

Abstract
Aims
Contraction of the heart is regulated by electrically evoked Ca2+ transients (CaTs). H+ ions, the end products of metabolism, modulate CaTs through direct interactions with Ca2+-handling proteins and via Na+-mediated coupling between acid-extruding proteins (e.g. Na+/H+ exchange, NHE1) and Na+/Ca2+ exchange. Restricted H+ diffusivity in cytoplasm predisposes pH-sensitive Ca2+ signalling to becoming non-uniform, but the involvement of readily diffusible intracellular Na+ ions may provide a means for combatting this.
Methods and results
CaTs were imaged in fluo3-loaded rat ventricular myocytes paced at 2 Hz. Cytoplasmic [Na+] ([Na+]i) was imaged using SBFI. Intracellular acidification by acetate exposure raised diastolic and systolic [Ca2+] (also observed with acid-loading by ammonium prepulse or CO2 exposure). The systolic [Ca2+] response correlated with a rise in [Na+]i and sarcoplasmic reticulum Ca2+ load, and was blocked by the NHE1 inhibitor cariporide (CO2/HCO3−-free media). Exposure of one half of a myocyte to acetate using dual microperfusion (CO2/HCO3−-free media) raised diastolic [Ca2+] locally in the acidified region. Systolic [Ca2+] and CaT amplitude increased more uniformly along the length of the cell, but only when NHE1 was functional. Cytoplasmic Na+ diffusivity (DNa) was measured in quiescent cells, with strophanthidin present to inhibit the Na+/K+ pump. With regional acetate exposure to activate a local NHE-driven Na+-influx, DNa was found to be sufficiently fast (680 µm2/s) for transmitting the pH–systolic Ca2+ interaction over long distances.
Conclusions
Na+ ions are rapidly diffusible messengers that expand the spatial scale of cytoplasmic pH–CaT interactions, helping to co-ordinate global Ca2+ signalling during conditions of intracellular pH non-uniformity.
Keywords: Acidosis, Calcium, Diffusion, E–C coupling, Na+–H+ exchange
1. Introduction
A brief rise of cytoplasmic [Ca2+] in cardiac myocytes (the Ca2+ transient, CaT) couples the action potential to cellular contraction.1 This CaT displays considerable plasticity. An important example is during changes of intracellular pH (pHi), where the CaT can increase or decrease in amplitude, depending on conditions.2 In healthy isolated myocytes, pHi is typically close to 7.2 (equivalent to ∼60 nM [H+]). This value represents a balance among metabolic acid/base production within the cell and H+-equivalent transport across the sarcolemma. pHi decreases by 0.1–0.2 units with increasing cardiac work-load and can vary during neurotransmitter and hormonal stimulation.2 The pHi also decreases dramatically (up to ∼0.8 units) during clinical conditions such as myocardial ischaemia, where it contributes to acute contractile failure and electrical arrhythmia.2–4 In all these examples, a coupling between pHi and intracellular Ca2+ signalling plays a role in shaping the myocardial response.
Cardiomyocytes have a propensity to develop pHi non-uniformity. Intracellular gradients of 0.1–0.5 units have been measured in ventricular myocytes under various experimental conditions.5–7 Key factors that predispose to non-uniformity are the low myoplasmic H+ diffusivity8 (∼100 µm2/s), high sarcolemmal H+-equivalent flux during acid/base disturbances,9 the expression of H+-equivalent transporters in spatially segregated sarcolemmal domains,6 and regional heterogeneity in the extracellular concentration of membrane-permeant weak acids, such as lactic acid and CO2 (e.g. at the borders of regionally ischaemic zones).10–12 While the effects of whole-cell pHi changes on intracellular Ca2+ signalling have been well characterized, far less is known about the effects of spatial pHi non-uniformity.
There are multiple routes that couple intracellular [Ca2+] ([Ca2+]i) with pHi in the mammalian cardiomyocyte. Resting [Ca2+]i increases during acidosis, because of Ca2+-unloading from pH-sensitive intracellular buffers, such as troponin C, histidyl dipeptides (HDPs), and ATP,5 an effect reinforced by H+-induced slowing of Ca2+ extrusion on sarcolemmal Na+/Ca2+ exchange (NCX).13 Modulation of the CaT by low pHi also relies on the effect of H+ ions on L-type Ca2+ current,14 on Ca2+ re-uptake into the sarcoplasmic reticulum (SR) by the SR Ca ATPase (SERCA),15,16 and on SR Ca2+ release channels (ryanodine receptor channels, RyRs).17,18 The integrated influence of all these effects during a global acidosis is a reduction in SR Ca2+ release, and hence a decrease in the amplitude of the CaT (for convenience, defined here collectively as a direct inhibitory H+ effect on the CaT). Low pHi, however, exerts an additional influence on the CaT. This occurs through stimulation of sarcolemmal Na+/H+ exchange (NHE1) and Na+–HCO3− co-transport (NBC). These membrane transporters extrude intracellular acid from cardiomyocytes in exchange for the entry of extracellular Na+. As a result, cytoplasmic [Na+]i can rise rapidly by several mM during a global fall of pHi. By acting on sarcolemmal NCX, the [Na+] rise then causes retention of intracellular Ca2+, which facilitates greater SR Ca2+ loading via SERCA, leading ultimately to enhanced SR Ca2+ release, and hence an increase in CaT amplitude19 (for convenience, defined here as an indirect stimulatory H+ effect on the CaT). The modulation of CaT amplitude by global acidosis therefore reflects a balance between the direct inhibitory and indirect stimulatory effect of H+ ions on Ca2+ signalling. Depending on which effect is the greater, reducing pHi can either increase or decrease CaT amplitude. The increase, when it occurs, can be physiologically advantageous as it helps to maintain myocardial contraction during acidosis, which would otherwise be depressed owing to decreased Ca2+ binding to the regulatory subunit, troponin C.2
Although a global acidosis can powerfully modulate the CaT, it is not known whether localized pHi changes, when they occur, produce local or more global effects within the cell. We have recently shown5 that a localized acidic microdomain, when induced experimentally in the bulk cytoplasmic compartment can, within seconds, instigate a localized and stable microdomain of elevated resting [Ca2+]i. This phenomenon is mediated by cytoplasmic HDP and ATP molecules acting as diffusible Ca2+/H+ exchangers. It ensures that any spatial non-uniformity of [H+]i is matched by a comparable non-uniformity in resting [Ca2+]i. It is not known, however, whether the characteristics of the dynamic CaT signal can similarly be localized by pHi. If so, this would lead to spatial dyssynchrony in the CaT, during local acid/base disturbances, potentially compromising the contractile efficiency of the cell.
In the present work, we have investigated the effects of local pHi changes on the spatial characteristics of the CaT in the isolated rat ventricular myocyte. We have induced a local intracellular acid-load by using a dual microperfusion apparatus to expose one end of the cell to a permeant weak acid. Our results show that although the direct inhibitory response of the CaT to H+ ions occurs locally within the cell, the indirect stimulatory response, which is driven by sarcolemmal NHE1/NBC activity, is dominant and manifested globally. We have investigated the possible reason for this latter effect. Global stimulation of the CaT by a localized intracellular acidosis appears to be dependent on a high myoplasmic diffusivity of intracellular Na+ ions, contrary to an earlier suggestion for a low value.20 High myoplasmic Na+ diffusivity permits a locally activated influx of Na+ into an acidic region of the cell to spread rapidly downstream, and facilitate the stimulation of SR Ca2+ loading and subsequent release in non-acidic regions, thus promoting a more global CaT response. We have used confocal fluorescence imaging to track the spatial movement of intracellular Na+ under these conditions, to confirm its high myoplasmic mobility. Thus, in addition to emphasizing the importance of intracellular Na+ for enhancing the CaT during acidosis, our work identifies an unexpected role for the Na+ ion as a spatial intracellular messenger, involved in the global regulation of Ca2+ signalling.
2. Methods
2.1. Isolation of ventricular myocytes
Enzymatic isolation of myocytes from rat heart ventricles was performed using a published method.7 Animals were sacrificed by Schedule 1 killing (cervical dislocation) approved by the UK Home Office.
2.2. Solutions
Normal Tyrode (NT; in mM): 135 NaCl, 4.5 KCl, 1 CaCl2 (or as indicated otherwise), 1 MgCl2, 11 glucose, 20 HEPES, and pH 7.4 (37°C). Acetate-containing solutions had equivalently reduced [Cl−]. Ammonium-containing solutions had equivalently reduced [Na+]. Where indicated, Ca2+ in acetate-containing solutions was raised 23% to compensate for Ca2+-acetate binding.14 CO2/HCO3−-buffered solutions contained 22 mM NaHCO3− in place of HEPES and were bubbled with 5% CO2 (for pH 7.4) or 20% CO2 (for pH 6.8). 0Na+/0Ca2+ solutions contained N-methyl-d-glucamine (NMDG) in place of Na+ and EGTA in place of Ca2+. All experiments were performed at 37°C.
2.3. Dual microperfusion
Dual microperfusion was performed in a Perspex superfusion (37°C) chamber mounted on a Leica IRBE microscope.7 One of the two microstreams contained 15 mM sucrose to visualize the interstream boundary. The position of the boundary was adjusted to change between dually microperfusing a myocyte with two microstreams (i.e. boundary across the middle of the cell) and uniformly superfusing with either microstream. Two platinum wires delivered a 2 ms pulse of field stimulation at 2 Hz to evoke contractions.
2.4. Ionic fluorescence imaging
Loading of acetoxymethyl esters of dyes was performed at room temperature (10 min for cSNARF1, 10 min for fluo3, and 2 h for SBFI). Fluorescence was measured using a Leica TCS NT confocal system7 with the following settings: cSNARF1 excitation 514 nm, emission 580 ± 20 and 640 ± 20 nm (ratiometrically); fluo3 excitation 488 nm and emission >520 nm; SBFI excitation 361 nm and emission 440 ± 40 nm and >550 nm (ratiometrically). cSNARF and SBFI images were taken in a X-Y mode (2 s/frame). Fluo3 images were taken in a linescan mode (2 ms/line). Photobleaching was minimized by exciting fluo3 along the raster line only with low laser power (yet sufficient for a good signal/noise ratio). Compared with UV-excitable (ratiometric) dyes, fluo3 offers superior spatio-temporal resolution of Ca2+ dynamics. Images were analysed with ImageJ to determine fluorescence time courses in regions of interest (ROIs) along length of myocyte. Myocyte movement and contraction were corrected by edge detection in the direction of the long axis (cell outline defined as threshold of 5% mean intracellular fluorescence).
2.5. Calibration of fluorescent dyes
cSNARF1 was calibrated using nigericin (10 µM).7 Fluo3 Kd was measured21 in situ at pHi = 7.25 to be 0.84 µM by pipette-loading cells, with 10 µM Ca2+ to measure maximal fluorescence (Fmax) relative to F0 (Fmax/F0 = 9.3 ± 0.3). The mild pH sensitivity of fluo3 was characterized previously5 and was accounted for in the calibration equation by adjusting Kd: (0.4048 × [H+] + 813.3 [nM]). For validation in situ, see Swietach et al.5 SBFI ratio was calibrated using monensin (40 µM) and gramicidin D (2 µM).22
2.6. Statistical analysis
Paired t-tests performed at 5% significance level. Where multiple comparisons are being made for statistical testing (e.g. data from adjacent ROIs), the critical P-value was corrected by the Holm–Bonferroni method. The number of observations is reported as ‘X/Y rats’ (X cells from Y hearts).
3. Results
3.1. Global intracellular acidification affects diastolic Ca2+ and CaT amplitude
Figure 1Ai shows specimen CaTs (2 Hz field stimulation) from a cell bathed in NT(CO2/HCO3−-free, HEPES-buffered). Figure 1Aii displays the CaT averaged, for several cells, in 14 serial ROIs (mean cell length 137.6 ± 3.0 µm and mean ROI length 9.8 µm), confirming that the Ca2+ signal displays good spatial (longitudinal) uniformity (CaT time courses re-plotted with error bars in Supplementary material online, Figure S1).
Figure 1.

Intracellular acid-loading on CaTs. (Ai) Time course of two CaTs reported by fluo3 fluorescence (F/F0) in rat ventricular myocyte paced at 2 Hz superfused in NT. Arrow indicates 2 ms field stimulation voltage pulse. (ii) Visualization of CaT time courses (n = 20 myocytes from four rats) plotted in 14 equally spaced ROIs of the myocyte (corrected for movement and contraction). (Bi) Time courses of pHi (cSNARF1; n = 5/2 rats) and diastolic and systolic [Ca2+] (fluo3; n = 7/3 rats) in response to 20 mM NH4+ pre-pulse solution manoeuvre (HEPES-buffered solutions). Removal of NH4+ imposes an acid load that is then extruded by NHE1. (ii) Experiment repeated in the presence of 30 µM cariporide to inhibit NHE1 (n = 7/3 rats). Dotted lines show time courses in the absence of cariporide to highlight the effect of NHE activity on pHi and CaTs. (C) Effect of raising superfusate CO2 from 5% (pH 7.4) to 20% (pH 6.8) on CaTs (fluo3; n = 8/3 rats). Media contain 22 mM HCO3−. (D) Effect of applying 80 mM acetate (HEPES-buffered media; CO2/HCO3−-free) on CaT (fluo3; n = 6/2 rats) when total extracellular [Ca2+] was 1 mM. Binding of Ca2+ to acetate reduces [Ca2+]o by 23%.
To manipulate pHi, myocytes were subjected to the NH4+ pre-pulse technique. Global superfusion with 20 mM NH4+ transiently raised pHi, and the return to NT produced a transient acidification (Figure 1Bi). pHi recovery from acidosis was blocked by 30 µM cariporide (a high affinity NHE1 inhibitor; Figure 1Bii), confirming that it was mediated by sarcolemmal acid extrusion on NHE1 (lack of CO2/HCO3− ensures negligible NBC activity). Baseline (diastolic) [Ca2+] ([Ca2+]dia) was largely unaffected by the alkalosis, but it reversibly increased during the acidic phase (Figure 1Bi). When NHE1 was functional (absence of cariporide), [Ca2+]dia returned to control levels in parallel with pHi but, when NHE1 was inhibited (with cariporide), it remained elevated, along with a sustained fall of pHi. The rise of [Ca2+]dia at low pHi was thus not dependent on NHE1 activity per se.
Systolic [Ca2+] ([Ca2+]sys) reversibly increased during both intracellular alkalosis (NH4+ superfusion) and acidosis (NH4+ removal; Figure 1B). The rise during alkalosis was cariporide-insensitive, and is probably caused by increased SERCA and RyR activity.15–18 In contrast, the [Ca2+]sys-rise during acidosis was inhibited by cariporide, and was thus dependent on NHE1 activity (cf. Figure 1Bi and ii). An acid-induced rise of [Ca2+]dia and [Ca2+]sys was also obtained using two other methods known to decrease pHi. (i) Raising the partial pressure of CO2 (pCO2) from 5 to 20% in 22 mM HCO3−-containing superfusates reversibly increased [Ca2+]dia and [Ca2+]sys (Figure 1C). (ii) Superfusing 80 mM acetate (in CO2/HCO3−-free, HEPES-buffered solutions; Figure 1D), promptly elevated [Ca2+]dia, and, after an initial reduction, produced a slow rise in [Ca2+]sys, as reported in rat myocytes under superfusion with other weak acids, such as butyrate.23 Adding 80 mM acetate (or butyrate) in NT, however, is known to bind14 ∼20% of the free Ca2+, which itself will affect [Ca2+]sys. In further experiments, free extracellular [Ca2+] ([Ca2+]o) was therefore maintained constant during acetate superfusion (by supplementing CaCl2 in the solution). When this was done, as shown in Figure 2Ci, the initial fall of [Ca2+]sys was prevented, while the slower rise still occurred. The initial fall was thus an artefact caused by acetate binding of extracellular Ca2+. Thus, three different methods for reducing pHi (NH4+ removal, pCO2 elevation, and acetate addition) elevated both [Ca2+]dia and [Ca2+]sys.
Figure 2.

Acid-evoked rise in systolic [Ca2+] is [Na+]i-dependent. (A) Time course of pHi (cSNARF1) in response to superfusion with 80 mM acetate (HEPES-buffered; CO2/HCO3−-free) under conditions of constant free extracellular [Ca2+]. (i) Control conditions, under which NHE is activated by fall in pHi (n = 12/4 rats). (ii) In the presence of 30 µM cariporide to block NHE (n = 12/4 rats). (B) Time course of [Na+]i (SBFI) in response to intracellular acidification by acetate under (i) control conditions (n = 6/2 rats) and (ii) in the presence of cariporide (n = 7/2 rats). (C) Time course of diastolic and systolic [Ca2+] in response to intracellular acidification by acetate under (i) control conditions (n = 20/5 rats) and (ii) in the presence of cariporide (n = 17/5 rats). Insets show calibrated CaT amplitude before and during acetate exposure. (D) Visualization of CaT time courses in 12 ROIs along the length of cell, measured before exposure to acetate (labelled as ‘NT’ in C) or after 90 s of exposure to 80 mM acetate (labelled as ‘Ac’ in C) under (i) control conditions and (ii) in the presence of cariporide. (E) Response to rapid application of 10 mM caffeine (evokes release of SR Ca2+), quantified in terms of fluo3 F/F0 amplitude and half-time of recovery from peak F/F0. Experiments under (i) control conditions (n = 9/3 rats) or (ii) in the presence of cariporide (n = 10/3 rats). *P < 0.05 (vs. NT).
3.2. CaT amplitude, but not [Ca2+]dia, tracks [Na+]i
Figure 2Ai quantifies the rapid intracellular acidification produced by 80 mM acetate superfusion. This was followed by a slow pHi recovery and accompanied by a rise of [Na+]i (reported by intracellular SBFI fluorescence; Figure 2Bi), both of which were inhibited by cariporide (Figure 2Aii and Bii), confirming the activity of NHE1 (NB: the small acetate-induced decrease in SBFI ratio in the presence of cariporide reflects the pH-sensitivity of the dye). [Ca2+]dia, again, increased during the acidosis, both in the presence and absence of cariporide (cf. Figure 2Ci and ii), indicating that the effect was independent of NHE1 activity, and thus of the rise in [Na+]i. In contrast, [Ca2+]sys, and hence CaT amplitude, increased more slowly during the acidosis, in parallel with [Na+]i (Figure 2Ci). Notably, this rise of [Ca2+]sys failed to occur when the [Na+]i elevation was inhibited by cariporide (Figure 2Cii). Thus, the enhancement of CaT amplitude during acidosis tracked the NHE1-dependent rise of [Na+]i, whereas the rise of [Ca2+]dia was independent of [Na+]i changes. Furthermore, when NHE1 had been inhibited, the fall of pHi during acetate superfusion decreased CaT amplitude (see calibrated and averaged time courses of individual CaTs shown as insets in Figure 2C). These observations match those seen earlier with a NH4+ rebound acidosis (Figure 1B). Results shown in Figures 1 and 2 thus indicate that intracellular acidosis raises [Ca2+]dia and CaT amplitude via different mechanisms, only the latter correlating with the rise of [Na+]i driven by NHE1 activity.
The spatial effect of globally reducing pHi on Ca2+ signalling, visualized along the length of the cell, is shown in Figure 2D (time courses re-plotted with error bars in Supplementary material online, Figures S2 and S3). After 90 s of the acid-load, [Ca2+]dia and [Ca2+]sys had increased uniformly. Thus, the response of Ca2+ signalling to a global acidosis is spatially the same throughout the bulk cytoplasmic compartment. The slower relaxation of the CaT during acidosis is consistent with the known inhibitory effects of H+ ions on SERCA and NCX.
3.3. Intracellular acidification raises SR Ca2+ content
Under conditions of active NHE1, the rise in [Ca2+]sys at low pHi is believed to reflect greater SR Ca2+ loading.14,23 To confirm this, the SR load was probed by rapid application of 10 mM caffeine, following a 10 s suspension of pacing. Caffeine superfusion was performed either under control conditions or after 2 min of acetate exposure. The SR load, assessed from the peak fluo3 fluorescence response, was increased at low pHi when NHE1 was active (Figure 2Ei), but not when NHE1 was inhibited (Figure 2Eii). Note also that the subsequent relaxation of this caffeine response (an approximate indicator of NCX activity in rat ventricular myocytes) was slowed at reduced pHi (Figure 2Eii), as expected from the inhibitory effect of H+ ions on NCX.
3.4. Local acidosis regulates [Ca2+]dia and [Ca2+]sys over different spatial domains
To induce a local microdomain of acidity, acetate exposure was restricted to one end of the myocyte, using a dual microperfusion device7 (see Supplementary material online, Methods for justification of using acetate to induce acidosis locally). This releases two parallel microstreams (Figure 3Ai), one containing 80 mM acetate, separated by a sharp interstream boundary of <10 µm. Within ∼20 s, subjecting half the cell length (∼60 ± 7 µm) to acetate produced a smooth longitudinal gradient of pHi, with the majority of acidosis confined to the acetate-exposed end.7 The pHi gradient was stable over time (for several minutes5), and unaffected by adding cariporide (∼0.45 pH units end-to-end; Figure 3Aii and iii), confirming that the technique can be used to clamp a spatial pHi gradient, while probing effects of NHE1 activity on Ca2+. The effect of the pHi gradient on Ca2+ signalling was assessed in 12 adjoining ROIs along the cell length. Figure 3Bi (HEPES-buffered superfusates) shows the time course of [Ca2+]dia and [Ca2+]sys changes, averaged in the three outermost ROIs in the acetate-exposed end where NHE1 is strongly activated by low pHi, and in the three outermost ROIs at the opposite (alkaline) end where NHE1 activity is at a low level. The insets show calibrated time courses of individual CaTs (offset to baseline). Within 90 s of dual microperfusion, [Ca2+]dia increased in the more acidic end, whereas in the more alkaline end it was unchanged (Figure 3Bi). Strikingly, [Ca2+]sys and CaT amplitude increased significantly (P < 10−3, paired t-test) in both regions. Thus, although the acidic microdomain caused an increase in [Ca2+]dia locally, it increased the CaT amplitude globally over a distance of ∼130 µm.
Figure 3.

Acidic microdomain of cytoplasm can influence CaTs remotely. (A) Generating pHi gradient along the length of myocyte. (i) Cartoon of dual microperfusion device releasing NT microstream (lower) and acetate-containing microstream (upper). (ii) pHi gradient along cell length after 90 s of dual microperfusion. (iii) Experiments repeated in the presence of 30 µM cariporide to block NHE. (Bi) Time course of diastolic and systolic [Ca2+] recorded in three adjacent ROIs at either end of the cell (proximal denotes the acetate-exposed end) (n = 24/6 rats). Note the transmission of the systolic [Ca2+] response to the distal end of the cell (P < 0.01; paired t-test). Insets show calibrated CaT amplitude measured before dual microperfusion (dashed line) and after 90 s of dual microperfusion (continuous line). (ii) Visualization of CaT time courses in 12 equally spaced ROIs along cell length at times labelled I and II. (C) Experiments repeated in the presence of 30 µM cariporide to block NHE (n = 14/5 rats). Note the absence of systolic [Ca2+] response distally.
The spatial effects of regional acidosis on CaTs are visualized in Figure 3Bii, where control CaTs (non-shaded transients) are compared with measurements made at 90 s of dual microperfusion (shaded transients; time courses re-plotted with error bars in Supplementary material online Figures S4 and S5). Whereas the [Ca2+]dia-rise co-localized with the acidic microdomain, the enhancement of CaT amplitude was more uniform along the whole-cell length. Furthermore, elevation of [Ca2+]sys, but not [Ca2+]dia, was blocked by 30 µM cariporide (Figure 3Ci). In the presence of the inhibitor, the local acidic microdomain now induced a local reduction in CaT amplitude without significantly affecting the CaT at the far, non-acidic end of the cell. Figure 4 quantifies these effects of local and global acidosis on Ca2+ signalling. The global response of CaT amplitude to both whole-cell and local acidosis (Figure 4A) required functional NHE1 activity. With NHE1 inhibition (Figure 4B), the CaT response became local (i.e. CaT amplitude declined only in acidic zones). In contrast, [Ca2+]dia elevation during acidosis was always local, and was independent of NHE1 activity. Diastolic [Ca2+] and the CaT amplitude therefore respond very differently to acidosis.
Figure 4.

Summary of the effects of global and local acidosis on diastolic and systolic Ca2+ in the presence and absence of acid-evoked Na+ entry. (A) Response of (i and iii) diastolic [Ca2+] (triangles) and systolic [Ca2+] (circles) and (ii and iv) CaT amplitude to regional (i and ii) or whole-cell (iii and iv) exposure to acetate. Filled symbols denote control conditions (uniform exposure to NT). Open symbols denote response to regional or whole-cell exposure to 80 mM acetate. *denotes significance P < 0.05 (paired t-test), corrected for multiple comparisons by the Holm–Bonferroni method. (B) Analysis of experiments performed in the presence of cariporide to block NHE. Key shows the direction of significant change in diastolic Ca2+ (dia-[Ca2+]), systolic Ca2+ (sys-[Ca2+]), or CaT amplitude (|CaT|). Light symbols: effects in acetate-exposed regions; black symbols: effects in acetate-free regions.
3.5. Local acidosis globally enhances CaT via cytoplasmic Na+ diffusion rather than intra-SR Ca2+ diffusion
CaT amplitude is determined largely by Ca2+ release from the SR. During a global acidosis, this amplitude rises because of enhanced SR Ca2+ loading. When acidification is restricted regionally, the global increase in CaT amplitude may arise from a rapid re-distribution of the SR Ca2+ load from acidic to non-acidic regions, via lumenal Ca2+ diffusion. Alternatively, it may arise from a rapid transmission of Na+ ions in cytoplasm. Since the delay in CaT response between acidic and non-acidic ends of myocytes is of the order of seconds (e.g. Figure 3Bi), the underlying mechanism would need to be adequately fast, characterized by diffusivity of the order of ∼103 µm2/s.
The feasibility of the first postulated mechanism (rapid redistribution of SR Ca2+) was tested by radically changing the SR release pattern locally, and observing whether this had a rapid effect on release in remote regions of the cell. Using dual microperfusion, one half of a myocyte (distal half) was exposed to NT solution containing normal (1 mM) Ca2+, and the other half (proximal half) to a microstream containing 80 mM acetate with low (100 µM) Ca2+. Both microstreams contained 30 µM cariporide to prevent NHE1 activation. Figure 5A shows the time course of [Ca2+]dia and [Ca2+]sys changes averaged in three outermost ROIs at the proximal and distal ends of the cells. At the proximal (acidic/low-[Ca2+] exposed) end of the cell, [Ca2+]dia was raised (due to the acidification), whereas [Ca2+]sys was reduced (low [Ca2+]o as well as low pHi greatly reduces SR Ca2+ release when NHE1 is inhibited). Upon return to uniform superfusion with NT, [Ca2+]sys at the proximal end greatly overshot control levels for a period of ∼20 s, which reflects a time-dependent resetting of SR and surface membrane Ca2+ fluxes. At the distal end (where NT had been present throughout the experiment), [Ca2+]sys remained at control levels, despite the major CaT changes <100 µm away. Figure 5B plots the shape of individual CaTs measured longitudinally in 12 ROIs along the length of the myocyte, during the dual microperfusion (unshaded CaTs), and then after 10 s of returning to uniform NT exposure (shaded CaTs). Proximal CaT amplitude increased by nearly 10-fold while at the distal end the CaT was unaffected (time courses re-plotted with error bars in Supplementary material online, Figure S6). Results indicate that the myocyte behaves as a series of adjoining compartments that do not rapidly communicate changes in SR Ca2+. This is consistent with low global intra-SR Ca2+ diffusivity (∼10 µm2/s).24
Figure 5.

Local disturbances to SR fluxes do not affect function remotely. (A) Diffusive coupling in SR along the length of myocyte during local disturbance to Ca2+ fluxes. (i) Myocyte dually microperfused with microstream containing 80 mM acetate and 0.1 mM Ca2+ (proximal end) and microstream of NT (distal end). Time course of diastolic and systolic [Ca2+] (n = 7/2 rats) demonstrates major changes in Ca2+ fluxes at the proximal end that are not transmitted distally. (B) Visualization of CaT time courses in ROIs along cell length, measured at times indicated in (i) by * and #.
Results shown in Figure 5 demonstrate that intra-SR Ca2+ diffusion is too slow to explain fast global systolic Ca2+ responses to local acidosis. Thus, the alternative hypothesis of rapid Na+ transmission was tested. Cytoplasmic Na+ diffusivity (DNa) was measured in myocytes equilibrated initially in Na+-free/Ca2+-free solution to deplete intracellular Na+ without causing Ca2+ overload via reverse mode NCX. A local influx of Na+ was then triggered, by exposing one end of the cell (proximal end, ∼30% of cell length; Figure 6Ai) to solution containing 140 mM Na+, with 80 mM acetate (to stimulate NHE1), and 100 µM strophanthidin (to block Na+-extrusion by the Na+/K+ pump). The remaining 70% (distal) length of the cell was exposed to a Na+-free/Ca2+-free solution, containing 100 µM strophanthidin to block the Na+/K+ pump, and 30 µM cariporide to block NHE1. This protocol ensured that any observed [Na+]-rise in distal regions would necessarily be due to cytoplasmic Na+ diffusion from the proximal end. [Na+]i was imaged by using intracellular SBFI. As shown in Figure 6Aii, SBFI reported a large and rapid proximal rise of [Na+]i upon locally evoking Na+ influx. A similar rise was observed distally, but after a short time-delay of ∼6 s. Best-fitting these data to a diffusion model25 suggested a DNa of 682 ± 184 µm2/s (n = 8). This is 40 ± 10% of the value for DNa in pure water26 at 37°C. The rapid rate of cytoplasmic Na+ diffusion is thus consistent with short longitudinal Na+ diffusion delays, of the order of seconds. To test whether intracellular Na+ diffusion was also fast under more physiological conditions (i.e. starting from a more normal [Na+]i and with a functional sarcolemmal Na+/K+ pump), electrically paced myocytes were exposed proximally to a microstream containing 140 mM Na+, with 80 mM acetate (to reduce pHi locally), but with no strophanthidin in any solution (Figure 6B). The proximal activation of NHE1 by acetate increased [Na+]i near-uniformly along the whole length of the cell, consistent with a high value for DNa (Figure 6Bi). The [Na+]i-rise was blocked completely by cariporide (Figure 6Bii), showing that it was due to NHE1 stimulation. Results suggest that bulk Na+ ion diffusivity in cytoplasm is fast enough to produce a near-uniform rise in releasable Ca2+ (despite slow intra-SR Ca2+ ion diffusion), and therefore a more coordinated global rise in CaT amplitude, even when the source of Na+ influx is highly localized.
Figure 6.

Na+ ions are rapidly diffusible messengers. (A) Measuring cytoplasmic Na+ diffusivity using dual microperfusion to produce local Na+-influx. (i) Cartoon of dual microperfusion device. Myocytes were pre-treated in Na+/Ca2+-free media to deplete intracellular Na+. One end (30%) of a quiescent myocyte was exposed to Ca2+-free solution containing 140 mM Na+ (source of Na+ influx) and 80 mM acetate (to stimulate NHE); the remainder of the myocyte was exposed to Na+/Ca2+-free media containing 100 µM strophanthidin and 30 µM cariporide. (ii) Rise in cytoplasmic [Na+] (SBFI) during dual microperfusion (n = 7/2 rats). (iii) Averaged data shown with superimposed best-fit for diffusivity 680 µm2/s. (Bi) Cytoplasmic [Na+] response during dual microperfusion with 80 mM acetate (along mid-point of the cell) in the presence of normal extracellular [Na+], [Ca2+], and Na+/K+ pump activity. (ii) Experiments repeated in the presence of 30 µM cariporide. Result also demonstrates that the SBFI ratio is not substantially pH-sensitive.
4. Discussion
4.1. Cytoplasmic H+ ions spatially modulate [Ca2+]dia
It is well established that pHi modulates Ca2+ signalling in ventricular myocytes, and strongly influences cardiac function, by regulating CaTs, and even triggering aberrant forms of signalling, such as Ca2+ waves. Most experimental studies have considered the effect of global (uniform) changes in pHi on Ca2+ signalling. With the discovery that cardiac pHi need not always be uniform (see Introduction), effects of pHi heterogeneity should also be considered. In a previous study, we demonstrated that a pHi gradient in quiescent rat ventricular myocytes produces an overlying spatial gradient of cytoplasmic [Ca2+]5. H+ ions displace Ca2+ ions from cytoplasmic buffers, such as histidyl residues on proteins, resulting in a rise in baseline [Ca2+]i that is independent of NHE activity and hence [Na+]i. Importantly, when a pHi gradient evokes a baseline [Ca2+] gradient, the latter can be stable because Ca2+ ions are recruited uphill into acidic regions via a spatial cytoplasmic Ca2+/H+ exchange process, mediated by small diffusible buffer molecules, such as HDPs.5 This process offsets diffusive dissipation of the Ca2+ microdomain. The present study confirms that [Ca2+]dia in electrically paced myocytes (Figure 4Ai and Bi) behaves in the same way as resting [Ca2+]i in quiescent cells, in response to an imposed pHi non-uniformity. The process of cytoplasmic Ca2+/H+ exchange, which accumulates Ca2+ in acidic microdomains, is functional only when pHi is non-uniform. For this reason, it had not been observed in experiments where myocytes were acid-loaded uniformly. Since cytoplasmic H+ ion diffusivity is low2 and the factors that affect pHi (metabolism, blood perfusion, and acid/base membrane transport) can be highly heterogeneous and compartmentalized,27 the effects of non-uniform pHi on Ca2+ signalling should be considered as physiologically relevant. The spatial coupling between pHi and [Ca2+]dia may be important for overcoming the inhibitory effects of H+ ions on Ca2+-sensitive proteins regulated by basal [Ca2+], for example, forms of calmodulin/calcineurin-dependent signalling.28
4.2. Local acidosis modulates CaT amplitude both locally and globally
To furnish a more complete understanding of pH–Ca2+ coupling, the present study has explored the spatial effects of pHi on the CaT. In agreement with previous findings,23,29,30 we observe that the influence of low pHi on CaT amplitude can be both inhibitory (when sarcolemmal NHE1 and NBC are not active) and excitatory (when these transporters are active, leading to a rise of [Na+]i, which ultimately, via NCX, enhances SR Ca2+ loading). Interestingly, the inhibitory effect, which is largely attributable to attenuation of SERCA, RyR, and L-type Ca2+ channel activity, is closely localized to acidic cytoplasmic regions, rather like the local effects of pHi on [Ca2+]dia. The surprising finding, however, is that this local inhibitory CaT response is transformed into a global excitatory response, when Na+-dependent acid extrusion is functional. In the present experiments using HEPES-buffered NT, such extrusion was via NHE1, although NBC will also have contributed in CO2/HCO3−-buffered conditions (e.g. Figure 1C). We have shown that an acidic cytoplasmic microdomain, which activates NHE1 locally, raises CaT amplitude both in the acidified domain and more remotely in non-acidic regions (Figure 4Ai and Bi). Thus, local acidosis is capable of increasing CaT amplitude over a much greater volume of the cell, provided an H+-activated Na+-entry pathway is present (Figure 7). This remote coupling between intracellular H+ and Ca2+ signals is unprecedented, given that high intracellular buffering greatly restricts their effective ionic diffusivity. The functional significance of expanding the spatial scale over which H+ ions affect systolic [Ca2+] may be to even out CaT heterogeneity under conditions of non-uniform pHi. This may help to decrease the incidence of aberrant forms of Ca2+ signalling.31,32
Figure 7.
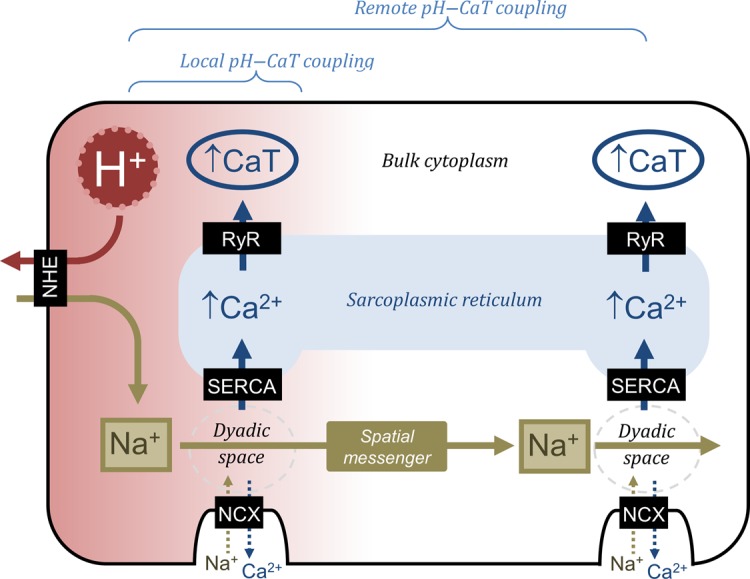
Schematic diagram showing local pH–Ca2+ interactions that influence diastolic [Ca2+] and Na+-mediated pH–Ca2+ coupling that influences systolic [Ca2+] locally and remotely.
A key factor affecting Ca2+ signalling uniformity is the distribution of Ca2+ in the SR. Luminal Ca2+ ions are buffered, but the extent to which their diffusivity is restricted is controversial,33 with reports of fast,34 slow,24 and spatially heterogeneous Ca2+ mobility.35 This study provides additional evidence that the SR content is weakly coupled by Ca2+ diffusion over distances of multiple sarcomeres. In the absence of an H+-activated Na+ influx, manipulating the CaT at one end of a myocyte does not affect Ca2+ signalling at the other (Figure 5), arguing that global apparent intra-SR Ca2+ diffusivity is low. Thus, Ca2+ signalling is predisposed to becoming non-uniform, in part because of slow Ca2+ diffusion in both the cytoplasm and SR. A means of coordinating SR Ca2+ content, in order to unify Ca2+ signalling, would thus be helpful for maintaining contractile synchrony within the myocyte.
4.3. Na+ ions are rapidly diffusible messengers for Ca2+ signalling
Among the three ionic species of the H+–Na+–Ca2+ signalling triumvirate,19 Na+ ions are the least buffered and therefore the most likely to diffuse rapidly in cytoplasm. The present work measured cytoplasmic Na+ diffusivity by restricting Na+ influx to one region of the cell under conditions of sarcolemmal Na+/K+ pump inhibition (i.e. limiting the transmembrane ‘loss’ of Na+ that may otherwise lead to an erroneous estimate of Na+ diffusivity). Our measured value for DNa is 40% of that in pure water, i.e. near its physical limit for a tortuous cytoplasmic compartment, considering that macromolecular crowding increases diffusion paths by approximately two-fold.25,26 The present estimate of DNa is in agreement with measurements made on skeletal myoplasm26 and with the fast mobility of a similar-sized monovalent cation, NH4+, estimated in cardiac myocytes.7 The magnitude of DNa is sufficient to produce a near-uniform rise in [Na+]i, when evoked locally by transporters such as NHE1. In contrast, an earlier estimate20 of DNa, which was 60-fold lower, would not be compatible with our observations of [Na+]i near-uniformity during regional NHE1 stimulation. Since intra-SR Ca2+ mobility is substantially lower than DNa, remote pHi–Ca2+ coupling must be dependent on fast cytoplasmic Na+ transmission. The rapid spread of Na+ ions, even when influx is localized, will therefore assist in coordinating the SR Ca2+ load, helping to unify the electrically evoked CaT.
4.4. Potential for Na+ diffusion in multicellular myocardium
In the present work, the principle of remote pHi–Ca2+ coupling was demonstrated by experimentally imposing a large pHi gradient (∼0.5 units) along an isolated cell (∼100 µm). Although physiological pHi gradients are typically smaller than this (e.g. up to 0.2 units over ∼30 µm during stimulated NHE1 activity2), the functional importance of Na+i diffusion will remain the same, helping to even out local variations of CaT amplitude caused by the pHi non-uniformity. This spatial role of Na+i may become particularly prominent in the intact myocardium. Here, the spatial range over which Na+ diffuses is likely to exceed the dimensions of a single myocyte, because Na+ ions readily permeate gap-junctional channels at intercalated discs36 (rather than requiring a permeant carrier molecule, as is the case for H+ ions37). Thus, Na+ diffusion may regulate CaTs in cells adjacent to acidified myocytes. Within the myocardium, local acidosis, evoked for example by compromised capillary perfusion, can greatly exceed 0.2 pHi units2 resulting in considerable spatial pHi non-uniformity. Since Na+ ions diffuse ∼7 times faster than H+ ions in the coupled syncytium,8,37 a large acid-induced rise of [Na+]i may extend beyond the acidified domain, provided that gap junctions remain open during acidosis,37 and Na+-coupled acid-extruders remain active. In this case, local myocardial acidity may promote significant pHi–Ca2+ coupling remotely. Under conditions of excessive Na+ influx into a restricted region, cell-to-cell transmission of Na+ may also predispose neighbouring cells to SR Ca2+ overload, Ca2+ waves, and delayed after-depolarizations. This behaviour would be the spatial equivalent of events occurring during cardiac ischaemia–reperfusion, where a substantial Ca2+ overload, driven by [Na+]i accumulation, and persisting at normal (recovered) pHi, is pro-arrhythmogenic and injurious.3 The border zones of regionally ischaemic areas38 may thus be particularly vulnerable to spreading pro-arrhythmogenic Na+ signals. It is notable that 23Na NMR imaging studies of regional myocardial ischemia39 have demonstrated [Na+]i gradients of sufficient magnitude to drive a lateral diffusive Na+ flux and plausibly modulate Ca2+ handling in remote regions, whereas Na+i overloading of one myocyte of a coupled pair, using a patch-pipette, has been shown to drive hypercontracture in the adjacent cell.36
4.5. Conclusions
We have shown that, within the ventricular myocyte, rapid diffusion of Na+ ions in cytoplasm can couple Ca2+ signalling to spatially remote H+ signals. The different spatial domains for the pHi sensitivity of diastolic and systolic [Ca2+] add a novel and important degree of complexity to the regulation of Ca2+ signalling in the heart. During pHi disturbances, high Na+ ion mobility will assist in the unification of Ca2+ signalling, by coordinating the degree of SR Ca2+ load, and overcoming the potential for heterogeneity caused by low intra-SR Ca2+ diffusivity. This novel role for Na+ ions need not be limited to the acid response, as any local Na+ influx should have a comparable effect.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by British Heart Foundation Programme Grant (to R.D.V.-J.), National Institutes of Health (to K.W.S. R37HL042873), and Royal Society University Research Fellowship (to P.S.). Funding to pay the Open Access publication charges for this article was provided by the British Heart Foundation.
Acknowledgements
The authors thank Philip Cobden for excellent technical assistance in isolating myocytes.
Conflict of interest: none declared.
References
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. doi: 10.1016/j.yjmcc.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 3.Orchard CH, Cingolani HE. Acidosis and arrhythmias in cardiac muscle. Cardiovasc Res. 1994;28:1312–1319. doi: 10.1093/cvr/28.9.1312. [DOI] [PubMed] [Google Scholar]
- 4.Steenbergen C, Deleeuw G, Rich T, Williamson JR. Effects of acidosis and ischemia on contractility and intracellular pH of rat heart. Circ Res. 1977;41:849–858. doi: 10.1161/01.res.41.6.849. [DOI] [PubMed] [Google Scholar]
- 5.Swietach P, Youm JB, Saegusa N, Leem CH, Spitzer KW, Vaughan-Jones RD. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci USA. 2013;110:E2064–E2073. doi: 10.1073/pnas.1222433110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garciarena CD, Ma YL, Swietach P, Huc L, Vaughan-Jones RD. Sarcolemmal localisation of Na+/H+ exchange and Na+-HCO3− co-transport influences the spatial regulation of intracellular pH in rat ventricular myocytes. J Physiol. 2013;591:2287–2306. doi: 10.1113/jphysiol.2012.249664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swietach P, Leem CH, Spitzer KW, Vaughan-Jones RD. Experimental generation and computational modeling of intracellular pH gradients in cardiac myocytes. Biophys J. 2005;88:3018–3037. doi: 10.1529/biophysj.104.051391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swietach P, Spitzer KW, Vaughan-Jones RD. pH-Dependence of extrinsic and intrinsic H+-ion mobility in the rat ventricular myocyte, investigated using flash photolysis of a caged-H+ compound. Biophys J. 2007;92:641–653. doi: 10.1529/biophysj.106.096560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leem CH, Lagadic-Gossmann D, Vaughan-Jones RD. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J Physiol. 1999;517(Pt 1):159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer WR, Hiller KH, Galuppo P, Neubauer S, Kopke J, Haase A, Waller C, Ertl G. Fast high-resolution magnetic resonance imaging demonstrates fractality of myocardial perfusion in microscopic dimensions. Circ Res. 2001;88:340–346. doi: 10.1161/01.res.88.3.340. [DOI] [PubMed] [Google Scholar]
- 11.Cascio WE, Yan GX, Kleber AG. Early changes in extracellular potassium in ischemic rabbit myocardium. The role of extracellular carbon dioxide accumulation and diffusion. Circ Res. 1992;70:409–422. doi: 10.1161/01.res.70.2.409. [DOI] [PubMed] [Google Scholar]
- 12.Decking UK. Spatial heterogeneity in the heart: recent insights and open questions. News Physiol Sci. 2002;17:246–250. doi: 10.1152/nips.01393.2002. [DOI] [PubMed] [Google Scholar]
- 13.Boyman L, Hagen BM, Giladi M, Hiller R, Lederer WJ, Khananshvili D. Proton-sensing Ca2+ binding domains regulate the cardiac Na+/Ca2+ exchanger. J Biol Chem. 2011;286:28811–28820. doi: 10.1074/jbc.M110.214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saegusa N, Moorhouse E, Vaughan-Jones RD, Spitzer KW. Influence of pH on Ca2+ current and its control of electrical and Ca2+ signaling in ventricular myocytes. J Gen Physiol. 2011;138:537–559. doi: 10.1085/jgp.201110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulme JT, Orchard CH. Effect of acidosis on Ca2+ uptake and release by sarcoplasmic reticulum of intact rat ventricular myocytes. Am J Physiol. 1998;275:H977–H987. doi: 10.1152/ajpheart.1998.275.3.H977. [DOI] [PubMed] [Google Scholar]
- 16.Mandel F, Kranias EG, Grassi de Gende A, Sumida M, Schwartz A. The effect of pH on the transient-state kinetics of Ca2+-Mg2+-ATPase of cardiac sarcoplasmic reticulum. A comparison with skeletal sarcoplasmic reticulum. Circ Res. 1982;50:310–317. doi: 10.1161/01.res.50.2.310. [DOI] [PubMed] [Google Scholar]
- 17.Balnave CD, Vaughan-Jones RD. Effect of intracellular pH on spontaneous Ca2+ sparks in rat ventricular myocytes. J Physiol. 2000;528(Pt 1):25–37. doi: 10.1111/j.1469-7793.2000.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L, Mann G, Meissner G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ Res. 1996;79:1100–1109. doi: 10.1161/01.res.79.6.1100. [DOI] [PubMed] [Google Scholar]
- 19.Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H+-activated Na+ influx in the ventricular myocyte couples Ca2+-signalling to intracellular pH. J Mol Cell Cardiol. 2013;61:51–59. doi: 10.1016/j.yjmcc.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Despa S, Kockskamper J, Blatter LA, Bers DM. Na/K pump-induced [Na]i gradients in rat ventricular myocytes measured with two-photon microscopy. Biophys J. 2004;87:1360–1368. doi: 10.1529/biophysj.103.037895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trafford AW, Diaz ME, Eisner DA. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Arch. 1999;437:501–503. doi: 10.1007/s004240050808. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto T, Swietach P, Rossini A, Loh SH, Vaughan-Jones RD, Spitzer KW. Functional diversity of electrogenic Na+-HCO3− cotransport in ventricular myocytes from rat, rabbit and guinea pig. J Physiol. 2005;562:455–475. doi: 10.1113/jphysiol.2004.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi HS, Trafford AW, Orchard CH, Eisner DA. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J Physiol. 2000;529(Pt 3):661–668. doi: 10.1111/j.1469-7793.2000.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swietach P, Spitzer KW, Vaughan-Jones RD. Ca2+-mobility in the sarcoplasmic reticulum of ventricular myocytes is low. Biophys J. 2008;95:1412–1427. doi: 10.1529/biophysj.108.130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swietach P, Zaniboni M, Stewart AK, Rossini A, Spitzer KW, Vaughan-Jones RD. Modelling intracellular H+ ion diffusion. Prog Biophys Mol Biol. 2003;83:69–100. doi: 10.1016/s0079-6107(03)00027-0. [DOI] [PubMed] [Google Scholar]
- 26.Kushmerick MJ, Podolsky RJ. Ionic mobility in muscle cells. Science. 1969;166:1297–1298. doi: 10.1126/science.166.3910.1297. [DOI] [PubMed] [Google Scholar]
- 27.Swietach P, Leem CH, Spitzer KW, Vaughan-Jones RD. Pumping Ca2+ ions up H+ gradients: a cytoplasmic Ca2+/H+ exchanger without a membrane. J Physiol. 2014;592:3179–3188. doi: 10.1113/jphysiol.2013.265959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saucerman JJ, Bers DM. Calmodulin binding proteins provide domains of local Ca2+ signaling in cardiac myocytes. J Mol Cell Cardiol. 2012;52:312–316. doi: 10.1016/j.yjmcc.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisner DA, Lederer WJ, Vaughan-Jones RD. The quantitative relationship between twitch tension and intracellular sodium activity in sheep cardiac Purkinje fibres. J Physiol. 1984;355:251–266. doi: 10.1113/jphysiol.1984.sp015417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison SM, Frampton JE, McCall E, Boyett MR, Orchard CH. Contraction and intracellular Ca2+, Na+, and H+ during acidosis in rat ventricular myocytes. Am J Physiol. 1992;262:C348–C357. doi: 10.1152/ajpcell.1992.262.2.C348. [DOI] [PubMed] [Google Scholar]
- 31.Miura M, Wakayama Y, Endoh H, Nakano M, Sugai Y, Hirose M, Ter Keurs HE, Shimokawa H. Spatial non-uniformity of excitation-contraction coupling can enhance arrhythmogenic-delayed afterdepolarizations in rat cardiac muscle. Cardiovasc Res. 2008;80:55–61. doi: 10.1093/cvr/cvn162. [DOI] [PubMed] [Google Scholar]
- 32.Wakayama Y, Miura M, Stuyvers BD, Boyden PA, ter Keurs HE. Spatial nonuniformity of excitation-contraction coupling causes arrhythmogenic Ca2+ waves in rat cardiac muscle. Circ Res. 2005;96:1266–1273. doi: 10.1161/01.RES.0000172544.56818.54. [DOI] [PubMed] [Google Scholar]
- 33.Bers DM, Shannon TR. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J Mol Cell Cardiol. 2013;58:59–66. doi: 10.1016/j.yjmcc.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Bers DM. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res. 2006;99:283–291. doi: 10.1161/01.RES.0000233386.02708.72. [DOI] [PubMed] [Google Scholar]
- 35.Picht E, Zima AV, Shannon TR, Duncan AM, Blatter LA, Bers DM. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Meana M, Garcia-Dorado D, Hofstaetter B, Piper HM, Soler-Soler J. Propagation of cardiomyocyte hypercontracture by passage of Na+ through gap junctions. Circ Res. 1999;85:280–287. doi: 10.1161/01.res.85.3.280. [DOI] [PubMed] [Google Scholar]
- 37.Swietach P, Rossini A, Spitzer KW, Vaughan-Jones RD. H+ ion activation and inactivation of the ventricular gap junction: a basis for spatial regulation of intracellular pH. Circ Res. 2007;100:1045–1054. doi: 10.1161/01.RES.0000264071.11619.47. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka H, Oyamada M, Tsujii E, Nakajo T, Takamatsu T. Excitation-dependent intracellular Ca2+ waves at the border zone of the cryo-injured rat heart revealed by real-time confocal microscopy. J Mol Cell Cardiol. 2002;34:1501–1512. doi: 10.1006/jmcc.2002.2096. [DOI] [PubMed] [Google Scholar]
- 39.Ouwerkerk R, Bottomley PA, Solaiyappan M, Spooner AE, Tomaselli GF, Wu KC, Weiss RG. Tissue sodium concentration in myocardial infarction in humans: a quantitative 23Na MR imaging study. Radiology. 2008;248:88–96. doi: 10.1148/radiol.2481071027. [DOI] [PMC free article] [PubMed] [Google Scholar]