:6 | 9:3 | |||
| Recanalization | 4 (28.6%) | 3 (25%) | |||
| Outcome scales | Mean ± SD | Median (IQR) | Mean ± SD | Median (IQR) | |
| NIHSS | 9.6 ± 8.6 | 6.5 (17.0) | 16.3 ± 12.3 | 15.5 (22.0) | 0.12 |
| mRS | 3.1 ± 1.8 | 3.5 (3.3) | 3.7 ± 1.7 | 4.5 (3.0) | 0.40 |
| BI | 48.9 ± 39.9 | 35.0 (83.8) | 37.1 ± 42.7 | 15.0 (87.5) | 0.47 |
| Infarct volume, cm3 | 91.1 ± 118.1 | 126.3 ± 76.6 | 0.39 | ||
| Laboratory data | |||||
| Valproate level, mcg/mL | 61.5 ± 24.9 | ||||
| GOT, U/L | 32.7 ± 18.4 | ||||
| GPT, U/L | 29.9 ± 21.6 |
one death.
two deaths and one withdrawal.
two deaths and three withdrawals.
BI, Barthel index; F, female; GOT, glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; IQR = Interquartile Range; M, male; mRS, modified Rankin scale; NIHSS, National Institutes of Health Stroke Scale. Statistically significant differences were evaluated using Fisher’s exact test for the categorical variables and Mann-Whitney U test for continuous variables between the valproate and non-valproate groups.
For all patients, changes in infarct volume were measured by a series of brain MRI scans including brain DWI at study entry and T2-FLAIR imaging at two weeks and three months post-stroke (see Figure 2). Eight (47.7%) patients from the valproate group and 10 (58.8%) from the non-valproate group showed an increase in lesion volume at the two-week follow-up, but there were no significant differences in infarct volume between the two groups (Table 2). The delayed increase observed here in infarct volume after cerebral ischemia echoes previous reports, which found that irreversible damage occurs relatively rapidly in the core of severe ischemia, but that damage in peripheral regions of less severe ischemia may develop over the course of many hours or even days [21-24].
In Table 2, no significant difference was observed between the valproate and non-valproate groups with regard to functional outcome measures—including the NIHSS, mRS, and BI—at either the two-week or three-month follow-up. Further statistical analysis for the estimate of patient numbers needed to treat by using Stata 9.0 sample-size and power determination demonstrated that each group needs the patient numbers of 53 in the item of NIHSS, 184 in mRS, 256 in BI and 169 in infarct volume for reaching 95% statistical power. Serum valproate levels in the valproate group were within the therapeutic range at week two (64.60 ± 25.19; N: 50-100 mcg/mL) and month three (61.53 ± 24.87) of follow-up. Similarly, no significant change in infarct volume was observed between the valproate and non-valproate groups, as measured at either the two-week or three-month follow-up. In the valproate group, no differences were observed in serum levels of glutamate oxaloacetate transaminase and glutamate pyruvate transaminase at the two-week or three-month follow-up compared with baseline measures.
Notably, a comparison of all functional outcomes within the individual groups found significant improvements between onset and the three-month follow-up for those patients in the valproate group (NIHSS: 17.0 ± 2.3 vs. 9.6 ± 2.3, p = 0.004; mRS: 4.3 ± 0.3 vs. 3.1 ± 0.5, p = 0.007; Figure 3A and 3B; BI: 11.1 ± 4.0 vs. 48.9 ± 10.7, p = 0.001; Figure 3C) as no significant change of infarct volume (Figure 3D). Although no such improvements were seen in the non-valproate group for the NIHSS and mRS scales, at the three-month follow-up, BI scores had improved significantly in the non-valproate group (9.2 ± 3.5 vs. 37.1 ± 12.3, p = 0.022); however, this improvement was significantly more pronounced in the valproate group (Figure 3C).
Figure 3.
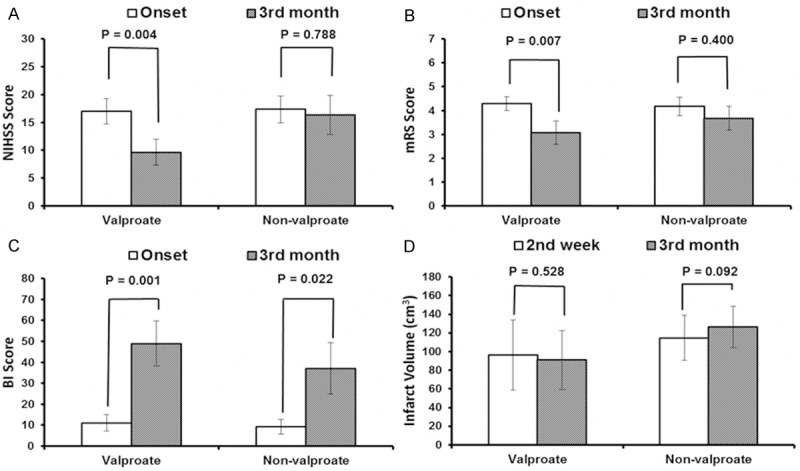
Effects of valproate treatment on functional stroke outcome and infarct volume. Comparisons of functional stroke outcomes (A-C) and infarct volume (D) were made at the three-month follow-up for patients in the valproate (n = 14) and non-valproate (n = 12) groups. Significant improvements were seen for on all scale scores for the valproate group (A) National Institutes of Health stroke scale (NIHSS), (B) modified Rankin scale (mRS), and (C) Barthel index (BI). No such improvement was seen in the non-valproate group for the NIHSS and mRS, and only modest improvement was seen in the BI scale (A-C). Meanwhile, no significant change of infarct volume (D) was found as comparison of the stroke lesion in FLAIR imaging between the two-week and three-month of follow up at both individual group. Vertical bars represent standard errors of the means. Statistical significance was determined by paired t-test.
Discussion
This open-label controlled study is the first to explore the clinical use of valproate in patients with acute MCA infarction. Two notable findings emerged. First, at three months post-stroke, no difference was observed in either functional outcome or infarct volume between patients who received valproate in addition to standard care, and those who received standard care alone. However, at the three-month follow-up, patients in the valproate group demonstrated remarkable improvements on all three functional outcome measures (NIHSS, mRS, and BI). In contrast, improvements in functional outcome were rather trivial for patients in the non-valproate group. These preliminary data from this small group of patients with acute MCA infarction suggest that valproate’s neuroprotective effects may translate successfully from preclinical studies to clinical settings, and may provide a much-needed future treatment for acute ischemic stroke.
Age, pre-treatment NIHSS scores, and final infarct size have typically been regarded as three major determinants of functional outcome in patients with acute ischemic stroke. Specifically, stroke outcome is significantly worse in the elderly because these patients tend to have higher rates of in-hospital medical complications [25]. In addition, a study of Medicare beneficiaries with acute ischemic stroke found a strong relationship between increasing NIHSS score and mortality at 30 days [26]. A substudy of the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET) showed initial growth in cerebral infarct volume in untreated acute ischemic stroke victims, as well as a significant correlation between functional outcome scale scores and lesion volume [27]. Similarly, Yoo and colleagues found that final infarct volume is a critical determinant of functional outcome three months post-insult [24].
In the present study, all recruited patients with acute MCA infarctions were elderly, had a high pre-treatment NIHSS score (76.5% of patients had an NIHSS > 10), had large infarct volumes at admission, and had delayed increase in infarct volume at the two-week follow-up. The severity of these clinical indicators suggests that the recruited patients were a broadly homogenous population with acute MCA infarction, severe neurological deficits, and a predicted poor prognosis if not administered aggressive therapy. None of the patients received rtPA thrombolytic therapy within the optimum time frame. As noted above, brain MRI at the two-week and three-month follow-ups also observed stroke lesion growth; valproate had no treatment effect on lesion growth, nor reperfusion was observed compared to patients in the non-valproate group (Table 2). This persistent inability to reduce infarct volume in acute stroke victims may be attributable to the relatively delayed administration of valproate (three to 24 hours after ischemia), a notion supported by preclinical results suggesting that the optimal time window for valproate to reduce infarct volume is about three hours after ischemic onset [18].
The goal of treatment for patients with acute ischemic stroke is to restore blood flow to the affected area of the brain as soon as possible, and to rescue the penumbral zone from brain ischemic damage [28]. Thrombolytic therapy with rtPA is the current standard of treatment for acute ischemic stroke patients who present within three hours of symptom onset. The benefits of thrombolytic treatment slowly decrease with time [29,30], and if recanalization occurs too late to benefit ischemic brain tissues, it may exacerbate tissue injury by promoting reperfusion injury, excessive brain edema, and hemorrhagic transformation. These harmful consequences underscore the need to develop neuroprotective therapies for stroke patients who are not eligible to receive rtPA thrombolytic therapy or post rtPA thrombolytic therapy. To our knowledge, the current study suggests that a neuroprotective agent that demonstrated effectiveness in preclinical setting of cerebral ischemia models also potentially has clinical efficacy in patients with acute ischemia stroke, but more studies are needed.
Valproate has a long record of safe and effective use in treating patients with bipolar mood disorder as well as seizures. In addition, a wide variety of CNS disease model studies—including those for stroke—have observed that valproate has remarkable neuroprotective properties, leading some to suggest that its use could be expanded to treat other brain disorders (for a review, see [13-15,19]). It is generally believed that valproate’s neuroprotective effects stem from its ability to inhibit class I and class IIa HDACs to modulate gene transcription (for a review, see [14]). In animal ischemic stroke models using middle cerebral artery occlusion (MCAO), post-insult valproate treatment robustly suppresses MCAO-induced neuroinflammation in the brain [15]; this, in turn, is reminiscent of valproate’s anti-inflammatory effects via inhibiting microglial activation in cellular models [31,32]. Post-stroke valproate also protects against MCAO-induced brain edema and disruption of blood-brain barrier by HDAC inhibition-mediated blockade of NF-kB activation and MMP-9 overexpression [16]. In addition, valproate stimulates MCAO-induced angiogenesis by inducing vascular endothelial growth factor (VEGF) and MMP-2/9, thereby increasing cerebral blood flow and functional recovery in rats. Relatedly, the HDAC inhibitor sodium butyrate, which is structurally and functionally similar to valproate, promotes neurogenesis after MCAO by activating the brain-derived neurotrophic factor (BDNF) signaling pathway, thus contributing to behavioral improvements in a rodent model [33]. Indeed, valproate appears to induce a number of key neurotrophic and neuroprotective molecules, such as BDNF, VEGF, glial cell line-derived neurotrophic factor (GDNF), heat shock protein-70, and brain cell lymphoma 2 (Bcl-2), among others, by transcriptional activation through HDAC inhibition [16,17,20,32-34]. It is conceivable that these molecular events identified in preclinical studies may be involved in the improvement of functional outcome demonstrated in the present clinical setting.
One strength of this study was that strict selection criteria—including clinical characteristics and brain MRI—were used to recruit patients from a broadly homogenous population with severe neurological deficits and with predicted worse stroke outcomes. However, a key limitation is the small sample size. As a result, the data comparing the valproate and non-valproate groups could not reach statistical significance at the three-month follow-up. The estimate of patient numbers needed to treat in the study by using Stata 9.0 sample-size and power determination is necessary to recruit at least 106 patients (53 patients for each valproate and non-valproate group) for reaching 95% statistical power. However, the estimated 106 patients with acute MCA infarction may need to be recruited from a large cohort in excess of 4000 stroke patients in order to test the clinical significance of valproate in the treatment of acute MCA infarction in a future full-scale clinical trial. Another limitation is that the investigators scoring the neurobehavioral outcomes were not blind to treatment group, which might increase study bias. Future double-blind studies with much larger sample sizes that increase statistical power will further help to clarify the causal benefits of valproate treatment in stroke outcome. Although investigators were blind to infarct volume measurement, it should also be noted that the reliability of semi-manual outlining of infarct size in brain MRI, which has better infarct visualization in the period of acute stroke, lacks a gold standard for lesion measurement. Additional validation is required for methods that compare pre- and post-treatment acute stroke measures. Fully automated software to measure lesion volumes may produce fewer sources of error.
Conclusion
This pilot study is the first to show that valproate treatment in patients with acute MCA infarction improved functional outcome at three-month follow-up compared to baseline. Our results highlight the clinical utility of valproate as a potential neuroprotective agent, either alone or in combination with rtPA, for treating patients with acute ischemic stroke. Future clinical trials are warranted to test the usefulness of this agent.
Acknowledgements
The authors thank Chia Ping Tu, Hui Chen Lin, Pei-Min Hsiao, and Wei Tsai of the Tri-Service General Hospital for their professional care of the patients during the course of this study. Ioline Henter of the NIMH, NIH, USA provided excellent editorial assistance. This work was supported by grants from the Tri-Service General Hospital (TSGH-C98-11-S01-04; TSGH-C99-11-S01-04; TSGH-C100-11-S01-3; TSGH-C102-076; TSGH-C103-86), the Teh-Tzer study group for the Human Medical Research Foundation (A1001028-2), and the Intramural Research Program, National Institutes of Health (IRP-NIH).
Disclosure of conflict of interest
No conflict declared.
References
- 1.Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J, Chen S, Chopp M. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142:623–35. [PMC free article] [PubMed] [Google Scholar]
- 2.Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557–65. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- 3.Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–7. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 4.Jauch EC, Saver JL, Adams HP Jr, Bruno A, Connors JJ, Demaerschalk BM, Khatri P, McMullan PW Jr, Qureshi AI, Rosenfield K, Scott PA, Summers DR, Wang DZ, Wintermark M, Yonas H American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870–947. doi: 10.1161/STR.0b013e318284056a. [DOI] [PubMed] [Google Scholar]
- 5.Cheng YD, Al-Khoury L, Zivin JA. Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx. 2004;1:36–45. doi: 10.1602/neurorx.1.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–77. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 7.Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, Hurn P, Liebeskind DS, Nogueira RG, Saver JL STAIR VII Consortium. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–50. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- 8.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH, Group S. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–50. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johannessen CU. Mechanisms of action of valproate: a commentatory. Neurochem Int. 2000;37:103–10. doi: 10.1016/s0197-0186(00)00013-9. [DOI] [PubMed] [Google Scholar]
- 10.Tunnicliff G. Actions of sodium valproate on the central nervous system. J Physiol Pharmacol. 1999;50:347–65. [PubMed] [Google Scholar]
- 11.Henry TR. The history of valproate in clinical neuroscience. Psychopharmacol Bull. 2003;37(Suppl 2):5–16. [PubMed] [Google Scholar]
- 12.Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16:695–714. doi: 10.2165/00023210-200216100-00004. [DOI] [PubMed] [Google Scholar]
- 13.Wang ZF, Fessler EB, Chuang DM. Beneficial effects of mood stabilizers lithium, valproate and lamotrigine in experimental stroke models. Acta Pharmacol Sin. 2011;32:1433–45. doi: 10.1038/aps.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65:105–42. doi: 10.1124/pr.111.005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fessler EB, Chibane FL, Wang Z, Chuang DM. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr Pharm Des. 2013;19:5105–20. doi: 10.2174/1381612811319280009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–67. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F, Leeds P, Chuang DM. Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke. 2012;43:2430–6. doi: 10.1161/STROKEAHA.112.652545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J Cereb Blood Flow Metab. 2011;31:52–7. doi: 10.1038/jcbfm.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinn DI, Kim SJ, Chu K, Jung KH, Lee ST, Song EC, Kim JM, Park DK, Kun Lee S, Kim M, Roh JK. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–72. doi: 10.1016/j.nbd.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Pantano P, Caramia F, Bozzao L, Dieler C, von Kummer R. Delayed increase in infarct volume after cerebral ischemia: correlations with thrombolytic treatment and clinical outcome. Stroke. 1999;30:502–7. doi: 10.1161/01.str.30.3.502. [DOI] [PubMed] [Google Scholar]
- 22.Markus R, Reutens DC, Kazui S, Read S, Wright P, Chambers BR, Sachinidis JI, Tochon-Danguy HJ, Donnan GA. Topography and temporal evolution of hypoxic viable tissue identified by 18F-fluoromisonidazole positron emission tomography in humans after ischemic stroke. Stroke. 2003;34:2646–52. doi: 10.1161/01.STR.0000094422.74023.FF. [DOI] [PubMed] [Google Scholar]
- 23.Schellinger PD, Fiebach JB, Jansen O, Ringleb PA, Mohr A, Steiner T, Heiland S, Schwab S, Pohlers O, Ryssel H, Orakcioglu B, Sartor K, Hacke W. Stroke magnetic resonance imaging within 6 hours after onset of hyperacute cerebral ischemia. Ann Neurol. 2001;49:460–9. [PubMed] [Google Scholar]
- 24.Yoo AJ, Chaudhry ZA, Nogueira RG, Lev MH, Schaefer PW, Schwamm LH, Hirsch JA, Gonzalez RG. Infarct volume is a pivotal biomarker after intra-arterial stroke therapy. Stroke. 2012;43:1323–30. doi: 10.1161/STROKEAHA.111.639401. [DOI] [PubMed] [Google Scholar]
- 25.Denti L, Scoditti U, Tonelli C, Saccavini M, Caminiti C, Valcavi R, Benatti M, Ceda GP. The poor outcome of ischemic stroke in very old people: a cohort study of its determinants. J Am Geriatr Soc. 2010;58:12–7. doi: 10.1111/j.1532-5415.2009.02616.x. [DOI] [PubMed] [Google Scholar]
- 26.Fonarow GC, Saver JL, Smith EE, Broderick JP, Kleindorfer DO, Sacco RL, Pan W, Olson DM, Hernandez AF, Peterson ED, Schwamm LH. Relationship of national institutes of health stroke scale to 30-day mortality in medicare beneficiaries with acute ischemic stroke. J Am Heart Assoc. 2012;1:42–50. doi: 10.1161/JAHA.111.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebinger M, Christensen S, De Silva DA, Parsons MW, Levi CR, Butcher KS, Bladin CF, Barber PA, Donnan GA, Davis SM Echoplanar Imaging Thrombolytic Evaluation Trial Investigators. Expediting MRI-based proof-of-concept stroke trials using an earlier imaging end point. Stroke. 2009;40:1353–8. doi: 10.1161/STROKEAHA.108.532622. [DOI] [PubMed] [Google Scholar]
- 28.Saver JL, Levine SR. Alteplase for ischaemic stroke--much sooner is much better. Lancet. 2010;375:1667–8. doi: 10.1016/S0140-6736(10)60634-4. [DOI] [PubMed] [Google Scholar]
- 29.Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, Brott T, Frankel M, Grotta JC, Haley EC Jr, Kwiatkowski T, Levine SR, Lewandowski C, Lu M, Lyden P, Marler JR, Patel S, Tilley BC, Albers G, Bluhmki E, Wilhelm M, Hamilton S ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363:768–74. doi: 10.1016/S0140-6736(04)15692-4. [DOI] [PubMed] [Google Scholar]
- 30.Lees KR, Bluhmki E, von Kummer R, Brott TG, Toni D, Grotta JC, Albers GW, Kaste M, Marler JR, Hamilton SA, Tilley BC, Davis SM, Donnan GA, Hacke W ECASS, ATLANTIS, NINDS and EPITHET rt-PA Study Group. Allen K, Mau J, Meier D, del Zoppo G, De Silva DA, Butcher KS, Parsons MW, Barber PA, Levi C, Bladin C, Byrnes G. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695–703. doi: 10.1016/S0140-6736(10)60491-6. [DOI] [PubMed] [Google Scholar]
- 31.Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, Yang S, Hong JS. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res. 2005;134:162–9. doi: 10.1016/j.molbrainres.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 32.Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–25. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- 33.Kim HJ, Leeds P, Chuang DM. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem. 2009;110:1226–40. doi: 10.1111/j.1471-4159.2009.06212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51–9. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]