

Abstract
Estrogen receptor α (ER)-positive breast cancers initially respond to antiestrogens but eventually become estrogen-independent and recur. ER+ breast cancer cells resistant to long-term estrogen deprivation (LTED) exhibit hormone-independent ER transcriptional activity and growth. A kinome-wide siRNA screen using a library targeting 720 kinases identified Polo-like kinase 1 (PLK1) as one of the top genes whose downregulation resulted in inhibition of estrogen-independent ER transcriptional activity and growth of LTED cells. High PLK1 mRNA and protein correlated with a high Ki67 score in primary ER+ breast cancers after treatment with the aromatase inhibitor letrozole. RNAi-mediated knockdown of PLK1 inhibited ER expression, estrogen-independent growth and ER transcription in MCF7 and HCC1428 LTED cells. Pharmacological inhibition of PLK1 with volasertib, a small molecule ATP-competitive PLK1 inhibitor, decreased LTED cell growth, ER transcriptional activity and ER expression. Volasertib in combination with the ER antagonist, fulvestrant, decreased MCF7 xenograft growth in ovariectomized mice more potently than each drug alone. JUNB, a component of the AP-1 complex, was expressed 16-fold higher in MCF7/LTED compared to parental MCF7 cells. Further, JUNB and BCL2L1 (which encodes anti-apoptotic BCL-xL) mRNA levels were markedly reduced upon volasertib treatment in MCF7/LTED cells while they were increased in parental MCF7 cells. Finally, JUNB knockdown decreased ER expression and transcriptional activity in MCF7/LTED cells, suggesting that PLK1 drives ER expression and estrogen-independent growth via JUNB. These data support a critical role of PLK1 in acquired hormone-independent growth of ER+ human breast cancer and is therefore a promising target in tumors that have escaped estrogen deprivation therapy.
Keywords: Estrogen-independent, PLK1, RNAi screen, volasertib
INTRODUCTION
Most breast cancers express ERα and are driven by estrogen (1–3). Targeting the function of ER with receptor antagonists such as tamoxifen, fulvestrant and aromatase inhibitors (AI), such as anastrozole and letrozole, are effective treatments for patients with this subtype of breast cancer (4–7). However, a significant number of patients with ER+ tumors, particularly those with advanced disease, exhibit primary and acquired resistance to antiestrogens (8–11). Most mechanisms that mediate this resistance are not yet fully understood. Therefore, identifying those mechanisms and strategies to interfere with them is needed in order to prevent or delay acquired endocrine resistance and improve patient outcome. We and others have modeled secondary resistance to treatment with aromatase inhibitors (AIs), the equivalent of estrogen deprivation in postmenopausal patients, by generating long-term estrogen-deprived (LTED) cells, whereby ER+ human breast cancer cells have adapted to estrogen deprivation and acquired robust growth under hormone-free conditions (12–14). In this study, we used ER+ MCF7/LTED and HCC1428/LTED breast cancer cells, which overexpress ER and retain estrogen-independent ER binding to DNA and ER transcriptional activity (15).
To identify targetable molecules that drive hormone-independent growth and ER transcription in LTED cells, we performed a screen using a library with siRNA pools targeting 720 kinases. We used two metrics in this assay: 1) luciferase activity as assessed by using an estrogen-response-element (ERE)-regulated reporter, and 2) cell viability measured by the Alamar Blue assay. In this screen, we identified Polo-like kinase 1 (PLK1) as the top hit whose downregulation decreased both ER transcriptional activity and cell viability. PLK1 is a serine/threonine kinase that is highly expressed during the G2 phase of the cell cycle where it controls the metaphase-to-anaphase transition and mitotic exit (16–18). PLK1 is one of three isoforms of PLKs (PLK2 and PLK3) (19, 20) and has been shown to be overexpressed in several human cancers (21–25). Currently, PLK1 specific small molecule inhibitors are in clinical trials in patients with advanced cancer (26, 27). Herein, we discovered that genetic and pharmacological inhibition of PLK1 decreased ER expression, estrogen-independent ER-mediated transcriptional activity and growth of ER+ xenografts alone and in combination with fulvestrant. These results provide a basis for combinations of PLK1 inhibitors with ER downregulators in patients with hormone-independent ER+ breast cancer.
MATERIALS AND METHODS
Cell lines and reagents
Parental cancer cell lines were from ATCC and maintained in improved minimum essential medium (IMEM)/10% FBS (Gibco) as previously described (28). LTED cells were generated in and maintained in phenol red–free IMEM with 10% dextran/charcoal-treated FBS (DCC-FBS) as previously described (28). BI2536 and volasertib were obtained from Selleckchem (Houston, TX). Fulvestrant was obtained from the Vanderbilt Hospital Outpatient Pharmacy. FOXM1-EE construct was a gift from Junjie Chen (Yale University) (29). The myristoylated-PLK1 construct (Addgene plasmid 20859) was a gift from William Hahn (Dana Farber Cancer Institute) (30).
RNAi screen
MCF-7/LTED cells were transfected with the Dharmacon RTF Protein Kinase siRNA library (14) as described in Supplementary Methods. Secondary validation was performed with four independent siRNAs against GSG2, RPS6KA2 and PLK1 (Qiagen).
Quantitative RT-PCR
Cells maintained in 10% Dextran-coated charcoal treated fetal bovine serum (DCC-FBS) were transfected with control or PLK1 siRNA oligonucleotides (Thermo Scientific) with Lipofectamine RNAiMAX for 48–72 h. Cells were harvested and their RNA extracted using the RNeasy Mini Kit (Qiagen). Using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA), 1 μg of RNA was reverse transcribed to cDNA and real-time PCR reactions were conducted in 96-well plates using the iCycler iQ (Bio-Rad) and primers obtained from SA Biosciences (Qiagen). RNA was extracted from MCF7 and MCF7/LTED cells that had been treated with volasertib or transfected with PLK1 siRNA; cDNA was applied to the Estrogen Receptor Signaling PCR Array (SA Biosciences). Fold changes in gene expression was determined using the web-based software from SA Biosciences (http://www.qiagen.com/us/products/genes%20and%20pathways/data-analysis-center-overview-page/). Genes identified in the array were verified by qRT-PCR.
Cell proliferation assays
Cells seeded in triplicate in 12-well plates (2.5×104 cells/well for MCF-7/LTED and 4×104 cells/well for other lines) were treated in 10% DCC-FBS. Media and inhibitors were replenished every 3 days; after 5–10 days, adherent cells were trypsinized and counted using a Coulter Counter or fixed/stained with crystal violet. Crystal violet-stained plates were scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences) and the staining intensity was quantified using the manufacturer’s analysis software. For the combination studies of volasertib and fulvestrant, MCF7 and MCF7/LTED cells were seeded in 96-well black plates followed by the Alamar Blue Assay after 72 h as described in the Supplementary Methods.
Immunoblot analysis
Cells were lysed with NP40 lysis buffer (150 mM Tris, pH 7.4, 100 mM NaF, 120 mM NaCl, 0.5% NP-40, 100 μM sodium vanadate, and 1× protease inhibitor cocktail [Roche]). Lysates (40 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes; these were first incubated with primary antibodies at 4°C overnight or at room temperature for 2 h, followed by incubation with HRP-conjugated anti-rabbit and anti-mouse secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence (Thermo Scientific). All primary antibodies were from Cell Signaling except the ERα antibody (Santa Cruz Biotechnology).
Transcriptional reporter assays
Cells were plated as above and transfected with pGLB-MERE (encodes estrogen response element-regulated Firefly luciferase), pGL4.23 vectors (Peak2 or Peak5 Firefly luciferase) (28) and pTK-Renilla (encodes TK-driven Renilla luciferase; Promega) plasmids. Cells were then treated as above; luciferase activity was measured 16–20 h later using the Dual Luciferase Kit (Promega; Madison, WI) according to the manufacturer’s instructions utilizing a Moonlight 3010 Luminometer (Analytical Luminescence Laboratory). The same procedure was used for the pCAGA (provided by J.-M. Gauthier, Laboratoire GlaxoSmithKline, Les Ulis Cedex, France), pGL2-E-cadherin(31) and pGL-ErbB3(32) Firefly Luciferase reporters.
Xenograft studies
Animal experiments were approved by the Vanderbilt Institutional Animal Care and Use Committee. Female ovariectomized athymic mice (Harlan Sprague Dawley) were implanted s.c. with a 14-day-release, 0.17-mg, 17β-estradiol pellet (Innovative Research of America, Sarasota, FL). Twenty-four h later, 5×106 MCF7 cells suspended in IMEM and matrigel (BD Biosciences, San Jose, California, USA) at 1:1 ratio were injected s.c. into the right flank of each mouse. Approximately 4 weeks later, mice bearing tumors measuring ≥150 mm3 were randomized to treatment with vehicle (control), volasertib (10 mg/kg/day via orogastric gavage), fulvestrant (5 mg/week s.c.), or both drugs. Animal weight and tumor diameters (with calipers) were measured twice weekly and tumor volume was calculated with the formula: volume = width2 x length/2. After 6 weeks, tumors were harvested and snap-frozen in liquid nitrogen or fixed in 10% neutral buffered formalin followed by embedding in paraffin for immunohistochemical analysis.
RESULTS
PLK1 siRNA oligonucleotides inhibit ER transcriptional activity and cell growth
Initially, we transfected cells with ERE firefly-luciferase and renilla-luciferase constructs. Transfection with ERα siRNA decreased ERE-firefly luciferase activity. Importantly, the renilla reading was markedly decreased (93%) resulting in a greater firefly/renilla ratio compared to control siRNA transfected cells (Suppl. Table 1). In the Alamar Blue assay, ER siRNA decreased cell viability only by 62% (Suppl. Fig. 1B). These results suggested that RNAi oligonucleotides reducing ER expression had a non-specific effect on renilla expression in MCF7/LTED cells, thus skewing the results. For this reason, we could not use renilla expression as a control in cells transfected with the siRNA pools. We next assessed whether LTED cell viability (Alamar Blue) and ERE luciferase activity can be measured consecutively. Firefly luciferase activity was similar in cells transfected with MERE-luc in the presence or absence of Alamar Blue dye (Suppl. Figs. 1A, C). Therefore, MCF7/LTED cells were next transfected with an ERE-luciferase construct and with siRNA pools targeting 720 kinases (schema in Suppl. Fig. 1A). Both cell viability (Alamar Blue) and ER reporter activity for each siRNA relative to nonsilencing controls (siCTL) were transformed to a Z-score; the median Z-score across 3 independent experiments was then calculated (Fig. 1A). Knockdown of 58 and 36 kinases was observed to significantly decrease cell viability and ER reporter activity, respectively (Fig. 1B; Suppl. Table 2). Of these, 10 kinases scored positive in both assays. Statistical analysis identified Polo-like kinase 1 (PLK1), RPS6KA2 and GSG2 as the top hits inhibiting both ER transcriptional activity and viability of MCF7/LTED cells (Table 1). We next validated the effect of these 3 genes using 4 independent siRNA oligonucleotides for each of them (Suppl. Fig. 2). Only in the case of PLK1, all 4 independent siRNA oligos decreased both ERE luciferase activity and cell viability.
Figure 1. RNAi screen identifies PLK1 is required for hormone-independent ER transcriptional activity and growth.

(A) MCF-7/LTED cells were screened with a siRNA library targeting 720 kinases. Ligand-independent cell growth and ER transcriptional activities were sequentially measured 72 h later using a high-throughput Alamar Blue and Luciferase Reporter assays, respectively. Both cell growth and ER transcriptional activity for each kinase siRNA relative to nonsilencing controls (siCTL) were transformed to a Z-score; the data are presented as median Z-score across 3 independent experiments. (B) Knockdown of 48 and 26 kinases significantly decreased MCF7/LTED cell proliferation and ER transcription, respectively. Knockdown of 10 kinases was found to decrease both cell viability and ER transcription. (C) MCF7/LTED and HCC1428/LTED cells transfected with RNAi targeting PLK1 and cultured for 5 days. Cells were trypsinized and their number determined in a Coulter Counter (*p<0.01). (D) MCF7/LTED and HCC1428/LTED cells were transfected with control (CTL) or PLK1 siRNA and re-seeded in 12-well plates in full growth media. Media was replenished every 3 days and the cells were stained with crystal violet after 7 days (*p<0.0002). (E) RNAi-mediated PLK1 downmodulation was validated by qRT-PCR in MCF7/LTED and HCC1428/LTED cells (*p<0.01). (F) MCF7/LTED cells were transfected with CTL or PLK1 siRNA for 72 h. Cell lysates were prepared and subjected to immunoblot analysis for ER, PLK1 and actin.
Table 1.
Ranking of the top 10 hits on Cell proliferation and ER transcription in the RNAi screen of MCF7/LTED cells
| Gene | Cell Proliferation Rank | ER transcription Rank |
|---|---|---|
| ADCK2 | 22 (−1.38) | 22 (−1.07) |
| CSNK1A1 | 34 (−1.21) | 15 (−1.16) |
| FN3KRP | 41 (−1.13) | 20 (−1.10) |
| GSG2 | 15 (−1.51) | 28 (−1.03) |
| NRBP | 12 (−1.55) | 16 (−1.15) |
| PLK1 | 17 (−1.46) | 5 (−1.32) |
| PRKCB1 | 42 (−1.13) | 9 (−1.24) |
| RPS6KA2 | 1 (−2.73) | 35 (−1.0) |
| STK35 | 23 (−1.36) | 17 (−1.15) |
| TTK | 9 (−1.61) | 8 (−1.30) |
PLK1 downmodulation with siRNA resulted in inhibition of proliferation and focus formation in monolayer of both MCF7/LTED and HCC1428/LTED cells (Fig. 1C, D). Knockdown of PLK1 was validated by both qRT-PCR and immunoblot analysis. Interestingly, PLK1 siRNA also decreased ERα protein levels (Fig. 1E, F), suggesting that PLK1 has a role in ligand-independent ER transcriptional activity and ER+ breast cancer cell growth via regulation of ERα expression.
To investigate the potential clinical significance of these findings, we correlated Ki67 levels (a marker of cell proliferation) with PLK1 protein and mRNA expression in tumor biopsies from patients with an operable ER+/HER2− breast cancer that had been treated with the aromatase inhibitor letrozole for 10–21 days prior to surgery. In this setting, Ki67 levels are used as a surrogate of the hormone independence since ER+ breast cancers that retain a high Ki67 score after therapy are considered resistant to estrogen deprivation (with letrozole) and vice versa, tumors with a low post-treatment Ki67 are considered highly hormone-dependent and, thus, antiestrogen sensitive (11, 33). In tumor biopsies from patients treated with letrozole, PLK1 protein levels measured by RPPA positively correlated with the Ki67 score (Fig. 2A). In addition, PLK1 mRNA transcript levels measured by RNA-seq correlated directly with Ki67 mRNA expression (Figs. 2B–C). Further, both MCF7 and HCC1428 LTED cells displayed increased PLK1 expression compared to their parental cell lines (Fig. 2D). These results suggest that high PLK1 expression associates with and could be used as a marker of hormone-independent ER+ breast cancer.
Figure 2. High PLK1 expression correlates with antiestrogen resistance in primary ER+ breast cancers.
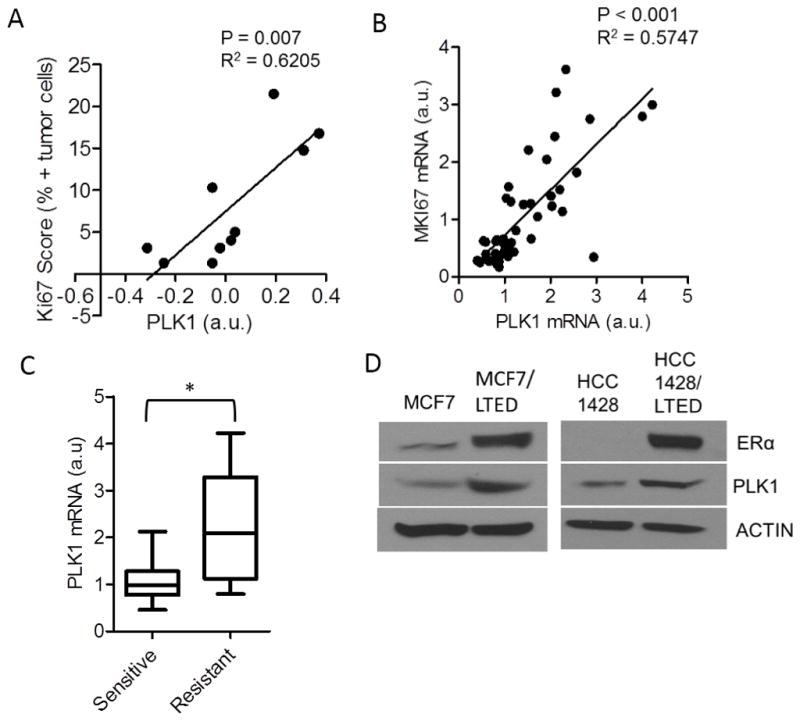
(A) Correlation PLK1 protein levels measured by RPPA with the Ki67 score in 10 post-letrozole breast tumor biopsies (n=10; p=0.007). (B) Correlation of PLK1 and MKI67 expression from RNA-seq analysis of RNA extracted from post-letrozole biopsies of primary ER+ breast cancers (n=47; p<0.001). (C) PLK1 expression was analyzed in patients’ tumors that were categorized as drug-sensitive or -resistant based on the post-letrozole Ki67 mRNA expression as described in Supplementary Methods (*p=0.002; a.u., arbitrary units). (D) Lysates from MCF7, HCC1428 cells and their LTED counterparts were assessed for ER and PLK1 expression by immunoblot analysis. Actin was used as a loading control.
PLK1 downmodulation decreases hormone-independent ER transcriptional activity
In MCF7/LTED cells, PLK1 siRNA decreased MERE-Luciferase activity (Fig. 3A) and expression of the ER-responsive genes progesterone receptor (PR), Trefoil factor 1 (TFF1) and growth regulation by estrogen in breast cancer 1 (GREB1; Fig. 3B). A more modest reduction in PR and TFF1 was observed in HCC1428/LTED cells transfected with PLK1 siRNA. To confirm that the effects of PLK1 siRNA were on ligand-independent ER transcription, we generated two luciferase reporters (Peak2 and Peak5) with ER binding regions proximal to genes reported to be downregulated by ER siRNA or fulvestrant in the absence of estrogen (28). Notably, the effect of PLK1 siRNA on the ligand-independent reporters was similar to that of ER siRNA oligonucleotides (Fig. 3C). In addition, MCF7/LTED cells were transfected with both Peak5 firefly luciferase and renilla luciferase plasmids two days after transfection with two independent PLK1siRNAs to avoid the effects of RNAi on renilla activity. We observed that both PLK1 siRNA oligonucleotides decreased estrogen-independent ER reporter activity (Suppl. Fig 3) when measured as a ratio of firefly to renilla luciferase. Conversely, overexpression of PLK1 in MCF7/LTED cells increased >4-fold Peak5-Luc reporter activity (Fig. 3D). To show that the effect of PLK1 downregulation on ER transcription was specific to ER and not secondary to an overall effect on chromatin segregation during mitosis, we examined other transcriptional reporters. PLK1 and ER siRNA did not affect the activity of the transcriptional reporters driven by SMAD, E-cadherin and ERBB3 promoter binding regions in MCF7/LTED cells (Figs. 3E–G).
Figure 3. PLK1 downmodulation abrogates estrogen-dependent and independent ER transcriptional activity.

(A) MCF7/LTED cells were transfected with CTL, PLK1 or ER siRNA in triplicate wells. After 48 h, cells were transfected with a MERE-Luciferase construct; a Luciferase Assay was performed the following day (*p<0.02). (B) MCF7/LTED and HCC1428/LTED cells were transfected with CTL or PLK1 siRNA for 72 h. RNA was extracted, reverse transcribed and subjected to quantitative PCR with specific primers to PR (*p=0.005), TFF1 (*p=0.04) and GREB1 (*p=0.0007). (C) MCF7/LTED cells transfected with CTL, PLK1 or ER siRNA. After 48 h, cells were transfected with hormone-independent ER reporter (Peak2-Luc and Peak5-Luc) constructs; a Luciferase Assay was performed the following day (*p<0.002; **p 0.03) (D) MCF7/LTED cells were transfected with myristoylated PLK1 or control DNA (pcDNA) for 72 h. Cells were then transfected with Peak5-Luc and TK-Renilla and the Dual Luciferase assay was performed 18 h later (*p<0.0001). MCF7/LTED cells were transfected with CTL, PLK1 or ER siRNA or treated with 1 nM estradiol (E2). After 48 h, cells were transfected with (E) pCAGA-Luc, (F) Ecad-Luc, (G) ErbB3-Luc reporter constructs; a Luciferase assay was performed the following day as described in Methods.
PLK1 inhibitor decreases hormone-independent growth and ER transcriptional activity in LTED cells
We next used BI2536 and volasertib, small molecule inhibitors derived from the dihydropteridinone class of compounds that target the ATP binding pocket in the PLK1 kinase domain (34). Growth of MCF7/LTED and HCC1428/LTED cells was inhibited by low nM concentrations of both BI2536 and volasertib in dose-dependent fashion (Figs. 4A, B) whereas parental MCF7 cells were only inhibited by concentrations in excess of 10 nM (Suppl. Fig. 4A). In both LTED cell lines, treatment with volasertib abrogated hormone-independent ER reporter activity (Fig. 4C) and reduced ER protein levels (Fig. 4D; Suppl. Fig. 4B). Furthermore, volasertib increased expression of cleaved PARP, an apoptotic marker, and phosphorylated Histone H2AX, a marker of DNA damage and fragmentation (Fig. 4E and Suppl. Fig. 4B).
Figure 4. PLK1 inhibitor decreases hormone-independent growth and ER transcriptional activity in LTED cells.

MCF7 LTED and HCC1428 LTED cells were treated with increasing concentrations of the PLK1 inhibitors (A) BI2536 or (B) BI6727 (volasertib) for 5 days (*p<0.05). (C) MCF7/LTED and HCC1428/LTED cells transfected with Peak5-luciferase and TK-Renilla luciferase constructs were treated with increasing concentrations of volasertib for 24 h (*p<0.05). Dual luciferase assay was performed and fold luciferase activity to control was calculated as described in Methods. (D, E) MCF7/LTED cells were treated with increasing concentrations of volasertib for 24–72 h. Lysates were collected and after SDS-PAGE, subjected to immunoblot analyses for ER, PARP and phospho-Histone H2AX as described in Methods.
Volasertib enhances the anti-tumor effect of fulvestrant in vivo
We next investigated whether combined inhibition of PLK1 and ER with volasertib and the ER antagonist fulvestrant, respectively, would have a synergistic antitumor effect. Volasertib enhanced the anti-proliferative effect of fulvestrant in MCF7 and MCF7/LTED cells (Figs. 5A, B and Suppl. Fig 4C). The combination of fulvestrant and volasertib also increased expression of cleaved PARP and phospho-Histone H2AX whereas each drug alone did not (Fig. 5C). To examine the effect of the combination in vivo, we established MCF7 xenografts in ovariectomized nude mice. Mice with tumors measuring ≥150 mm3 were treated with vehicle, volasertib, fulvestrant or the combination of both inhibitors. After 6 weeks of treatment, the combination induced a statistically superior antitumor effect compared to fulvestrant or volasertib alone (Fig. 5D). During this period, the mice in all four treatment groups displayed stable body weight (data not shown) and no signs of toxicity. Treatment with volasertib reduced ER levels but the combination of volasertib and fulvestrant more potently reduced both ER and PR levels as measured by IHC of tumor sections (Figs. 5E–G).
Figure 5. Volasertib enhances the anti-tumor effect of fulvestrant.

(A) MCF7 and (B) MCF7/LTED cells were treated with vehicle, fulvestrant (1 μM) or combinations of volasertib and fulvestrant for 72 h. Cell viability was assessed using the MTT assay. (C) Lysates of MCF7 cells treated with vehicle, fulvestrant or combinations of volasertib and fulvestrant for 48 h were prepared and subjected to ER, PARP and p-Histone H2AX immunoblot analyses. (D) MCF7 (5×106) cells were inoculated s.c. in the dorsum of ovariectomized athymic mice supplemented with a 2-week release estradiol pellet. Approximately 4 weeks later, mice with tumors ≥150 mm3 were randomized to treatment with vehicle, volasertib, fulvestrant, and both drugs for 6 weeks. Tumors were measured twice weekly with calipers; tumor volumes in mm3 are shown (*p=0.005). Each data point represents the mean tumor volume in mm3 ± SD (n=9/group). (E) IHC analysis of ER and PR in formalin-fixed paraffin-embedded xenograft sections from each group was performed and scored as described in Methods. Images of ER and PR IHC in representative xenografts are shown. (F) H-scores of ER (*p =0.04) and (G) PR expression in xenografts from all 4 groups are displayed on the right (n=7–8/group).
PLK1 inhibition decreases BCL-xL and JUNB expression in LTED cells
It has been shown that PLK1 may have a tumor suppressive role in cancer including MCF7 breast cancer cells (21, 35, 36). Based on the findings from the RNAi screen, we sought to elucidate mechanistic differences between parental MCF7 and estrogen-independent MCF7/LTEDs in response to PLK1 loss. Using an Estrogen Receptor Signaling PCR Array with 84 ER-associated genes, a 24-h treatment with each volasertib and PLK1 siRNA oligonucleotides altered (>1.4-fold) the expression of 7 genes, including ERα, in MCF7 and MCF7/LTED cells (Suppl. Fig. 5A). Expression of BCL2L1, the gene encoding BCL-xL, and JUNB were also increased in hormone-dependent parental MCF7 cells following treatment with each volasertib and PLK1 siRNA. In MCF7/LTED cells, however, volasertib and PLK1 siRNA decreased the expression of both BCL-xL and JUNB (Figs. 6A, B). HCC1428/LTED cells treated with volasertib also displayed a modest reduction in BCL-xL and JUNB whereas PLK1 siRNA markedly decreased BCL-xL and JUNB transcript levels (Fig. 6C). BCL-xL and JUNB expression were approximately 5-fold and 17-fold higher, respectively, in MCF7/LTED cells compared to the parental MCF7 cells (Fig. 6D). We next confirmed that treatment with volasertib and downmodulation of PLK1 with two independent siRNAs decreased protein levels of ER, BCL-xL and JUNB (Fig. 6E). mRNA and protein levels of BCAR1 (Breast cancer antiestrogen resistance 1), which has been associated with poor prognosis and resistance to tamoxifen (37), were also decreased upon treatment with volasertib (Suppl. Figs. 5A,B). JUNB is a component of the AP-1 transcriptional complex that drives the expression of genes involved in cellular growth and development (11). In addition, estrogen-activated ER is known to bind to AP-1 promoter sites in ER+ breast cancer cells (38). Therefore, we examined whether PLK1-mediated regulation of JUNB affected ER expression and transcriptional activity. MCF7/LTED cells transfected with JUNB siRNA exhibited reduced hormone-independent ER transcriptional activity (Fig. 6F; Suppl. Fig. 5C) and ER expression (Fig. 6F). Conversely, JUNB siRNA increased ER expression in the parental MCF7 cells (Fig. 6G). These observations suggest that PLK1 regulates ERα via transcriptional regulation of JUNB.
Figure 6. Inhibition of PLK1 decreases BCL2L1 and JUNB expression in LTED cells.

(A) MCF7 and MCF7/LTED cells were treated with vehicle (0.1% DMSO) or 100 nM volasertib for 24 h. An ER Signaling-specific PCR Array was performed with RNA extracted from these cells. The expression of JUNB and BCL2L1 was compared between parental and LTED MCF7 cells. (B) MCF7 and MCF7/LTED cells were transfected with control or PLK1 siRNA for 48 h. RNA was extracted and tested in an ER Signaling-specific PCR Array. The expression of JUNB and BCL2L1 was compared between the parental and LTED cells. (C) HCC1428/LTED cells were treated with vehicle or 100 nM volasertib for 24 h or transfected with control and PLK1 siRNA for 48 h. RNA was extracted and subjected to qRT-PCR analysis for BCL2L1 and JUNB (*p=0.024; **p=0.003). (D) BCL2L1, JUNB and ESR1 (ER) as determined in the Estrogen Signaling PCR Array described in A and B is shown as the ratio of vehicle-treated MCF7/LTED over MCF7 parental cells. (E) MCF7/LTED cells were treated with either vehicle or volasertib (24 h) or transfected with control or PLK1 siRNA (48 h). Cell lysates were prepared and separated by SDS-PAGE followed by immunoblot analysis using the indicated antibodies. (F) Left: MCF7/LTED cells were transfected with control or JUNB siRNA. After 48 h, cells were transfected with the ligand-independent Peak5-Luc reporter construct. A Luciferase Assay was performed 24 h later (*p=0.006). Right: Lysates from cells transfected with Control or JUNB siRNA were assessed for JUNB and ER expression. (G) MCF7 cells transfected with control or JUNB siRNA for 72 h were assessed for ER and JUNB protein levels by immunoblot analysis. (H) Model of PLK1 function in hormone-independent ER+ breast cancer. FOXM1 regulates the expression of one of its transcriptional targets PLK1. In estrogen-deprived cell lines, PLK1 regulates the expression of JUNB and BCL-xL. As shown by the RNAi studies shown herein, JUNB can regulate the expression of ER and ER transcriptional activity.
PLK1 transcription is driven by FOXM1, a member of the forkhead family of transcription factors (39). PLK1 and FOXM1 have an inter-regulatory relationship as part of a kinase-driven positive feedback loop that leads to the PLK1-mediated phosphorylation of FOXM1 and potentiation of its activity (29, 40). Consistent with this positive interaction, transfection of a vector encoding FOXM1 constitutively phosphorylated at the PLK1 site (FOXM1-EE) (29) increased ER expression and ER transcriptional activity (Suppl. Fig. 6A). Treatment with volasertib still reduced constitutive ER transcriptional reporter activity in the FOXM1-EE expressing cells, suggesting PLK1 mediates ER activity downstream of FOXM1 in LTED cells. Supporting an interaction between FOXM1 and PLK1 in hormone-independent primary tumors, FOXM1 transcript levels correlated positively with both Ki67 and PLK1 mRNA levels in patients with ER+ breast cancer treated with letrozole (Suppl. Figs. 6B,C). Finally, we determined the effect of PLK1 downmodulation on the expression of other nuclear receptors involved in breast cancer pathogenesis, androgen receptor (AR) and estrogen receptor beta (ERβ). In ER+ positive breast cancers, AR expression is associated with lower risk of recurrence and increased overall survival (41, 42). ERβ has been shown to antagonize ERα by decreasing growth and ERα-mediated transcriptional activity (43). Furthermore, it was recently shown that ERβ can be regulated by activated AR to decrease ER+ breast cancer cell growth (44). We observed that PLK1 downmodulation augmented the expression of both AR and ERβ while decreasing the expression of the ERα target gene PR (Suppl. Fig. 6D).
These results suggest that, in breast cancer cells that adapt to estrogen deprivation, PLK1 positively regulates anti-apoptotic genes (BCL-xL) and components of the AP-1 (JUNB) transcription complex (Fig. 6H). In turn, this promotes ERα expression and transcriptional activity and is therapeutically targetable with PLK1 inhibitors in hormone-independent, ER-driven breast cancers.
DISCUSSION
In this study, we identified PLK1 as a potential therapeutic target in hormone-independent ER+ breast cancer cells. Using a high-throughput RNAi screen approach to identify kinases that contribute to hormone-independent transcriptional activity and tumor cell viability, PLK1 was the top candidate gene associated with both of these phenotypes. In addition to ER transcriptional activity, PLK1 downmodulation decreased ER expression. Consistent with these results, treatment with the PLK1 inhibitor volasertib reduced LTED cell viability, ER levels and ER transcriptional activity. Further, volasertib enhanced the anti-tumor action of fulvestrant in MCF7 xenografts via enhanced ER downmodulation. Using an Estrogen Receptor Signaling PCR Array, we compared the effect of PLK1 inhibition on gene expression in parental and LTED MCF7 cells. Inhibition of PLK1 with volasertib and PLKsiRNA resulted in a reduction of BCL-xL and JUNB expression in LTED cells whilst these genes were upregulated in parental MCF7 cells. Taken together, these findings suggest that PLK1 inhibition may provide therapeutic benefit to patients with endocrine-resistant ER+ breast cancer.
RNAi kinome screens have previously identified PLK1 as a gene causally associated with growth of basal-like breast cancer and tumor-initiating cells (35, 45). This finding may reflect a crucial role of PLK1 in mitosis of rapidly proliferating cancer cells. PLK1 was also recently shown to cooperate with estrogen-dependent ER signaling (21). In this study, Wierer et al. showed that PLK1 phosphorylates the transcriptional regulator MLL2 which forms a complex with ER to drive transcription of tumor suppressive genes. Those findings suggest PLK1 drives proliferation via its role in interphase but it decreases growth via its interaction with ER. The findings in our report herein suggest PLK1 has lost this negative cell cycle and transcriptional regulatory function in estrogen-independent breast cancer cells that still depend on unliganded ER function.
PLKs exist in 3 different isoforms, PLK1, PLK2 and PLK3. Although PLK1 was shown to significantly affect both viability and ER transcription, PLK3 siRNA only decreased ER transcriptional activity in the siRNA screen (Suppl. Table. 2). PLK3 is not aberrantly expressed in breast cancer and maintains steady state levels during cell division (46). Volasertib has a high affinity for PLK1 (0.87 nM), PLK2 (5 nM) and PLK3 (56 nM) (34). Therefore, treatment of antiestrogen-resistant ER+ breast cancer cells with volasertib may have a very potent effect on growth and ER transcriptional activity by inhibition of both PLK1 and PLK3.
Blockade of PLK1 with volasertib or siRNA decreased ER transcriptional activity and ER expression. JUNB, a component of the AP-1 transcription complex was also decreased upon inhibition of PLK1 in LTED cell lines and knockdown of JUNB decreased ER transcriptional activity and expression. It has been shown that ER can cooperate with AP-1 complexes at AP-I sites to drive transcription, particularly E2F1 transcription (38, 47). Further, JUNB mRNA and protein expression have been reported as poor prognostic biomarkers in patients with ER+ breast cancer (48). We previously reported that an E2F1 transcriptional signature correlates with a resistance to estrogen deprivation in ER+ breast cancer cells (28). Treatment with volasertib also reduced expression of BCAR1 (Breast cancer anti-estrogen resistance) which encodes p130Cas (Crk-associated substrate; Suppl. Fig. 5A)(49). BCAR1 overexpression has also been shown to correlate with poor prognosis and resistance to tamoxifen in patients with ER+ breast cancer (37). p130Cas complexes with ER and drives non-genomic signaling such as activation of c-Src and p85 phosphoinositide 3- kinase (PI3K) (50). We have reported that hyperactivation of PI3K is causally associated with hormone-independent growth of ER+ breast cancer cells (15). Moreover, p130Cas has E2F and c-Jun binding elements in its promoter. Taken together, these data suggest inhibition of PLK1 may interfere with E2F transcription.
Finally, the combination of volasertib and fulvestrant exhibited synergistic anti-tumor efficacy and abrogated ER expression levels in vivo. Volasertib has currently completed phase I clinical trials where it showed a favorable pharmacokinetic profile with minor hematological toxicities (34). It is under active investigation in phase II studies addressing efficacy (www.clinicaltrials.gov). Therefore, based on the two-pronged effect of PLK1 inhibition on endocrine resistant ER+ breast cancer cell viability and ER transcriptional activity, we propose that PLK1 inhibitors like volasertib should be investigated in patients with ER+ breast cancer that have escaped estrogen deprivation therapy.
Supplementary Material
Acknowledgments
This work was supported by the NIH Breast Cancer Specialized Program of Research Excellence (SPORE) grant P50 CA98131; Vanderbilt-Ingram Cancer Center Support Grant P30 CA68485; Susan G. Komen for the Cure Foundation grants SAC100013 (CLA); a grant from the Breast Cancer Research Foundation; a Komen Post-Doctoral award PDF 12227859 (to NEB); and a NHGRI grant U54HG003067(EVA). We would like to acknowledge Genoptix Medical Laboratory for performing the quantification of Ki67 nuclei staining by AQUA methodology.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2013. Annals of Oncology. 2013;24:792–800. doi: 10.1093/annonc/mdt010. [DOI] [PubMed] [Google Scholar]
- 2.Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thürlimann B, Senn H-J, et al. Strategies for subtypes—dealing with the diversity of breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Annals of Oncology. 2011;22:1736–47. doi: 10.1093/annonc/mdr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Tanani MKK, Green CD. Interaction between estradiol and growth factors in the regulation of specific gene expression in MCF-7 human breast cancer cells. The Journal of Steroid Biochemistry and Molecular Biology. 1997;60:269–76. doi: 10.1016/s0960-0760(96)00226-9. [DOI] [PubMed] [Google Scholar]
- 4.Brenton JD, Carey LA, Ahmed AA, Caldas C. Molecular Classification and Molecular Forecasting of Breast Cancer: Ready for Clinical Application? Journal of Clinical Oncology. 2005;23:7350–60. doi: 10.1200/JCO.2005.03.3845. [DOI] [PubMed] [Google Scholar]
- 5.Mauri D, Pavlidis N, Polyzos NP, Ioannidis JPA. Survival With Aromatase Inhibitors and Inactivators Versus Standard Hormonal Therapy in Advanced Breast Cancer: Meta-analysis. J Natl Cancer Inst. 2006;98:1285–91. doi: 10.1093/jnci/djj357. [DOI] [PubMed] [Google Scholar]
- 6.Howell A, Robertson JFR, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, et al. Fulvestrant, Formerly ICI 182,780, Is as Effective as Anastrozole in Postmenopausal Women With Advanced Breast Cancer Progressing After Prior Endocrine Treatment. Journal of Clinical Oncology. 2002;20:3396–403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- 7.Howell A, Robertson JFR, Abram P, Lichinitser MR, Elledge R, Bajetta E, et al. Comparison of Fulvestrant Versus Tamoxifen for the Treatment of Advanced Breast Cancer in Postmenopausal Women Previously Untreated With Endocrine Therapy: A Multinational, Double-Blind, Randomized Trial. Journal of Clinical Oncology. 2004;22:1605–13. doi: 10.1200/JCO.2004.02.112. [DOI] [PubMed] [Google Scholar]
- 8.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of Tamoxifen Resistance: Increased Estrogen Receptor-HER2/neu Cross-Talk in ER/HER2–Positive Breast Cancer. J Natl Cancer Inst. 2004;96:926–35. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 9.Millar EKA, Graham PH, O’Toole SA, McNeil CM, Browne L, Morey AL, et al. Prediction of Local Recurrence, Distant Metastases, and Death After Breast-Conserving Therapy in Early-Stage Invasive Breast Cancer Using a Five-Biomarker Panel. Journal of Clinical Oncology. 2009;27:4701–8. doi: 10.1200/JCO.2008.21.7075. [DOI] [PubMed] [Google Scholar]
- 10.Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. The Lance. 365:1687–717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 11.Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet Oncol. 2008;9:45–53. doi: 10.1016/S1470-2045(07)70385-6. [DOI] [PubMed] [Google Scholar]
- 12.Santen RJ, Song RX, Zhang Z, Kumar R, Jeng M-H, Masamura A, et al. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocrine-Related Cancer. 2005;12:S61–S73. doi: 10.1677/erc.1.01018. [DOI] [PubMed] [Google Scholar]
- 13.W-S S, MC, SM, WY, J-P W, RK, et al. Estradiol Hypersensitivity and Mitogen-Activated Protein Kinase Expression in Long-Term Estrogen Deprived Human Breast Cancer Cells in Vivo. Endocrinology. 2000;141:396–405. doi: 10.1210/endo.141.1.7270. [DOI] [PubMed] [Google Scholar]
- 14.Fox EM, Miller TW, Balko JM, Kuba MG, Sánchez V, Smith RA, et al. A Kinome-Wide Screen Identifies the Insulin/IGF-I Receptor Pathway as a Mechanism of Escape from Hormone Dependence in Breast Cancer. Cancer Research. 2011;71:6773–84. doi: 10.1158/0008-5472.CAN-11-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller TW, Hennessy BT, Gonz xE, lez-Angulo AM, Fox EM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor–positive human breast cancer. The Journal of Clinical Investigation. 2010;120:2406–13. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–30. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 17.Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. The Journal of Cell Biology. 1995;129:1617–28. doi: 10.1083/jcb.129.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, Nishida E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410:215–20. doi: 10.1038/35065617. [DOI] [PubMed] [Google Scholar]
- 19.Donaldson MM, Tavares AAM, Hagan IM, Nigg EA, Glover DM. The mitotic roles of Polo-like kinase. Journal of Cell Science. 2001;114:2357–8. doi: 10.1242/jcs.114.13.2357. [DOI] [PubMed] [Google Scholar]
- 20.Nigg EA. Polo-like kinases: positive regulators of cell division from start to finish. Current Opinion in Cell Biology. 1998;10:776–83. doi: 10.1016/s0955-0674(98)80121-x. [DOI] [PubMed] [Google Scholar]
- 21.Wierer M, Verde G, Pisano P, Molina H, Font-Mateu J, Di Croce L, et al. PLK1 Signaling in Breast Cancer Cells Cooperates with Estrogen Receptor-Dependent Gene Transcription. Cell Reports. 3:2021–32. doi: 10.1016/j.celrep.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto Y, Matsuyama H, Kawauchi S, Matsumoto H, Nagao K, Ohmi C, et al. Overexpression of Polo-Like Kinase 1 (PLK1) and Chromosomal Instability in Bladder Cancer. Oncology. 2006;70:231–7. doi: 10.1159/000094416. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, et al. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Science. 2003;94:148–52. doi: 10.1111/j.1349-7006.2003.tb01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takai N, Miyazaki T, Fujisawa K, Nasu K, Hamanaka R, Miyakawa I. Polo-like kinase (PLK) expression in endometrial carcinoma. Cancer Letters. 2001;169:41–9. doi: 10.1016/s0304-3835(01)00522-5. [DOI] [PubMed] [Google Scholar]
- 25.Knecht R, Elez R, Oechler M, Solbach C, Ilberg Cv, Strebhardt K. Prognostic Significance of Polo-like Kinase (PLK) Expression in Squamous Cell Carcinomas of the Head and Neck. Cancer Research. 1999;59:2794–7. [PubMed] [Google Scholar]
- 26.Schoffski P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist. 2009;14:559–70. doi: 10.1634/theoncologist.2009-0010. [DOI] [PubMed] [Google Scholar]
- 27.Wolf G, Hildenbrand R, Schwar C, Grobholz R, Kaufmann M, Stutte H-J, et al. Polo-like kinase: a novel marker of proliferation: Correlation with estrogen-receptor expression in human breast cancer. Pathology - Research and Practice. 2000;196:753–9. doi: 10.1016/S0344-0338(00)80107-7. [DOI] [PubMed] [Google Scholar]
- 28.Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1:338–51. doi: 10.1158/2159-8290.CD-11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008;10:1076–82. doi: 10.1038/ncb1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative Genomic Approaches Identify IKBKE as a Breast Cancer Oncogene. Cell. 2007;129:1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 31.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The Two-Handed E Box Binding Zinc Finger Protein SIP1 Downregulates E-Cadherin and Induces Invasion. Molecular Cell. 2001;7:1267–78. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 32.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proceedings of the National Academy of Sciences. 2011;108:5021–6. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, A’Hern R, et al. Prognostic Value of Ki67 Expression After Short-Term Presurgical Endocrine Therapy for Primary Breast Cancer. J Natl Cancer Inst. 2007;99:167–70. doi: 10.1093/jnci/djk020. [DOI] [PubMed] [Google Scholar]
- 34.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15:3094–102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 35.Lu L-Y, Wood JL, Minter-Dykhouse K, Ye L, Saunders TL, Yu X, et al. Polo-Like Kinase 1 Is Essential for Early Embryonic Development and Tumor Suppression. Molecular and Cellular Biology. 2008;28:6870–6. doi: 10.1128/MCB.00392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellegrino R, Calvisi DF, Ladu S, Ehemann V, Staniscia T, Evert M, et al. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology. 2010;51:857–68. doi: 10.1002/hep.23467. [DOI] [PubMed] [Google Scholar]
- 37.Flier Svd, Brinkman A, Look MP, Kok EM, Meijer-van Gelder ME, Klijn JGM, et al. Bcar1/p130Cas Protein and Primary Breast Cancer: Prognosis and Response to Tamoxifen Treatment. J Natl Cancer Inst. 2000;92:120–7. doi: 10.1093/jnci/92.2.120. [DOI] [PubMed] [Google Scholar]
- 38.AP, CT, FG, CR-C, J-M R, HR, et al. FRA-1 Expression Level Modulates Regulation of Activator Protein-1 Activity by Estradiol in Breast Cancer Cells. Molecular Endocrinology. 1998;12:973–85. doi: 10.1210/mend.12.7.0133. [DOI] [PubMed] [Google Scholar]
- 39.Wang I-C, Chen Y-J, Hughes D, Petrovic V, Major ML, Park HJ, et al. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Molecular and Cellular Biology. 2005;25:10875–94. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Major ML, Lepe R, Costa RH. Forkhead Box M1B Transcriptional Activity Requires Binding of Cdk-Cyclin Complexes for Phosphorylation-Dependent Recruitment of p300/CBP Coactivators. Molecular and Cellular Biology. 2004;24:2649–61. doi: 10.1128/MCB.24.7.2649-2661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qu Q, Mao Y, Fei X-c, Shen K-w. The Impact of Androgen Receptor Expression on Breast Cancer Survival: A Retrospective Study and Meta-Analysis. PLoS ONE. 2013;8:e82650. doi: 10.1371/journal.pone.0082650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castellano I, Allia E, Accortanzo V, Vandone A, Chiusa L, Arisio R, et al. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res Treat. 2010;124:607–17. doi: 10.1007/s10549-010-0761-y. [DOI] [PubMed] [Google Scholar]
- 43.Lin C-Y, Strom A, Li Kong S, Kietz S, Thomsen J, Tee J, et al. Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Research. 2007;9:R25. doi: 10.1186/bcr1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rizza P, Barone I, Zito D, Giordano F, Lanzino M, De Amicis F, et al. Estrogen receptor beta as a novel target of androgen receptor action in breast cancer cell lines. Breast Cancer Research. 2014;16:R21. doi: 10.1186/bcr3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu K, Law JH, Fotovati A, Dunn SE. Small interfering RNA library screen identified polo-like kinase-1 (PLK1) as a potential therapeutic target for breast cancer that uniquely eliminates tumor-initiating cells. Breast Cancer Res. 2012;14:R22. doi: 10.1186/bcr3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simizu S, Osada H. Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat Cell Biol. 2000;2:852–4. doi: 10.1038/35041102. [DOI] [PubMed] [Google Scholar]
- 47.DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire M-D, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–2. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kharman-Biz A, Gao H, Ghiasvand R, Zhao C, Zendehdel K, Dahlman-Wright K. Expression of activator protein-1 (AP-1) family members in breast cancer. BMC Cancer. 2013;13:441. doi: 10.1186/1471-2407-13-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brinkman A, van der Flier S, Kok EM, Dorssers LCJ. BCAR1, a Human Homologue of the Adapter Protein p130Cas, and Antiestrogen Resistance in Breast Cancer Cells. J Natl Cancer Inst. 2000;92:112–20. doi: 10.1093/jnci/92.2.112. [DOI] [PubMed] [Google Scholar]
- 50.Cabodi S, Moro L, Baj G, Smeriglio M, Di Stefano P, Gippone S, et al. p130Cas interacts with estrogen receptor α and modulates non-genomic estrogen signaling in breast cancer cells. Journal of Cell Science. 2004;117:1603–11. doi: 10.1242/jcs.01025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.