

Abstract
TP53 mutations confer subgroup specific poor survival for children with medulloblastoma. We hypothesized that WNT activation which is associated with improved survival for such children abrogates TP53 related radioresistance and can be used to sensitize TP53 mutant tumors for radiation. We examined the subgroup-specific role of TP53 mutations in a cohort of 314 patients treated with radiation. TP53 wild-type or mutant human medulloblastoma cell-lines and normal neural stem cells were used to test radioresistance of TP53 mutations and the radiosensitizing effect of WNT activation on tumors and the developing brain. Children with WNT/TP53 mutant medulloblastoma had higher 5-year survival than those with SHH/TP53 mutant tumours (100% and 36.6% ± 8.7%, respectively (p < 0.001)). Introduction of TP53 mutation into medulloblastoma cells induced radioresistance (survival fractions at 2Gy (SF2) of 89% ± 2% vs. 57.4% ± 1.8% (p < 0.01)). In contrast, β-catenin mutation sensitized TP53 mutant cells to radiation (p < 0.05). Lithium, an activator of the WNT pathway, sensitized TP53 mutant medulloblastoma to radiation (SF2 of 43.5% ± 1.5% in lithium treated cells vs. 56.6 ± 3% (p < 0.01)) accompanied by increased number of γH2AX foci. Normal neural stem cells were protected from lithium induced radiation damage (SF2 of 33% ± 8% for lithium treated cells vs. 27% ± 3% for untreated controls (p = 0.05). Poor survival of patients with TP53 mutant medulloblastoma may be related to radiation resistance. Since constitutive activation of the WNT pathway by lithium sensitizes TP53 mutant medulloblastoma cells and protect normal neural stem cells from radiation, this oral drug may represent an attractive novel therapy for high-risk medulloblastomas.
Electronic supplementary material
The online version of this article (doi:10.1186/s40478-014-0174-y) contains supplementary material, which is available to authorized users.
Introduction
Medulloblastoma is the most common malignant brain tumor of childhood [1]. Survival of these children has improved in the last two decades due to collaborative studies utilizing aggressive surgical resection, high dose craniospinal irradiation and chemotherapy [2-4]. This aggressive approach to therapy has come at a severe cost with the majority patients suffering significant long-term neurocognitive, endocrine and other toxicities [5]. Recent progress in the molecular stratification of medulloblastoma has revealed striking heterogeneity that has led to stratification into several genetically distinct clinical subgroups (WNT, SHH, Group 3, Group 4) which respond differently to current therapies [6].
Of the four subgroups, patients with WNT subgroup medulloblastoma exhibit an excellent overall survival (OS), those with SHH medulloblastoma have an intermediate long-term survival, whereas those with Groups 3 and 4 disease have particularly decimal outcomes [7,8]. Recent studies highlighted the role of additional genetic alterations in identifying risk factors in each molecular subgroup. For example, iso17q together with MYC amplification define the high risk patients in group 3, but not group 4. In contrast, Group 4 patients with whole chromosome 11 loss, or chromosome 17 gain together with loss of chromosome 10p, demonstrate better survival. Absence of GLI2 amplification and 14q loss identify a low-risk patient population, chromothripsis is found in SHH patients with inferior outcomes, and monosomy of chromosome 6 carries favorable prognostic value only within the WNT subgroup [9].
Our group has recently uncovered the role of TP53 mutations in clinically distinct subgroups of medulloblastoma [10]. TP53 mutations are restricted to the SHH and WNT subgroups. In the former it is associated with poor survival while in the latter, this survival disadvantage is not seen [10].
TP53 is mutated or deleted in over 50% of human cancers resulting in loss of p53-associated apoptosis, cell cycle arrest or DNA brake/repair response [11]; Loss of normal p53 function and resultant impaired G1 check point control correlates with increased resistance to both low- and high-dose ionizing radiation in several cancers including medulloblastoma [12-21]. It is argued that, though tissue specific, TP53 mutation status can predict tumor radiosensitivity and a patient’s response to radiation therapy [19,20].
Craniospinal radiation therapy remains the cornerstone of treatment in childhood medulloblastoma. It is, therefore, reasonable to hypothesize that the poor survival of patients with TP53 mutant medulloblastoma is related to radioresistance of these cancers. In contrast, in 10-15% of medulloblastoma displaying activation of the WNT/β-catenin pathway TP53 mutant status does not negatively influence survival [18,22,23]. In the WNT subgroup, patients with either wild-type or mutant TP53 tumors respond equally well to radiotherapy. Most WNT subgroup tumors harbor activating mutations in exon 3 of the β-catenin gene, and this genetic alteration is a universally favorable prognostic marker for both average- and high-risk patients, and long term patient survival exceeds 90% [3,6,7,22,24-28].
Lithium is a non-competitive and specific inhibitor of GSK3β which is a negative regulator of the WNT pathway. Lithium mimics canonical WNT activation marked by nuclear translocation of β-catenin and transcriptional activation of downstream targets [29-31]. Since lithium can be safely administered to patients and crosses the brain–blood barrier [32], it might constitute a therapeutic means to induce radiosensitivity and improve survival in SHH/mutant TP53 patients.
To test our hypothesis we first assessed the impact of TP53 mutations on survival of children with medulloblastoma treated with craniospinal irradiation. We then examined the role of lithium-mediated WNT activation in abrogating the radioresistance observed in TP53 mutant medulloblastoma cells. Our results suggest that lithium administration with radiation therapy may be a safe and efficacious therapy for childhood medulloblastoma.
Materials and Methods
Patient and sample description: We collected clinical and biological data on 314 patients from the Hospital for Sick Children (Toronto, Canada), Division of Molecular Genetics and Department of Paediatric Oncology, Haematology and Immunology, University Hospital (DKFZ) (Heidelberg, Germany), Northern Institute for Cancer Research, Newcastle University (Newcastle, United Kingdom) [33], Institute of Neuropathology, University of Bonn Medical Centre (Bonn, Germany) [34] and the Medulloblastoma Advanced Genomics International Consortium (MAGIC) [35] cohorts. Patients between 5 and 19 years of age were eligible for this study. We excluded patients younger the 5 years of age at diagnosis under the assumption that they were not eligible for radiation therapy according to the most of the current medulloblastoma protocols. All patient samples were procured in accordance with the Research Ethics Board of their corresponding institution. For 300 (95.5%) patients complete clinical and survival data were available for analysis. Samples from all centers were either obtained as frozen tissue or formalin fixed paraffin embedded (FFPE) biopsies and processed to determine subgroup β-catenin and TP53 status as previously described [10].
Cell line work: Human medulloblastoma wild-type TP53 cells lines: ONS76, D283MED, MEB-MED-8A and TP53 mutant cell lines: UW228 and Daoy were cultured as previously described [36,37]. Normal neural stem cells (NNSC) Hf5205 were cultured as previously described [38]. ONS76, D283MED, MEB-MED-8A, UW228 and Daoy cells were provided by Taylor Lab (The Hospital for Sick Children, Toronto) and Brain Tumor Research Centre (BTRC) repository (The Hospital for Sick Children, Toronto). NNSC, Hf5205, were provided by Dirks Lab (The Hospital for Sick Children, Toronto). All cells were authenticated including verification of TP53 status. Lithium chloride (Sigma, 02685-1EA), was used at a final concentration of 2 mM in regular culture medium. Radiation exposure was performed using a Nordion Gammacell 40 irradiator with the central dose rate 1.24 Gy/min. Single discrete doses of 1 to 8Gy were delivered according to experimental design.
Clonogenic experiments: To generate radiation sensitivity curves cells were seeded at specific numbers in triplicates into 10 cm dishes to yield between 50 and 150 colonies. Cells were allowed to adhere overnight and then irradiated with the range of single doses from 1 to 5Gy. The medium was changed after irradiation and cells were maintained at 37°C for 14 days to allow colony formation. The colonies (50 cells/colony threshold) were then fixed and stained with Crystal violet (0.1%) and counted manually. The surviving fractions were calculated after adjustment for plating efficiency of non-irradiated controls as follows [39]:
Survival fraction at 2 Gray (SF2) calculations: To fit the clinical relevance, the radiation dose of 2Gy was selected to match the daily fraction size commonly used in paediatric clinical practice [40]. The SF2 was calculated as following:
Sensitizer enhancement ratio (SER) calculations: SER was calculated as the radiation dose needed for radiation alone divided by the radiation dose together with of lithium needed to eliminate 90% of cells [36].
Same corresponding cell lines simultaneously maintained without lithium chloride were used as controls. Data from three independent experiments performed in triplicate were each plotted as a function of radiation dose on a semi-logarithmic scale.
Transfection experiments: For each cell line, 3 × 105 cells were transiently transfected in parallel with 2 μg of the plasmid-of-interest DNA or 2 μg pcDNA3 control plasmid (either GFP- or FLAG-tagged) by nucleoporation, as per the manufacturer’s instructions, using the Amaxa Mouse Neural Stem Cell Kit (Nucleofector, Lonza, VPG-1004) and X001 program. At 24 hours post transfection the transfection efficiency of 59.9% ± 12.3% for the pcDNA3-TP53R175H/pcDNA3-GFP plasmid combination and 90% for the pcDNA3-S33Y/pcDNA3-FLAG plasmid combination was estimated by manual counting of GFP- and FLAG- positive cells under immunofluorescent microscopy. Subsequently, cells were irradiated between 24 and 48 hours and plated for clonogenic assay as described above. Plasmids used: pcDNA3-TP53R175H, pcDNA3 (courtesy of Dr. David Malkin, The Hospital for Sick Children, Toronto), and pcDNA3-S33Y β-catenin [41] obtained from Addgene: http://www.addgene.org/.
Clonogenic experiments with combination of lithium and radiation exposure: ONS76 and UW228 cells were cultured, as described above. Cells were exposed to 2 mM lithium chloride for 24 hours, and then irradiated with the range of doses from 2 to 8Gy. Drug-containing medium was replaced with normal growth medium. Clonogenic experiments were performed as described above.
Immunofluorescence – β-catenin nuclear translocation: Cells transfected with S33Y-CTNNB1 mutation and/or pre-treated with 2 mM of lithium chloride for 24 hours were cultured as described above. β-catenin expression was visualized by immunofluorescence using a Quorum Spinning Disk Confocal Microscope. The antibodies used were: a mouse anti-β-catenin (BD Transduction Labs, 6101153) and a donkey-anti-mouse IgG conjugated with Alexa Fluor 488 (Invitrogen, A21202); both were used at 1:500 dilution. All experiments were repeated three times.
β-catenin luciferase reporter assay: For the reporter assay, 1 × 105 cells were plated in 24-well plates in triplicate per sample and left to attach overnight. Cells were transiently transfected in parallel with either 0.5 μg of the TCF reporter construct (M50 Super 8xTOPFlash) DNA or 0.5 μg of the mutated reporter construct (M51 Super 8×FOPFlash) DNA [39,42] together with 1 ng of Renilla DNA. Plasmids were obtained from Addgene: http://www.addgene.org/. Transfections were performed using Lipofectamine 2000 (Invitrogen, 11668–027) according to manufacturer’s protocol. After 4 hr post transfection medium was replaced with the regular growth medium and cells were treated with 2 mM of lithium chloride for 24 hr. TCF-mediated transcriptional activity was determined by the ratio of TOPFlash/FOPFlash luciferase activity, normalized to the Renilla luciferase activity, using Promega Dual Luciferase Reporter Assay System, (Promega, E1960). 3 independent experiments were done.
Phospho-histone H2AX (γH2AX) foci immunofluorescent microscopy and imaging: Medulloblastoma and NNSC cells were cultured on cover slips in 6-well dishes and fixed as described above. Cells were pre-treated with 2 mM lithium chloride for 24 hours and then exposed to 5Gy single dose of radiation. Untreated and non-irradiated cells were used as control. Specimens were collected at 30 min, 4 and 24 hours post radiation. At appropriate time points, cells were fixed and processed as described above. The antibodies used were: a mouse anti-phospho-histone-H2AX (Ser139) (Millipore, 05–636), 1:800 dilution, and an Alexa Fluor 488 donkey-anti-mouse IgG (Invitrogen, A21202), 1:500 dilution. For quantification of γH2AX foci 3 independent experiments were performed. Between 100 and 250 nuclei were examined and foci were counted using Perkin Elmer Velocity 6.0.1 software (Perkin Elmer, USA) with 0.2um cut off for foci size. The following procedure was applied to decrease inter-experimental variability: 1) all 3 experiments were pooled together and individual values were used for analysis [43]; 2) average number of foci per nucleus was calculated; 3) outliers, which were estimated as nuclei containing > 2 standard deviation (2 SD) from the average for the treatment subgroup, were eliminated; 4) baseline number of foci per nucleus was estimated in the untreated sample as average plus 2 SD; 5) to remove background noise nuclei with baseline or less number of foci were eliminated from experimental samples and the remaining nuclei were considered γH2AX-positive for further calculations; 6) number of foci in γH2AX-positive nuclei was calculated and expressed as an average number of foci per nuclei; 7) comparison was done between treatments using average number of foci per nucleus and number of γH2AX-positive cells as defined above.
Normal neuronal stem cell viability experiments: NNSC (Hf5205) were cultured and pre-treated with lithium chloride as previously described. Cells were counted five days after radiation using a Cell Viability Analyzer (Vi-Cell XR, Beckman Coulter). Survival curves were generated as described above. The figure was plotted from three independent experiments performed in triplicate each.
Statistical analysis: Overall survival was estimated using the Kaplan-Meier method with significance (α = 0.05) based on long-rank test, using Stata 12 for OS X. OS was defined as the interval between the date of diagnosis and the date of death of any cause or the date of the last follow-up visit.
For biological data results were expressed as the mean ± SE of three or more independent experiments carried out in triplicate. Statistically significant differences between samples were determined using Student 2-tailed t-test and two-way ANOVA with the Bonferroni correction method, when appropriate, using GraphPad Prism version 5.00 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com; p value < 0.05 was considered significant. The error bars in the figures represent SEs.
Results
Impact of TP53 mutations on children treated with irradiation. In order to assess the role of TP53 mutations in medulloblastoma patients who received radiotherapy, we selected patients between 5 and 19 years old who received craniospinal radiation according to local standard of care protocols. The complete characteristic of the cohort is summarized in Table 1. We identified somatic TP53 mutations in 16.9% (53/314) of cases. Median age at diagnosis and gender distribution was not different between wild-type or mutant TP53 patients. We observed a higher proportion (p < 0.01) of metastatic disease in TP53 wild-type patients (Table 1). Anaplastic histology was more prevalent in TP53 mutant patients (p < 0.001).
Table 1.
Patient characteristics according to TP53 status
| Variable | TP53 wild-type | TP53 mutant | p-value |
|---|---|---|---|
| N | 261/314 (83.1%) | 53/314 (16.9%) | |
| Age (median, range) | 9.0 (5.0 – 18.5) | 10.0 (5.0 – 17.0) | p = 0.4 |
| Gender (male) | 154/261 (59%) | 25/53 (47.2%) | p = 0.1 |
| Histology | |||
| LCA | 31/261 (11.9%) | 23/53 (43.4%) | p = 0.0001* |
| Classic | 204/261 (78.2%) | 25/53 (47.1%) | |
| Desmoplastic/nodular | 21/261 (8.0%) | 2/53 (3.8%) | |
| Missing | 5 /261 (1.9%) | 3/53 (5.7%) | |
| M+ disease | 80/261 (30.7%) | 7/53 (13.2%) | p = 0.01 |
| Subgroups | |||
| WNT | 72/261 (27.6%) | 15/53 (28.3%) | p = 0.003** |
| SHH | 59/261 (22.6%) | 36/53 (67.9%) | |
| Group 3 | 37/261 (14.2%) | 0/53 (0%) | |
| Group 4 | 92/261 (35.2%) | 1/53 (1.9%) | |
| Missing | 1/261 (0.4%) | 1/53 (1.9%) | |
| Death | 54/261 (20.7%) | 24/53 (45.3%) | |
| WNT | 5/72 (6.9%) | 0/15 (0%) | p = 0.004*** |
| SHH | 9/59 (15.3%) | 24/36 (66.7%) |
*Incidence of LCA histological variant in TP53 wild-type and TP53 mutant patients; **Incidence of TP53 mutations in WNT and SHH patients; ***Incidence of death in TP53 mutant population in WNT and SHH patients.
As previously reported, no TP53 mutations were observed in groups 3 and 4 tumors while 17.2% (15/87) of WNT and in 37.9% (36/95) of SHH tumors harbored mutations.
Five-year OS for children with TP53 wild-type and mutant tumors was 80.6% ± 2.8% and 55.9% ± 7.4% respectively (p < 0.0001) (Figure 1A). For children with SHH medulloblastoma, the 5-year OS for TP53 wild-type patients was 86.8% ± 4.7% versus 36.6% ± 8.7% for TP53 mutant ones (p < 0.0001) (Figure 1B). In contrast, the 5-year OS in children with WNT medulloblastoma was 95.7% ± 3% for the TP53 wild-type and 100% for the TP53 mutant cases (p = 0.4887) (Figure 1C). The difference in patient survival between TP53 mutant and wild-type tumors outside the WNT subgroup was more striking here than in our previous report, because the latter included patients who did not receive radiotherapy [10]. TP53 mutations clustered with 66.7% of deaths of SHH patient in this age group. Furthermore, of the TP53 mutant cancers that recurred, 42% (22/53) were SHH and only 16% (1/6) were WNT (p < 0.0001). Together, these findings suggest that in children with SHH tumors receiving radiotherapy, TP53 mutation is a marker of poor outcome. This effect is entirely driven by SHH tumors, as it was not observed in WNT activated tumors. This clinical observation suggests that TP53 mutation confer poor survival in part due to radiation resistance in non WNT tumors while this is reversed in WNT activated medulloblastomas.
Figure 1.
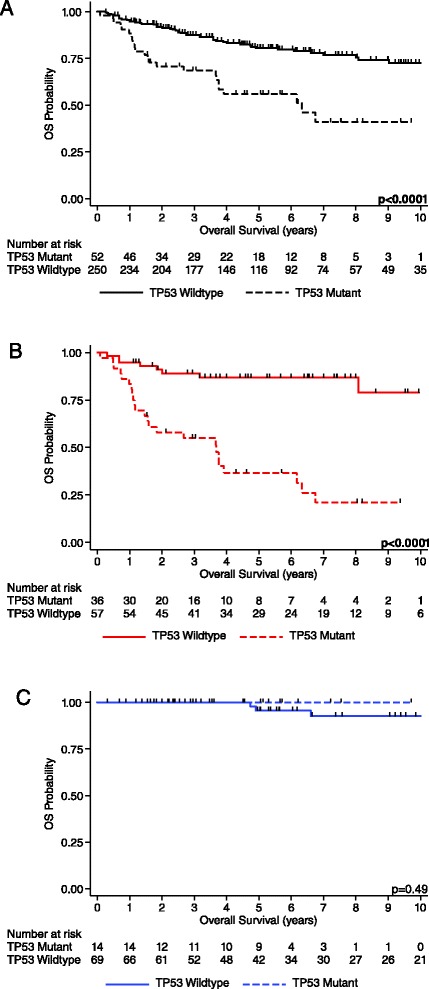
Survival estimates for medulloblastoma patients by TP53 status. In all figures TP53 status is demonstrated by solid line (wild-type) and dotted line (mutant). A) OS survival for all medulloblastoma patients. B) OS for patients with SHH medulloblastoma. C) OS for patients with WNT medulloblastoma.
TP53 mutations confer radiation resistance in medulloblastoma cell lines. In order to determine the response to radiation in TP53 wild-type and mutant medulloblastoma cells we used two approaches.
First, we performed a series of clonogenic assays post radiation treatment using a panel of TP53 wild-type and TP53 mutant non-isogenic medulloblastoma cell lines. We observed significantly higher survival in response to radiation treatment in TP53 mutant cell lines (Daoy and UW228), compared to TP53 wild-type cell lines (ONS76, MEB-MED-8A and D283MED) (p < 0.01) (Figure 2A). Specifically, survival fraction at clinically relevant dose of 2Gy (SF2) was 76% ± 2.5% for UW228, 67% ± 4.7% for Daoy, 64% ± 2.2% for ONS76, 54% ± 4.7% for D283MED and 33% ± 9.9% for MEB-MED 8A cells (p < 0.001) (Additional file 1: Figure S1A).
Figure 2.

Survival of medulloblastoma cells harboring alterations in TP53 and CTNNB1 after radiotherapy. A) Clonogenic assay on a panel of TP53 wild-type: D283MED, ONS76, MEB-MED-8A(blue) and TP53 mutant:UW228, Daoy (red) medulloblastoma cell lines. TP53 mutant cells exhibit superior radiation resistance (p < 0.01). B) Transfection of the TP53 wild-type (blue) medulloblastoma cells (ONS76) with R175H dominant-negative TP53 mutation (red) results in increased survival of the cells under the radiation treatment (p < 0.01). C) Transfection of TP53mut cells (UW228) (blue) with S33Y-CTNNB1 mutation (red) resulted in sensitization of cells to radiation (p < 0.05). D) Transfection of TP53 mutant medulloblastoma cells (UW228) with mutant S33Y-β-catenin construct results in nuclear translocation of β-catenin (bottom panel), no nuclear translocation of β-catenin is observed in the control cells, transfected with pcDNA3 construct (top panel): β-catenin (green), nucleus (red), co-localization of β-catenin to nucleus (yellow).
Second, to further confirm the pro-survival effect of TP53 mutation under radiation exposure in an isogenic system, the wild-type TP53 cell line (ONS76) was transiently transfected with an expression vector for the dominant-negative TP53-R175H mutation. Expression of the mutant p53 protein resulted in a significant increase (p < 0.01) in radiation resistance in transfected cells compared to the empty-vector control (Figure 2B). The SF2 was 89% ± 2% in TP53-R175H transfected cells versus 57.4% ± 1.8% in the parental TP53 wild-type cells (p < 0.001) (Additional file 1: Figure S1B).
Activation of WNT signaling via CTNNB1 mutation (S33Y) sensitizes TP53 mutant medulloblastoma cells to radiation. To test whether WNT pathway activation results in radiosensitivity in medulloblastoma cells, we examined the role of exon 3 mutations and subsequent WNT pathway activation in sensitizing TP53 mutant medulloblastoma cells to radiation. UW228, TP53 mutant/CTNNB1 wild-type medulloblastoma cells were transfected with vector containing the activating S33Y mutation (TP53-T155N/CTNNB1-S33Y) and the empty vector control (TP53-T155N/pcDNA3). Expression of the S33Y-mutant β-catenin protein in TP53 mutant medulloblastoma cells sensitizes these cells to radiation (p < 0.05) (Figure 2C). This was accompanied by marked nuclear translocation of β-catenin in the cells (Figure 2D), similar to what has been reported for CTNNB1-mutant tumors [22,26,28].
These findings suggest that TP53 mutations confer radioresistance in medulloblastoma cells while WNT activation results in radiosensitizsation.
Pharmacological activation of WNT by lithium reduces radioresistance in medulloblastoma with mutant TP53. Since lithium is known to activate WNT signaling via inhibition of GSK3β, we examined whether pharmacological intervention could mimic the radiosensitivity observed in the WNT group and sensitize TP53 mutant cells to radiation. Pre-treatment with lithium, resulted in significantly increased (p < 0.01) radiation sensitivity of the treated cells compared to the untreated parental cells (Figure 3A) The sensitizer enhancement ratio was 1.3 at a surviving fraction of 0.10 and the SF2 was 43.5% ± 1.5% for treated cells versus 56.6% ± 3% for treated untreated cells controls (p < 0.01) (Additional file 1: Figure S1C and 1D). Activation of the WNT signaling pathway by lithium was demonstrated by increased luciferase activity, nuclear translocation of β-catenin in treated cells and increased number of γH2AX foci post radiation (Figure 3B,C and D). Interestingly, lithium exposure also enhances radiation sensitivity of TP53 wild-type cells as demonstrated with ONS76 cells (Additional file 1: Figure S2A, B and C). In both TP53 mutant and TP53 wild-type cells we observed a higher number of γH2AX foci in lithium pre-treated cells in response to radiation treatment; at 24 hours post irradiation we found 36.6% of γH2AX foci increase in TP53 mutant cells and 10.4% increase in TP53 wild-type ones, suggestive of pharmacologically induced DNA break/repair mechanism impairment (Additional file 1: Figure S3).
Figure 3.
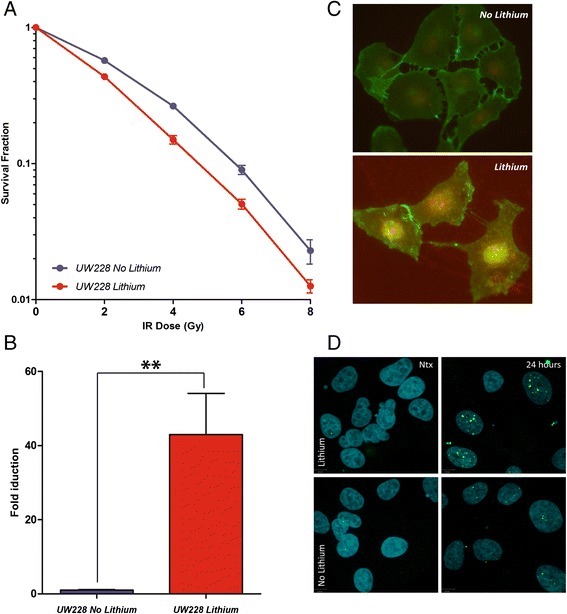
Survival of TP53 mutant medulloblastoma (UW228) cells after the combination treatment with 2 mM lithium and radiation. A) Survival curves for TP53 mutant cells given increasing radiation doses following 24 hours exposure to lithium (red) and untreated control (blue) (p < 0.01). B) Increase in luciferase activity in treated cells (red) over control (blue) in TP53 mutant cells (p < 0.01). C) Immunofluorescent imaging of the nuclear translocation of β-catenin (bottom panel); β-catenin (green), nucleus (DAPI-red), co-localization (yellow) following 24 hours exposure to lithium. D) γH2AX foci in non-irradiated cells and 24 hours post irradiation: γH2AX foci (green), nucleus (blue).
Lithium does not sensitize normal neuronal stem cells to radiation. Since lithium may have detrimental effects on the developing brain if it sensitizes normal neural cells to radiation, we used Hf5205 NNSCs as a surrogate to test a combination effect of lithium and radiation therapy on normal brain cells. Pre-treatment of NNSCs with lithium did not result in decreased survival. Pre-treated cells demonstrated a slight, though not statistically significant (p = 0.15), survival advantage over their untreated counterparts (Figure 4A). SF2 analysis for both treated and untreated cells revealed a somewhat increased survival for lithium pretreated cells (33% ± 8% vs. 27% ± 3%, respectively (p = 0.05) (Additional file 1: Figure S1E). Lithium pre-treatment did not result in nuclear translocation of β-catenin (Figure 4B), nor in increased gamma γH2AX foci formation: at 24 hours post irradiation we observed 3.1 ± 0.4 foci per nucleus in lithium treated NNSCs and 2.7 ± 0.3 foci per nucleus in untreated controls (Figure 4C and Additional file 1: Figure S4).
Figure 4.
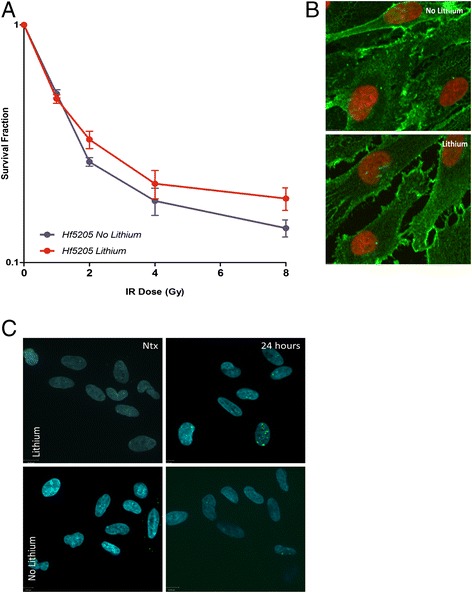
Survival of normal neuronal stem cells (Hf5205) after the combination treatment with 2 mM lithium and radiation. A) Survival curves for normal neuronal stem cells given increasing radiation doses following 24 hours of exposure to lithium (red) and untreated control (blue) (p = 0.054). B) Immunofluorescent imaging of normal neuronal stem cells; absence of the nuclear translocation of β-catenin (green) is observed in both control (top panel) and lithium treated (bottom panel) cells, nucleus (DAPI-red). C) γH2AX foci in non-irradiated cells and 24 hours post irradiation: γH2AX foci (green), nucleus (DAPI-blue).
Discussion
In this study, we confirmed that TP53 mutations remain a poor prognostic marker and account for a high proportion of the treatment failure in SHH subgroup medulloblastoma patients receiving radiation treatment, while TP53 mutant WNT patients demonstrate excellent outcomes. The importance of this finding is further highlighted through molecular studies in which we demonstrated that TP53 mutant medulloblastoma cells are more resistant to radiation. We further confirmed our results using an isogenic in vitro model, transfecting TP53 wild-type cells with a known dominant-negative TP53 mutation. These findings are in agreement with previously published data suggesting a context-dependent role of TP53 in radiation response [12,19-21,44].
Activation of WNT signaling either via increased levels of β-catenin or pharmacologically with lithium radiosensitized medulloblastoma cells, abrogated the pro-survival effects of TP53 mutations [21,45]. This effect seems to be highly tissue/tumor specific, as in the majority of tumor types increased WNT signaling is associated with tumor resistance to radiation. Recent studies suggested that WNT/β-catenin activation contributes to radiation resistance in the cancer stem cell population via the induction of chromosomal instability, deregulation of mitotic spindles and increased tolerance to DNA damage [46]. The WNT pathway has also been found to mediate radiation resistance in both mammary progenitor cells and human breast cancer cell lines [47,48], as well as in esophageal and colorectal cancers [49,50]. Furthermore, in glioblastoma multiforme mouse models and cell lines transcriptionally active β-catenin is found in the stem-like population of cells that display resistance to radiation [51].
Pharmacological radiosensitizsation of medulloblastoma cells by utilizing clinically tolerable doses of lithium prior and during radiation exposure adds a new and relatively simple dimension to the current treatment of childhood medulloblastoma. Since the decreased cell survival associated with the combination of lithium and radiation was accompanied by nuclear translocation of β-catenin and transcriptional activation of β-catenin TCF/LEF mediated signaling it is reasonable to suggest that this beneficial effect is due to WNT pathway activation in these tumors. Since lithium sensitizes all medulloblastoma cells to radiation, addition of lithium to current protocols may be beneficial to all patients but specifically to the lethal SHH TP53 mutant subgroup. In addition, combination of lithium with small-molecule inhibitors of the SHH pathway may represent an attractive regimen for high risk patients with SHH medulloblastoma.
Our results are especially exciting since, in our experiments lithium does not activate WNT signaling in normal neuronal stem cells, nor does it sensitize them to radiation. Moreover, we were able to observe marginal improvement in NNSC survival treated with lithium, which is in agreement with published reports demonstrating protective capacity of lithium against radiation in mouse models [52,53]. Taken together, our data suggests that lithium is a tolerable and safe pharmacological agent to be added to treatment of medulloblastoma.
Conclusion
In summary, we demonstrate here how combination of genomic and genetic analysis of brain tumors can predict differential responses to current therapies, and provide insight into ways that favorable signaling pathways may be mimicked by pharmacological intervention. We recognize that despite being safely used in humans for treatment of psychiatric disorders, lithium needs further pre-clinical evaluation prior to administration to humans for the purpose of medulloblastoma treatment. Nevertheless, our findings can also serve as proof of principle for implementation of other drugs which abrogate the detrimental effects of genetic events in the cancer therapeutic response.
Acknowledgements
A grant from B.R.A.I.N Child Canada.
Grants from Cancer Research UK, Action Medical Research and The Brain Tumour Charity.
Medulloblastomas investigated in this study include samples provided by the United Kingdom Children’s Cancer and Leukemia Group (CCLG) as part of CCLG-approved biologic study BS-2007-04”.
Hungarian Brain Research Program - Grant No. KTIA_13_NAP-A-V/3, the TÁMOP-4.2.2.A-11/1/KONV-2012-0025 project and A. Klekner was supported by the János Bolyai Scholarship of the Hungarian Academy of Sciences.
Postdoctoral fellowship from the German Cancer Aid/Dr. Mildred Scheel Foundation for Cancer Research (M.R.).
CIHR Fellowship and AIHS Clinical Fellowship (V.R.).
Grants from the Cure Childhood Cancer Foundation, St. Baldrick’s Foundation, and the Southeastern Brain Tumor Foundation (to EGVM).
KZ acknowledges research support from CZ.1.05/2.1.00/03.0101 (RECAMO) and CZ.1.07/2.3.00/20.0183 (CEB).
Additional file
Survival fractions at 2Gy (SF2) and SER for medulloblastoma cells treated with radiation and combination of lithium and radiation. A) SF2 for the panel of wild-type (blue) and mutant (red) medulloblastoma cells exposed to radiation. B) SF2 for the TP53 wild-type (blue) and R175H TP53 mutant (red) medulloblastoma cells. C) SF2 for lithium treated TP53 mutant cells (red) comparing to untreated control (blue). D) SER for lithium TP53 mutant cells (red) comparing to untreated control (blue). E) SF2 for untreated NNSC (red) compared to treated with lithium (blue); *p < 0.05, **p < 0.001. Figure S2. Survival of TP53 wild-type medulloblastoma cells after the combination treatment with lithium and radiation. A) Survival curves for TP53 wild-type cells given increasing radiation doses following 24 hours exposure to lithium (red) and untreated control (blue) (**p < 0.01). B) Increase in luciferase activity in treated cells (red) over control (blue) in TP53 wild-type cells (**p < 0.01). C) γH2AX foci in non-irradiated cells and 24 hours post irradiation: γH2AX foci (green), nucleus (DAPI-blue). Figure S3. DNA damage-repair response of TP53 wild-type (ONS76) and TP53 mutant (UW228) medulloblastoma cells to combined treatment with 2 mM lithium and radiation. Both TP53 wild-type A) and mutant cells B) demonstrate increased number of γH2AX foci (green) in response to a combination treatment comparing to control (p < 0.0001); nucleus (DAPI-blue). Figure S4. DNA damage-repair response of normal neuronal stem cells to combined treatment with 2 mM lithium and radiation. A and B) Normal neuronal stem cells demonstrate no increase in the number of γH2AX foci (green) in response to a combination treatment comparing to control (p < 0.0001); nucleus (DAPI-blue).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conception and design: ZN, TU. Financial support: TU. Provision of study materials or patients: BE, DP, vBAO, PE, KA, JDTW, NPA, KM, PT, PSL, CYJ, PT, GM, RS, BL, CBK, ECG, F-MM, FM, FPJ, GWA, GN, HP, JN, JA, JS, KSK, KA, KT, LB, LJR, LLM, ML, PIF, RYS, RJB, VMEG, WKC, WWA, ZK, AB, HCE, MD, CSC, PSM, TMD and TU. Collection and assembly of data: All authors. Data analysis and interpretation: ZN, RV, RM, MDC, C-BP, ZCH, FM, TK, PR, SDJH, BB, RP, BR, AB, HC, MD, TMD, TU. Manuscript writing: All authors. Final approval of manuscript: All authors.
Contributor Information
Nataliya Zhukova, Email: nataliya.zhukova@sickkids.ca.
Vijay Ramaswamy, Email: vijay.ramaswamy@sickkids.ca.
Marc Remke, Email: marc.remke@sickkids.ca.
Dianna C Martin, Email: di.martin2012@yahoo.ca.
Pedro Castelo-Branco, Email: castelobranco.pedro@gmail.com.
Cindy H Zhang, Email: cindy.zhang@sickkids.ca.
Michael Fraser, Email: michael.fraser@rmp.uhn.ca.
Ken Tse, Email: ken.tse@rmp.uhn.on.ca.
Raymond Poon, Email: raymond.poon@sickkids.ca.
David JH Shih, Email: david.shih@utoronto.ca.
Berivan Baskin, Email: berivan.baskin@akademiska.se.
Peter N Ray, Email: peter.ray@sickkids.ca.
Eric Bouffet, Email: eric.bouffet@sickkids.ca.
Peter Dirks, Email: peter.dirks@sickkids.ca.
Andre O von Bueren, Email: andre.vonbueren@med.uni-goettingen.de.
Elke Pfaff, Email: e.pfaff@dkfz-heidelberg.de.
Andrey Korshunov, Email: andrey.korshunov@med.uni-heidelberg.de.
David TW Jones, Email: david.jones@dkfz.heidelberg.de.
Paul A Northcott, Email: panorthcott@gmail.com.
Marcel Kool, Email: m.kool@dkfz.de.
Trevor J Pugh, Email: trevor.pugh@utoronto.ca.
Scott L Pomeroy, Email: Scott.Pomeroy@childrens.harvard.edu.
Yoon-Jae Cho, Email: yjcho1@stanford.edu.
Torsten Pietsch, Email: t.pietsch@uni-bonn.de.
Marco Gessi, Email: marco.gessi@ukb.uni-bonn.de.
Stefan Rutkowski, Email: s.rutkowski@uke.de.
Laszlo Bognár, bognarlaszlo@t-email.hu.
Byung-Kyu Cho, Email: bkcho@snu.ac.kr.
Charles G Eberhart, Email: ceberha@jhmi.edu.
Cecile Faure Conter, Email: cecile.conter@lyon.unicancer.fr.
Maryam Fouladi, Email: maryam.fouladi@cchmc.org.
Pim J French, Email: p.french@erasmusmc.nl.
Wieslawa A Grajkowska, Email: w.grajkowska@czd.pl.
Nalin Gupta, Email: guptan@neurosurg.ucsf.edu.
Peter Hauser, Email: hauserpeti@yahoo.com.
Nada Jabado, Email: nada.jabado@mcgill.ca.
Alexandre Vasiljevic, Email: alexandre.vasiljevic@chu-lyon.fr.
Shin Jung, Email: sjung@chonnam.ac.kr.
Seung-Ki Kim, Email: nsthomas@snu.ac.kr.
Almos Klekner, Email: aklekner@yahoo.com.
Toshihiro Kumabe, Email: ku.ma@nsg.med.tohoku.ac.jp.
Boleslaw Lach, Email: lach@hhsc.ca.
Jeffrey R Leonard, Email: jeffrey.leonard@nationwidechildrens.org.
Linda M Liau, Email: LLIAU@mednet.ucla.edu.
Luca Massimi, lmassimi@email.it.
Ian F Pollack, Email: ian.pollack@chp.edu.
Young Shin Ra, Email: ysra@amc.seoul.kr.
Joshua B Rubin, Email: rubin_j@kids.wustl.edu.
Erwin G Van Meir, Email: evanmei@emory.edu.
Kyu-Chang Wang, Email: kcwang@snu.ac.kr.
William A Weiss, Email: waweiss@gmail.com.
Karel Zitterbart, Email: kzitterbart@fnbrno.cz.
Robert G Bristow, Email: rob.bristow@rmp.uhn.on.ca.
Benjamin Alman, Email: benjamine.alman@sickkids.ca.
Cynthia E Hawkins, Email: cynthia.hawkins@sickkids.ca.
David Malkin, Email: david.malkin@sickkids.ca.
Steven C Clifford, Email: steve.clifford@ncl.ac.uk.
Stefan M Pfister, Email: s.pfister@dkfz.de.
Michael D Taylor, Email: mdtaylor@sickkids.ca.
Uri Tabori, Email: uri.tabori@sickkids.ca.
References
- 1.Packer RJ, Vezina G. Management of and prognosis with medulloblastoma: therapy at a crossroads. Arch Neurol. 2008;65(11):1419–1424. doi: 10.1001/archneur.65.11.1419. [DOI] [PubMed] [Google Scholar]
- 2.Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, Marymont MH, Muraszko K, Langston J, Sposto R. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24(25):4202–4208. doi: 10.1200/JCO.2006.06.4980. [DOI] [PubMed] [Google Scholar]
- 3.Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, Hale GA, Stewart CF, Dauser R, Sanford RA, Fuller C, Lau C, Boyett JM, Wallace D, Gilbertson RJ. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 4.Jakacki RI, Burger PC, Zhou T, Holmes EJ, Kocak M, Onar A, Goldwein J, Mehta M, Packer RJ, Tarbell N, Fitz C, Vezina G, Hilden J, Pollack IF. Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children’s Oncology Group Phase I/II study. J Clin Oncol. 2012;30(21):2648–2653. doi: 10.1200/JCO.2011.40.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong GT, Stovall M, Robison LL. Long-term effects of radiation exposure among adult survivors of childhood cancer: results from the childhood cancer survivor study. Radiat Res. 2010;174(6):840–850. doi: 10.1667/RR1903.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, Clifford SC, Pietsch T, von Bueren AO, Rutkowski S, McCabe M, Collins VP, Bäcklund ML, Haberler C, Bourdeaut F, Delattre O, Doz F, Ellison DW, Gilbertson RJ, Pomeroy SL, Taylor MD, Lichter P, Pfister SM. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfaff E, Remke M, Sturm D, Benner A, Witt H, Milde T, von Bueren AO, Wittmann A, Schöttler A, Jorch N, Graf N, Kulozik AE, Witt O, Scheurlen W, von Deimling A, Rutkowski S, Taylor MD, Tabori U, Lichter P, Korshunov A, Pfister SM, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 2010;28(35):5188–5196. doi: 10.1200/JCO.2010.31.1670. [DOI] [PubMed] [Google Scholar]
- 9.Shih DJ, Northcott PA, Remke M, Korshunov A, Ramaswamy V, Kool M, Luu B, Yao Y, Wang X, Dubuc AM, Garzia L, Peacock J, Mack SC, Wu X, Rolider A, Morrissy AS, Cavalli FM, Jones DT, Zitterbart K, Faria CC, Schüller U, Kren L, Kumabe T, Tominaga T, Shin Ra Y, Garami M, Hauser P, Chan JA, Robinson S, Bognár L, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32(9):886–896. doi: 10.1200/JCO.2013.50.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, von Bueren AO, Jones DT, Northcott PA, Kool M, Sturm D, Pugh TJ, Pomeroy SL, Cho YJ, Pietsch T, Gessi M, Rutkowski S, Bognar L, Klekner A, Cho BK, Kim SK, Wang KC, Eberhart CG, Fevre-Montange M, Fouladi M, French PJ, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31(23):2927–2935. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 12.Dahm-Daphi J. p53: biology and role for cellular radiosensitivity. Strahlenther Onkol. 2000;176(6):278–285. doi: 10.1007/s000660050010. [DOI] [PubMed] [Google Scholar]
- 13.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 14.Bristow RG, Hardy PA, Hill RP. Comparison between in vitro radiosensitivity and in vivo radioresponse of murine tumor cell lines. I: Parameters of in vitro radiosensitivity and endogenous cellular glutathione levels. Int J Radiat Oncol Biol Phys. 1990;18(1):133–145. doi: 10.1016/0360-3016(90)90277-Q. [DOI] [PubMed] [Google Scholar]
- 15.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97(19):10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24(4):679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 17.Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118(1):9–17. doi: 10.1016/j.cell.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 18.Bristow RG, Benchimol S, Hill RP. The p53 gene as a modifier of intrinsic radiosensitivity: implications for radiotherapy. Radiother Oncol. 1996;40(3):197–223. doi: 10.1016/0167-8140(96)01806-3. [DOI] [PubMed] [Google Scholar]
- 19.Williams JR, Zhang Y, Zhou H, Gridley DS, Koch CJ, Slater JM, Little JB. Overview of radiosensitivity of human tumor cells to low-dose-rate irradiation. Int J Radiat Oncol Biol Phys. 2008;72(3):909–917. doi: 10.1016/j.ijrobp.2008.06.1928. [DOI] [PubMed] [Google Scholar]
- 20.Williams JR, Zhang Y, Zhou H, Gridley DS, Koch CJ, Russell J, Slater JS, Little JB. A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int J Radiat Biol. 2008;84(4):253–264. doi: 10.1080/09553000801953342. [DOI] [PubMed] [Google Scholar]
- 21.Salaroli R, Di Tomaso T, Ronchi A, Ceccarelli C, Cammelli S, Cappellini A, Martinelli GN, Barbieri E, Giangaspero F, Cenacchi G. Radiobiologic response of medulloblastoma cell lines: involvement of beta-catenin? J Neurooncol. 2008;90(3):243–251. doi: 10.1007/s11060-008-9659-5. [DOI] [PubMed] [Google Scholar]
- 22.Fattet S, Haberler C, Legoix P, Varlet P, Lellouch-Tubiana A, Lair S, Manie E, Raquin MA, Bours D, Carpentier S, Barillot E, Grill J, Doz F, Puget S, Janoueix-Lerosey I, Delattre O. Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol. 2009;218(1):86–94. doi: 10.1002/path.2514. [DOI] [PubMed] [Google Scholar]
- 23.Ellison D. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol. 2002;28(4):257–282. doi: 10.1046/j.1365-2990.2002.00419.x. [DOI] [PubMed] [Google Scholar]
- 24.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsić A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, Clifford SC, Versteeg R. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, Kenney AM, Brat DJ, Perry A, Yong WH, Taylor RE, Bailey S, Clifford SC, Gilbertson RJ. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121(3):381–396. doi: 10.1007/s00401-011-0800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers HA, Miller S, Lowe J, Brundler MA, Coyle B, Grundy RG. An investigation of WNT pathway activation and association with survival in central nervous system primitive neuroectodermal tumours (CNS PNET) Br J Cancer. 2009;100(8):1292–1302. doi: 10.1038/sj.bjc.6604979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23(31):7951–7957. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 29.Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 30.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6(12):1664–1668. doi: 10.1016/S0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 31.Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol. 1997;185(1):82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- 32.Ormandzhieva KV, Petrova EB, Kadiysky DS. Lithium: specifics, toxicityand effects on the blood–brain and blood-cerebrospinal fluid barriers. Med Data Rev. 2014;4(1):37–42. [Google Scholar]
- 33.Lindsey JC, Hill RM, Megahed H, Lusher ME, Schwalbe EC, Cole M, Hogg TL, Gilbertson RJ, Ellison DW, Bailey S, Clifford SC. TP53 mutations in favorable-risk Wnt/Wingless-subtype medulloblastomas. J Clin Oncol. 2011;29(12):e344–e346. doi: 10.1200/JCO.2010.33.8590. [DOI] [PubMed] [Google Scholar]
- 34.Gessi M, von Bueren AO, Rutkowski S, Pietsch T. p53 expression predicts dismal outcome for medulloblastoma patients with metastatic disease. J Neurooncol. 2012;106(1):135–141. doi: 10.1007/s11060-011-0648-8. [DOI] [PubMed] [Google Scholar]
- 35.Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stütz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, Lionel AC, Mack S, Dubuc A, Yao Y, Ramaswamy V, Luu B, Rolider A, Cavalli FM, Wang X, Remke M, Wu X, Chiu RY, Chu A, Chuah E, Corbett RD, Hoad GR, Jackman SD, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhukova N. Prognostic and therapeutic implications of biological behavior of TP53 mutations in WNT and Sonic-Hedgehog medulloblastomas. MSc Thesis. Toronto, ON Canada: Institute of Medical Science. University of Toronto; 2012. [Google Scholar]
- 37.Provençal M, Berger-Thibault N, Labbé D, Veitch R, Boivin D, Rivard GE, Gingras D, Béliveau R. Tissue factor mediates the HGF/Met-induced anti-apoptotic pathway in DAOY medulloblastoma cells. J Neurooncol. 2010;97(3):365–372. doi: 10.1007/s11060-009-0041-z. [DOI] [PubMed] [Google Scholar]
- 38.Castelo-Branco P, Zhang C, Lipman T, Fujitani M, Hansford L, Clarke I, Harley CB, Tressler R, Malkin D, Walker E, Kaplan DR, Dirks P, Tabori U. Neural tumor-initiating cells have distinct telomere maintenance and can be safely targeted for telomerase inhibition. Clin Cancer Res. 2011;17(1):111–121. doi: 10.1158/1078-0432.CCR-10-2075. [DOI] [PubMed] [Google Scholar]
- 39.Buch K, Peters T, Nawroth T, Sänger M, Schmidberger H, Langguth P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay–a comparative study. Radiat Oncol. 2012;7:1. doi: 10.1186/1748-717X-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu CY, Chang HS, Chen IS, Chen CJ, Hsu ML, Fu SL, Chen YJ. Costunolide causes mitotic arrest and enhances radiosensitivity in human hepatocellular carcinoma cells. Radiat Oncol. 2011;6:56. doi: 10.1186/1748-717X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolligs FT, Hu G, Dang CV, Fearon ER. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol. 1999;19(8):5696–5706. doi: 10.1128/mcb.19.8.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13(8):680–685. doi: 10.1016/S0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 43.Kumareswaran R, Ludkovski O, Meng A, Sykes J, Pintilie M, Bristow RG. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J Cell Sci. 2012;125(Pt 1):189–199. doi: 10.1242/jcs.092262. [DOI] [PubMed] [Google Scholar]
- 44.von Bueren AO, Shalaby T, Oehler-Jänne C, Arnold L, Stearns D, Eberhart CG, Arcaro A, Pruschy M, Grotzer MA. RNA interference-mediated c-MYC inhibition prevents cell growth and decreases sensitivity to radio- and chemotherapy in childhood medulloblastoma cells. BMC Cancer. 2009;9:10. doi: 10.1186/1471-2407-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronchi A, Salaroli R, Rivetti S, Della Bella E, Di Tomaso T, Voltattorni M, Cammelli S, Ceccarelli C, Giangaspero F, Barbieri E, Cenacchi G. Lithium induces mortality in medulloblastoma cell lines. Int J Oncol. 2010;37(3):745–752. doi: 10.3892/ijo_00000724. [DOI] [PubMed] [Google Scholar]
- 46.Moncharmont C, Levy A, Gilormini M, Bertrand G, Chargari C, Alphonse G, Ardail D, Rodriguez-Lafrasse C, Magné N. Targeting a cornerstone of radiation resistance: cancer stem cell. Cancer Lett. 2012;322(2):139–147. doi: 10.1016/j.canlet.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 47.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM, et al. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104(2):618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, Rosen JM. Wnt/beta-catenin mediates radiation resistance of Sca1+ progenitors in an immortalized mammary gland cell line. J Cell Sci. 2007;120(Pt 3):468–477. doi: 10.1242/jcs.03348. [DOI] [PubMed] [Google Scholar]
- 49.Che SM, Zhang XZ, Liu XL, Chen X, Hou L. The radiosensitization effect of NS398 on esophageal cancer stem cell-like radioresistant cells. Dis Esophagus. 2011;24(4):265–273. doi: 10.1111/j.1442-2050.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- 50.Kendziorra E, Ahlborn K, Spitzner M, Rave-Fränk M, Emons G, Gaedcke J, Kramer F, Wolff HA, Becker H, Beissbarth T, Ebner R, Ghadimi BM, Pukrop T, Ried T, Grade M. Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo-) radiotherapy. Carcinogenesis. 2011;32(12):1824–1831. doi: 10.1093/carcin/bgr222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee SJ, Park K, Yang H, Jin J, Joo KM, Lee J, Nam DH, et al. Wnt activation is implicated in glioblastoma radioresistance. Lab Invest. 2012;92(3):466–473. doi: 10.1038/labinvest.2011.161. [DOI] [PubMed] [Google Scholar]
- 52.Yazlovitskaya EM, Edwards E, Thotala D, Fu A, Osusky KL, Whetsell WO, Jr, Boone B, Shinohara ET, Hallahan DE. Lithium treatment prevents neurocognitive deficit resulting from cranial irradiation. Cancer Res. 2006;66(23):11179–11186. doi: 10.1158/0008-5472.CAN-06-2740. [DOI] [PubMed] [Google Scholar]
- 53.Yang ES, Wang H, Jiang G, Nowsheen S, Fu A, Hallahan DE, Xia F. Lithium-mediated protection of hippocampal cells involves enhancement of DNA-PK-dependent repair in mice. J Clin Invest. 2009;119(5):1124–1135. doi: 10.1172/JCI34051. [DOI] [PMC free article] [PubMed] [Google Scholar]