

Abstract
Objectives
VIP is an immunomodulatory neuropeptide with therapeutic properties in multiple murine models of inflammatory disease including the trinitrobenzene-sulfonic acid (TNBS)-colitis model of Crohn's disease. Understanding the spectrum of biological actions of endogenously produced VIP may help us dissect the complex and multifactorial pathogenesis of such inflammatory diseases. Our goal was to determine the contribution of endogenously produced VIP to TNBS-colitis by using VIP knockout (KO) mice.
Methods
TNBS was intracolonically administered to wild type (WT) and VIP KO mice, and weight loss and colitis were assessed over time. Colon histopathological changes and myeloperoxidase (MPO) activities were analyzed and the levels of TNFα and IL-6 in colon and serum quantified. The proliferative response in vitro of splenocytes from TNBS WT and VIP KO administered mice to anti-CD3 and anti-CD28 was determined.
Results
VIP KO mice did not exhibit the predicted exacerbated response to TNBS. Instead, they developed a milder clinical profile than WT mice, with lower TNFα and IL-6 levels. Such potential defects seem selective, because other parameters such as the histopathological scores and the cytokine levels of in the colon did not differ between the two strains of mice. Moreover, splenocytes from TNBS-treated VIP KO mice exhibited an enhanced proliferative response to anti-CD3/CD28 stimulation in vitro.
Conclusion
Chronic loss of VIP in mice leads to a disruption of certain but not all immunological compartments, corroborating recent findings that VIP KO mice exhibit reduced mortality in the LPS-induced endotoxemia model and attenuated clinical development of experimental autoimmune encephalomyelitis while developing robust T cell responses.
Keywords: VIP, Crohn's disease, inflammation, neuropeptide, knockout, TNBS
Introduction
Crohn's disease (CD) is a relapsing inflammatory disease of unknown etiology that affects the gastrointestinal tract [1]. Together with ulcerative colitis, it belongs to the category of inflammatory bowel diseases (IBD). Although its pathogenesis is complex and not completely understood, it has been suggested that CD is caused by an excessive immune response against the normal microflora of the gut. This causes innate immune cells to release cytokines such as TNFα and IL-1β, which are elevated in the inflamed mucosa of CD patients, and triggers an unrestrained inflammatory response that leads to intestinal damage. Evidence suggests that chronic inflammation during the disease is maintained by Th1 and Th17 cells that massively invade the intestine [2,3]. Even though numerous novel therapeutic strategies have been designed to specifically target single or multiple immunological components of CD with some success, the incidence and prevalence of the disease is continuously increasing [1]. Treatments such as TNFα blockers have been shown to ameliorate its clinical symptoms, but complications including malignancies and infections have been reported [4,5]. Thus, it is imperative to investigate further the mechanisms involved in the disease in order to identify new molecules with potential therapeutic use.
Unraveling the pathogenesis of CD is an arduous task because multiple factors seem to contribute to its development or affect its course. Evidence suggests that interactions between the enteric nervous system and the immune system take place during the disease. In this sense, it has been shown that neuropeptides, including substance P, corticotropin-releasing hormone, neurotensin, and vasoactive intestinal peptide (VIP) may participate in the pathogenesis of CD [6]. Because multiple studies on the neuroimmune-endocrine interactions occurring during inflammatory bowel disease have revealed multiple cellular targets for several molecular mediators, further research in this field is warranted.
VIP is a 28 aminoacid neuropeptide that was originally isolated from the small intestine of the pig by Sami Said and Victor Mutt in 1970 [7]. It belongs to a family of neuropeptides that include PACAP (pituitary adenylate cyclase-activating polypeptide), which is structurally 70% identical to VIP. All major layers of each segment of the gastrointestinal tract contain VIP-immunoreactive fibers [8,9]. The effects of this neuropeptide in the gastrointestinal system have been extensively studied, and include alterations of motility in the bowel and gallbladder, stimulation of gastric acid and intestinal secretion, hormone/enzyme release from the exocrine and endocrine pancreas, and altered proliferation and survival of tumoral cells (e.g. human adenocarcinoma colonic cells) [9,10]. VIP exerts these actions by way of two receptors that belong to the G protein-coupled receptor family, VPAC1 and VPAC2, and which also bind PACAP with high affinity [11]. Each of these receptors is strongly coupled via Gs to adenylyl cyclase and cAMP production, although in some case they couple to other intracellular pathways.
Despite the classical concept of this peptide as a neurotransmitter or neuromodulator, it has been found that VIP is also produced by lymphocytes, and that almost all immune cells express one of more VPAC receptor types (reviewed in [12]).Through these receptors, VIP and PACAP exert multiple modulatory actions on adaptive and innate immunity. It is now documented that exogenously administered VIP and PACAP can exert powerful anti-inflammatory actions in vivo, promoting Th2 over Th1 responses (reviewed in [12]). VIP and PACAP have recently been shown to exhibit therapeutic activity in several experimental models of Th1-driven inflammatory human immune disease. In an established model of CD, with intracolonic administration of 2,4,6-trinitrobenzene sulfonic acid ((TNBS)-induced colitis), VIP reduced the clinical and histopathological severity of disease, abrogating body weight loss, diarrhea, and macroscopic and microscopic intestinal inflammation [13]. Treatment with VIP decreased the levels of a wide array of chemokines and proinflammatory cytokines [14]. In addition, VIP downregulated the expression of toll-like receptors (TLR)-2 and 4 in the colon and mesenteric lymph nodes of TNBS-treated animals [15,16]. These latter receptors have been depicted as innate immune sensors of bacterial and other pathogen components and have been implicated in inflammatory responses. Thus, several anti-inflammatory effects of exogenously administered VIP in murine colitis have been demonstrated. In addition, it has been shown that VIP regulates epithelial paracellular permeability in vitro and that VIP administration ameliorated bacterial-induced colitis by protecting the epithelial colonic barrier [17].
Given the high abundance of this peptide in the intestine, determining specific roles of the endogenously produced VIP may contribute to understand the pathogenesis of the disease. Here, we have investigated the impact of VIP gene deletion in mice in their response to TNBS-colitis.
Material and Methods
Colitis induction and clinical scoring
Both wild-type (WT) and VIP deficient (KO) [18] mice backcrossed for at least 6-12 generations (6-8 weeks old) on a C57BL/6 background were bred in-house. Mice were housed and fed ad libitum at the UCLA Neuroscience Research Building vivarium. All procedures were approved by UCLA's Animal Care and Use Committee and conducted in accordance with the guidelines in “NIH Guide for the Care and Use of Laboratory Animals”.
For colitis induction, a solution containing 5 mg of TNBS (Sigma Chemical Co., St. Louis, MO) diluted in 50% ethanol was rectally administered with a 4 cm catheter to mice lightly anesthetized with isofluorane. Control mice received a solution with only 50% ethanol (vehicle). Mice were held from the tail in inverted vertical position for 30 seconds, returned to their cages, and monitored for recovery from anesthesia. Mice were weighed every day and colitis was assessed from 0 to 4 according to the consistency of the stools (0, well-formed pellets; 2, loose stools; 4, watery diarrhea) as described [13].
Histology
Colons were collected from mice four days post-TNBS administration, flushed with PBS to eliminate feces, and cut into 0.5 cm pieces. Portions of the distal colon were fixed for 2 hours in Bouin's fixative, and transferred to 70% EtOH until dehydration with progressive alcohols for paraffin embedding. Six-μm sections were deparaffinized with xylene, rehydrated, and stained with alcian blue, hemalum and picroindigocarmine following standard procedures. Pathological changes in sections were scored in a blinded-fashion following a histological scoring system modified from Gay, J. et al [19] which considers separately the degrees of immune cell infiltration (0, no inflammatory cell infiltration; 1, mild inflammatory cell infiltration, few scattered cells; 2, moderate, distributed but not dense inflammatory cell infiltration; 3, dense inflammatory cell infiltration), epithelial damage/ulceration (0, no epithelial damage or ulcers; 1, minimal loss of goblet cells with erosions or single ulceration not exceeding lamina propria; 2, extensive loss of goblet cells and multifocal ulcerations not exceeding the mucosa; 3, extensive loss of crypts and multiple ulcerations not exceeding the submucosa), mucosal edema (0, normal thickness; 1, mild edema (submucosal expansion <10%); 2, moderate edema (submucosal expansion 1-100%); 3, severe edema (submucosal expansion >100%)) and fibrosis (0, no fibrosis; 1, fibrosis present in <50% area; 2, fibrosis present in >50% area), with maximum cumulative score of 11.
Myeloperoxidase (MPO) assay
As previously described [13], MPO activity was determined in colon extracts four days after EtOH or TNBS administration. Briefly, colons were dissected, flushed with PBS, minced and 50 mg pieces of the distal colon were homogenized with a hand-held homogenizer in phosphate buffer with 0.5% hexadecyltrimethylammonium bromide (HTAB) at 50 mg/ml. Homogenates were frozen and thawed three times, then centrifuged at 30.000 g for 20 minutes, and supernatants were collected and stored at -80°C until assay. Samples were distributed in 96-well plates, diluted 1:30 with assay buffer (50 mM phosphate buffer, pH 6.0, containing 0.167 mg/mL o-dianisidine (Sigma) and 0.0005% H2O2) and A450nm was measured in a microtiter reader. MPO activity (mU/g of tissue) was calculated based on a MPO activity standard curve.
ELISA
For cytokine measurement, blood and colons were collected from mice four days after TNBS instillation. Blood was clotted for 2 hours at room temperature, centrifuged for 10 minutes at 1800 rpm and serum was collected and stored at -80°C until assay. For colon extract preparation, 350 mg of the colons (proximal/distal region) were flushed with PBS and tissue was homogenized at 0.5 g/mL in 50 mmol/L Tris-HCl buffer (pH 7.4), containing a cocktail of protease inhibitors (0.5 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, and 10 μg/mL leupeptin (all from Sigma)). Samples were centrifuged at 30,000g for 20 minutes and supernatants were stored at -80°C until assay. TNFα and IL-6 levels were measured by sandwich ELISA with antibody pairs from Peprotech (Rocky Hill, NJ) following the manufacturer's instructions.
RNA isolation and real time PCR
For measurement of cytokine mRNA levels, colons were dissected four days after TNBS or vehicle treatment, flushed with PBS to remove the fecal material and tissue corresponding to the proximal and distal colon were homogenized in Ultraspec RNA isolation reagent (Biotecx, Houston, TX) with a hand held homogenizer. Samples were kept for 5 minutes at 4°C and 0.2 ml of chloroform per 1 ml of Ultraspec was added. Tubes were covered tightly, shaken vigorously for 15 seconds and placed on ice at 4°C for 5 minutes. The emulsion was then centrifuged at 12,000 g for 15 minutes, and the aqueous phase containing the RNA was carefully aspirated. An equal volume of isopropanol was added, centrifuged at 12,000 g for 10 minutes, and the pellet was washed with 70% ethanol. Samples were resuspended in RNAse-free water and A260nm was quantified with a spectrophotometer (Nanodrop, Thermo Scientific, Wilmington, DE). Then, 1μg of RNA was retrotranscribed to cDNA with the Superscript mix from BioRad (Hercules, CA) following the manufacturer's instructions. For Real time PCR analysis, the iQ™ SYBR Green Supermix from Bio-Rad was used in triplicate in 25 μL reaction volumes. The sequences of the primers used were: for IL-6, sense 5′-TTCCATCCAGTTGCCTTCTTG-OH and antisense 5′-TTGGGAGTGGTATCCTCTGTGA-OH (accession number NM_010551), For TNFα, sense 5′-CGATCACCCCGAAGTTCAGTA-OH and antisense 5′-GGTGCCTATGTCTCAGCCTCTT-OH (accession number NM_013693) for HPRT, sense 5′-TGGTGAAAAGGACCTCTCGAA-OH and antisense 5′-TCAAGGGCATATCCAACAACA-OH (accession number NM_013556). The PCR conditions were: 4 minutes at 96°C followed by 40 PCR cycles on the following conditions: for IL-6, denaturation at 96°C for 20 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 20 seconds; for TNFα denaturation at 96°C for 20 seconds, annealing at 62°C for 30 seconds, and extension at 72°C for 20 seconds. HPRT was used in all experiments as internal control gene. The formula used for relative quantitation used was 2-ΔΔCt, representing the n-fold differential expression of a specific gene in a treated sample compared with the control sample as described [14]. Melting curve analysis was used to determine the purity of the amplified products, which were also individually sequenced to confirm identity.
T cell proliferation assessment
Mice were sacrificed four days after TNBS treatment, and spleens were dissected. Cell suspensions were obtained by tapping the spleens through a 40μm nylon mesh. Samples were then treated with red blood lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4) for 5 minutes at 4°C. 2 × 105 cells were dispensed in 96-well plates and treated with anti-CD3 (10μg/ml; Becton Dickinson, Franklin Lakes, NJ) and anti-CD28 (5μg/ml; Becton Dickinson) antibodies in RPMI 1640 complete medium (Gibco, BRL, Grand Island, NY) containing 2% fetal bovine serum for FBS (HyClone, Logan, UT), 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco) for 48 hours. Then, [3H]-thymidine (1 μCi/well; Amersham Biosciences, Piscataway, NJ) was added and 24 hours later cells were collected. Cells were harvested, lysed, acid-precipitated, and [3H]-thymidine incorporation was determined by liquid β-scintillation counting (Beckman).
Statistical analysis
Differences between pairs of groups were assessed by Student's t-test with Welch's correction using the GraphPad Prism 5.01 program.
Results
VIP KO mice exhibited a milder clinical response to TNBS administration than WT mice
Intracolonic administration of TNBS to rodents causes a rapid loss of weight and colitis that is accompanied by acute inflammation. To determine the relevance of endogenously produced VIP in this model, we administered TNBS to WT and VIP KO mice and monitored their weight and colitis score over the time. Figure 1 represents the cumulative results of five experiments. As shown in Figure 1A, control (EtOH-treated) mice continued gaining weight steadily over time, but on average, WT mice gained more weight than VIP KO mice. In response to TNBS treatment, both WT and VIP KO mice began to lose weight one day after administration of the compound. However, the weight loss was slightly less pronounced in the VIP KO mice group treated with TNBS, with a percentage of original weight always higher than that observed in the TNBS-treated WT mice at each time point. Data on Figure 1B represents the differences in weight between each of the TNBS treated groups and their respective EtOH-treated controls at each time point as percentage of original weight. When treated with TNBS, VIP KO mice exhibited significantly less weight loss than WT mice at each time point, with an average difference from control weights over the four days analyzed of -13.18% in the KO vs -17.61% in the WT mice. Overall in our experiments, a total of 9 out of 40 WT (22.5%) vs 3 out of 38 VIP KO (7.9%) mice had died by the end of the study, indicating a lower mortality in the KO group. In agreement with their more pronounced weight loss, colitis scores tended to be were higher in WT mice than in VIP KO mice, although colitis scores were only significantly higher on day 2 post-TNBS administration(Figure 1C). Because sex differences have been shown in the sensitivity of VIP KO mice to inflammation, we further analyzed our results segregating female and male data (not shown). However, we found that both sexes of KO mice displayed a similar ameliorated response to TNBS.
Figure 1. Clinical parameters of TNBS-induced colitis are milder in VIP KO mice compared to WT mice.
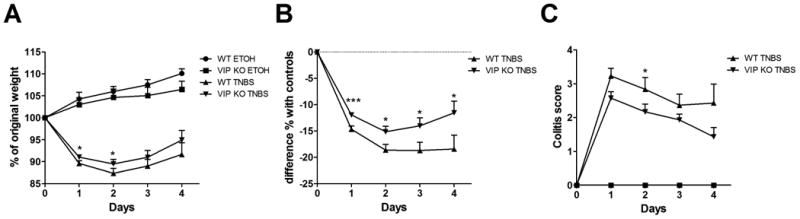
TNBS (5 mg) in 50% EtOH was intracolonically administered to 6-8 week-old WT and VIP KO mice. The results shown are a combination of five experiments for a total of n=14 for each group of vehicle-treated mice (50% EtOH), n=40 for TNBS –treated WT mice and n=38 for VIP KO mice. Mice were weighed every day for four days and colitis was scored from 0 to 4 as described in the Material and Methods section with 0 being no colitis and 4 severe colitis. A) represents % of original weight, B) represents the difference of weight between each TNBS-treated group and their respective EtOH-treated control group as percentage of original weight, and C) represents the colitis score. VIP KO mice exhibited significantly reduced weight loss and colitis compared to WT mice. Student's t-test *p<0.05, ***p<0.001.
Histological scores and MPO activities were similar in VIP KO and WT mice
TNBS-administration to rodents triggers an acute inflammatory response characterized by rapid infiltration of neutrophils, followed by macrophages and lymphocytes. These immune cells invade the lamina propria where they are activated and ultimately lead to the destruction of the colonic epithelium with the loss of goblet cells. Whereas minimal histopathology was observed in control ethanol-treated mice, inflammation and tissue damage were elevated in both groups four days after TNBS treatment (Figure 2A, B and Table 1). Trends to increased inflammation were observed in EtOH and TNBS VIP KO vs WT colons, although these differences were not statistically significant. In addition to the histological score, the level of MPO activity in tissue homogenates is frequently used as an indirect measurement of neutrophil infiltration. We found that the average levels of MPO activity were elevated in both VIP KO and WT TNBS-treated mice, suggesting that neutrophil infiltration took place in the two strains of mice (Figure 2C). Consistent with the histological scores, MPO levels tended to be higher in VIP KO mice both in EtOH and TNBS-treatment conditions.
Figure 2. Histopathological changes and MPO activities are not different between WT and VIP KO mice.
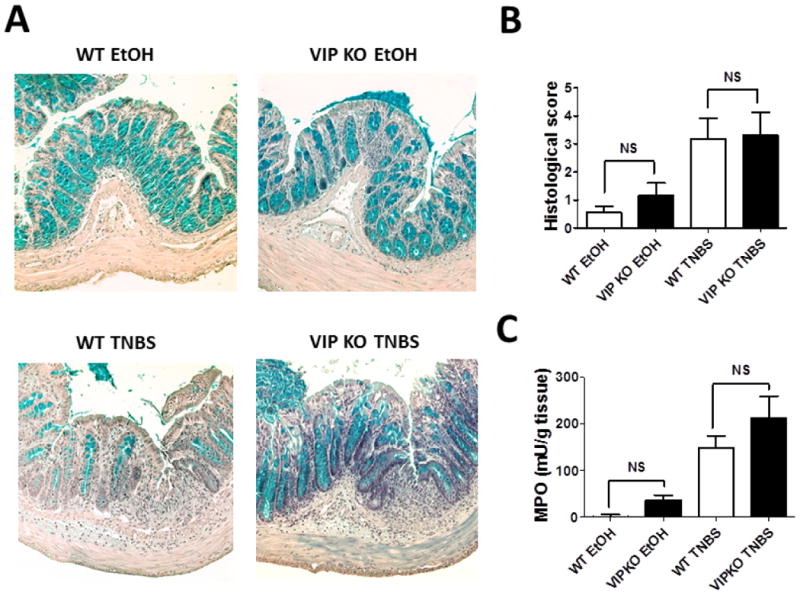
WT and VIP KO EtOH (control) and TNBS-treated mice (n=8 for each) were sacrificed four days after treatment, and colons were collected and flushed with PBS. A) shows representative micrographs of colons (20X) B) represents the average cumulative histological score (maximum score is 11) and C) the level of MPO activity on the tissue. We found no significant differences between the histological scores and MPO levels of WT and VIP KO EtOH groups or the WT and VIP KO TNBS groups (NS=non-significant).
Table 1. Histological scores of WT and VIP KO mice treated with EtOH or TNBS.
| Immune infiltration | Ulceration/epithelial damage | Edema | Fibrosis | Cumulative score | |
|---|---|---|---|---|---|
| WT ETOH | 0.6 ± 0.3 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.6 ± 0.2 |
| VIPKO ETOH | 0.8 ± 0.3 | 0.3 ± 0.2 | 0 ± 0 | 0 ± 0 | 1.2 ± 0.5 |
| WT TNBS | 1.3 ± 0.2 | 1.0 ± 0.3 | 0.7 ± 0.2 | 0.2 ± 0.1 | 3.2 ± 0.8 |
| VIPKO TNBS | 1.7 ± 0.2 | 0.9 ± 0.3 | 0.6 ± 0.3 | 0.2 ± 0.1 | 3.4 ± 0.9 |
VIP KO mice exhibit lower cytokine levels than WT mice after TNBS administration
The inflammatory response induced in the colon with TNBS, is characterized by a local and systemic elevation of proinflammatory cytokine levels. Although the pathogenesis of this model has multiple components, the better clinical outcome of VIP deficient mice compared to WT mice after TNBS treatment may be a consequence of ameliorated inflammation due to immunological defects in these mice. To test this hypothesis, we assessed the degree of inflammatory response at a molecular level by measuring the protein levels of the proinflammatory cytokines TNFα and IL-6 in sera and colon extracts from WT and VIP KO mice four days post-TNBS administration (Figure 3A, B, D, E). As expected, the protein levels of these cytokines were elevated in the sera and colon extracts of WT mice after TNBS administration compared to EtOH controls. However, in agreement with the lower severity of colitis in VIP KO mice, no significant increase was found for these cytokines in these mice. Similarly, the mRNA levels for these cytokines were significantly lower in the colons of VIP KO mice treated with TNBS than WT mice (Figure 3C,F).
Figure 3. IL-6 and TNFα levels are reduced in the sera and colons of TNBS-treated mice VIP KO mice.
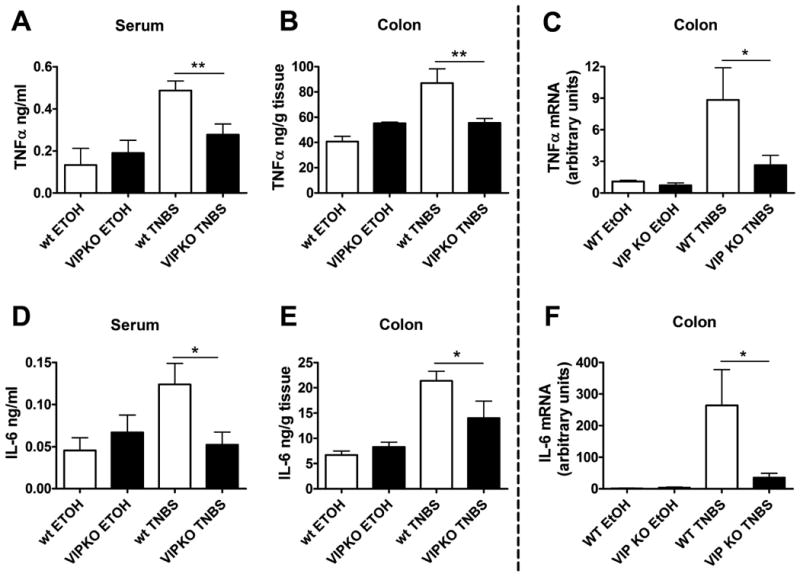
WT and VIP KO EtOH (control) and TNBS-treated mice (n=8 for each) were sacrificed four days after treatment, and blood and colons were collected. Sera and colon protein extracts were prepared as described in the Material and Methods section. Protein levels of TNFα (A, B) and IL-6 (D, E) were measured by ELISA. TNFα (C) and IL-6 mRNA (F) levels in the colons were determined by Real time RT-PCR. We found elevated cytokine levels in WT mice with TNBS treatment, but not in VIP KO mice. One of three representative experiments is shown. Student's t-test *p<0.05, **p<0.01.
Splenocytes from VIP KO mice exhibit robust proliferative response
It has been shown that VIP receptors are expressed on T cells, and that this neuropeptide modulates their functionality directly or indirectly through antigen presenting cells (such as macrophages and dendritic cells) or other cells [12,20]. Although the pathogenic involvement of T cells in TNBS-induced colitis is controversial, some reports describe a certain degree of T-cell activation in this model, even at early stages [21]. In order to determine if T cell responses at this level are impaired in VIP KO mice, we obtained splenocytes from control and TNBS treated mice and studied their proliferative index when activated in vitro with anti-CD3 and anti-CD28 antibodies. We found an increase of T cell proliferation in splenocytes from TNBS-treated WT mice comparison with EtOH-treated WT controls (Figure 4). Proliferation also increased in VIP KO mice with TNBS vs EtOH treatment, but in contrast to other inflammatory parameters this increase was at least 2-fold higher than in WT mice. This suggests that T cell responsiveness may be enhanced in the KO mice in this model, despite the reduced induction of proinflammatory cytokines.
Figure 4. Splenocyte proliferation in response to anti-CD3 and anti-CD28 antibodies is not impaired in VIP KO mice.
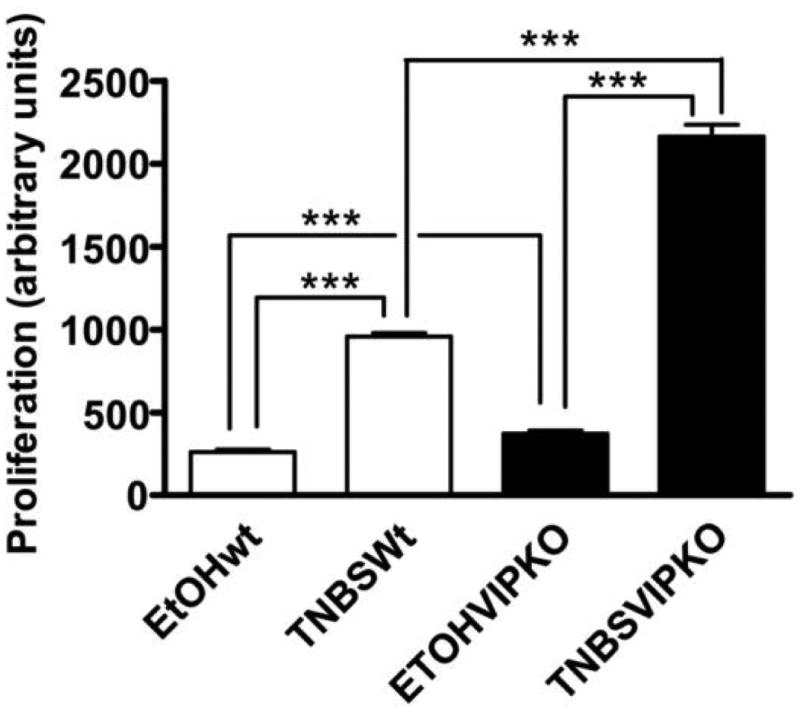
Spleens were dissected from WT and VIP KO mice (n=6 for each) four days after treatment with EtOH (controls) or TNBS. Cell suspensions were prepared as described in the Material and Methods section, and incubated with anti-CD3 and anti-CD28 antibodies for 72 hours. [3H]-thymidine (1 μCi/well) was added in the last 24 hours of culture and [3H]-thymidine incorporation in cell lysates was determined by liquid β-scintillation counting. Both WT and VIP KO TNBS-treated mice presented robust proliferative responses. One representative experiment out of three is shown. Student's t-test: ***p<0.001.
Discussion
The effects of exogenous VIP on several murine models of inflammatory diseases have been repeatedly tested and at least some of its main mechanisms of action have been described. On the other hand, endogenous VIP is widely distributed in the organism, and its levels have been reported to be elevated in response to inflammatory challenges [22-24]. The relevance of the endogenous source of this peptide in normal and abnormal conditions has only begun to be characterized. The goal of our study was to address this question by examining the response of VIP KO mice to chemically induced colitis. Despite the well-known anti-inflammatory actions of exogenous VIP, VIP KO mice treated with TNBS did not develop the exacerbated clinical response that would be expected due to the lack of this protective peptide. Instead, they exhibited slightly reduced weight loss and clinical disease and decreased local and systemic levels of TNFα and IL-6. Nevertheless, the degree of immune cell infiltration in the colon and the splenocyte proliferative index in response to CD3/CD28 stimulation were robust and even enhanced in the KO mice, suggesting that not all immunological responses are similarly affected in these mice.
VIP is normally expressed in all layers of the gastrointestinal system [9]. In rat and guinea pig, a large proportion of all cell bodies in the submucosal plexus of the ileum and colon are VIP immunoreactive. The myenteric plexus also contains VIP-immunoreactive neuronal cell bodies, although in lower proportions. In addition, there is a tendency for increasing proportions of VIP immunoreactive cells to occur in the distal parts of the gastrointestinal tract. VIP fibers extend into all the intestinal layers. Among them, the VIP innervation of the lamina propria is particularly rich, constituting a dense network of fibers that often come very close to the surface, seemingly innervating the epithelium. The levels of VIP have been reported to fluctuate in disease situations, although some results in this respect seem to be discordant. In humans for example, there are studies describing an increase, decrease or no significant changes in this neuropeptide levels. In one report, a positive correlation was found between plasma VIP levels and clinical disease activity, with two-fold increases in neuropeptide concentration during active periods of disease [25]. In the same line, other reports demonstrated that in the colon of CD patients compared to healthy controls, the number of VIP-producing neurons was increased [26,27], and that these neurons exhibited an hypertrophic appearance [28]. On the contrary, a reduction in the abundance of VIP-immunoreactive nerve fibers in the lamina propria and submucosa of both CD and UC patients was reported in a different study [29]. There are also conflicting results regarding the expression of VIP in animal models of IBD. It has been shown that VIP levels in the serum, colon and brain increased during TNBS-induced colitis in mice with a peak two days post-disease induction [30]. Nevertheless, a reduction in the number VIP-containing neurons has been reported in the same model [31]. The variability in results reporting the expression of VIP during human IBD or animal models of colitis may be due to the collection of samples on different stages of the disease. In fact, due to the tissue damage consequent to TNBS administration, a reduction has been found to be common for several types of neuropeptide-containing nerves [32]. In any case, alterations in VIP levels are likely to be relevant in the development of the disease, as this might contribute to the disruption of local immunoregulation.
Despite the well-known anti-inflammatory properties of VIP, the reduced levels of proinflammatory cytokines in TNBS-treated VIP KO mice compared to WT mice were not unexpected in view of recent findings in which VIP KO mice have been subjected to other models of inflammatory disease. For example, we have demonstrated that mice deficient in VIP (VIP KO) exhibit ameliorated responses to experimental autoimmune encephalomyelitis (EAE) and to lipopolysaccharide (LPS)-induced endotoxemia [33,34]. In addition, VIP KO mice displayed a milder weight loss and intestinal damage than WT mice in a dextran sodium sulphate (DSS) murine model of disease [35]. These reports hint that long term lack of VIP is somehow protective in the development of this disease or that VIP is necessary for the development of certain inflammatory responses. Further research is needed to determine the mechanisms involved in these phenotypes. Because VIP and its receptors are abundantly present in the intestine where they control multiple functions, other alterations independent of inflammation are likely to appear in VIP KO mice. The intestines of naïve VIP-deficient mice exhibit thicker smooth muscle layers, longer villi, and more goblet cells than WT mice, although these latter were deficient in mucus production [36]. In addition, these mice exhibit deficits in intestinal transit [36]. Intestinal mucus constitutes a physical barrier against toxins and pathogens. Hence, a protective role for intestinal mucus against chemically-induced models of colitis has been suggested [37-39]. Therefore, it would be expected that the reduction in mucus found in the lumen of VIP KO intestines would lead to enhanced colitis. Likewise, the reduction in the KO intestinal motility and mucus could alter the exposure and/or responsiveness of the mucosal immune system to commensal or pathogenic microbiota, potentially resulting in increased immune responses. Unexpectedly, we found significantly reduced proinflammatory cytokines in VIP KO vs. WT mice after TNBS treatment, indicating that loss of these potentially-protective mechanisms in VIP-deficient mice did not, in fact, heighten inflammation. Finally, the lack of VIP secretagogue actions in the intestine may predict and contribute to the reduced colitis symptomology in the KO mice, for example, the diminished weight loss.
Our previous study showing an increased survival of VIP KO mice after LPS-administration, and the reduced production of proinflammatory cytokines by peritoneal cells of these mice point to an impairment in the innate arm of immunity of these mice. However, this impairment may not affect all populations of innate immune cells or all innate immune processes as we found here that histopathological scores and MPO levels, which indicate the degree of neutrophil infiltration, were not different between the colons of WT and VIP KO TNBS-treated mice. A potential defect in innate immunity may be relevant in IBD and its TNBS-colitis experimental model, as it has been hypothesized that the disease is triggered by an abnormal activation of intestinal innate immune cells by commensal flora that surpasses the natural control mechanisms [40]. Alternatively, it is possible that control mechanisms that naturally dampen inflammation may be enhanced in the knockout mice as a compensatory response to the chronic lack of VIP, which may paradoxically promote tolerance of these mice to inflammatory stimuli.
We found that splenocytes from both WT and KO mice exhibited an increased proliferative response when stimulated in vitro with anti-CD3/anti-CD8 antibodies. In addition, the splenocytes from TNBS-administered VIP KO mice proliferated more than those from WT mice. The involvement of adaptive immunity in the TNBS-colitis experiments is uncertain due to the acute character of the inflammatory response in our model and the short course of symptoms. However, it has been suggested that general T cell activation is required for neutrophil recruitment during the early stages of the TNBS-induced disease [21]. Our results suggest that T cell activation is not impaired and may be enhanced in VIP KO mice. This is in agreement with our previous finding that lymphocytes from VIP KO mice with EAE exhibited hyper-proliferation in response to myelin oligodendrocyte glycoprotein (MOG) the protein administered for EAE induction, and were capable to induce the disease when transferred upon naïve WT mice, demonstrating their enhanced encephalitogenic potential to promote inflammation in the brain [33]. In addition, these cells responded to MOG by producing as much or higher levels of cytokines than WT mice [33]. This suggests that T cells may not be intrinsically impaired, but that their action in VIP KO mice may be ultimately affected by a defective innate immune response. In fact, we found that although VIP KO mice were highly resistant to EAE, T cells accumulated in the meninges of their brains and spinal cords, but did not penetrate in the parenchyma like in the WT mice. Thus, alterations in T cells secondary to other defects in VIP KO mice cannot be ruled out at this point.
The opposing actions of VIP may be explained in part by the presence of its two different receptors. A recent report by Yadav et al. [41] suggested that the VPAC1 receptor may mediate the proinflammatory actions of VIP, and VPAC2 may mediate its anti-inflammatory actions in DSS-induced colitis. In that study, weight loss, colitis and intestinal damage after DSS administration were milder in VPAC1 deficient mice compared to WT mice. On the contrary, VPAC2 deficient mice exhibited enhanced symptoms of disease than WT mice. Thus, how VIP acts in a certain situation may depend on the combined action of these receptors or the cell types exposed to the ligand. In addition, it has been shown that the expression level of VPAC receptors changes with immune activation. For example, whereas resting T cells and macrophages do not express the VPAC2 receptor, it is highly upregulated and functional after in vitro stimulation [42-44]. Moreover, a downregulation of the VPAC1 receptor in human peripheral blood T CD4+ lymphocytes activated with anti-CD3 and PMA has been reported [45]. Thus, it seems that developing drugs acting on specific VPAC receptors may be promising. In addition, investigating the cellular expression and time-course of VPAC receptors during inflammatory diseases may be crucial to develop precise treatment strategies.
Acknowledgments
This work was supported by a Pilot and Feasiblity Study Grant from the CURE: Digestive Disease Research Center (P30DK41301), and by National Multiple Sclerosis Society RG4859 and National Institutes of Health Grant HD04612.
References
- 1.Baumgart DC, Sandborn WJ. Crohn's disease. Lancet. 2013;380:1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 2.Monteleone I, Pallone F, Monteleone G. Th17-related cytokines: New players in the control of chronic intestinal inflammation. BMC Med. 2011;9:122. doi: 10.1186/1741-7015-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahluwalia JP. Immunotherapy in inflammatory bowel disease. Med Clin North Am. 2012;96:525–544. x. doi: 10.1016/j.mcna.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Ngo B, Farrell CP, Barr M, Wolov K, Bailey R, Mullin JM, Thornton JJ. Tumor necrosis factor blockade for treatment of inflammatory bowel disease: Efficacy and safety. Curr Mol Pharmacol. 2010;3:145–152. doi: 10.2174/1874467211003030145. [DOI] [PubMed] [Google Scholar]
- 6.Margolis KG, Gershon MD. Neuropeptides and inflammatory bowel disease. Curr Opin Gastroenterol. 2009;25:503–511. doi: 10.1097/MOG.0b013e328331b69e. [DOI] [PubMed] [Google Scholar]
- 7.Said SI, Mutt V. Polypeptide with broad biological activity: Isolation from small intestine. Science. 1970;169:1217–1218. doi: 10.1126/science.169.3951.1217. [DOI] [PubMed] [Google Scholar]
- 8.Lakhan SE, Kirchgessner A. Neuroinflammation in inflammatory bowel disease. J Neuroinflammation. 2010;7:37. doi: 10.1186/1742-2094-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fahrenkrug J. Comprehensive physiology. John Wiley & Sons, Inc.; 2011. Vasoactive intestinal peptide; pp. 611–629. [Google Scholar]
- 10.Moody TW, Chan D, Fahrenkrug J, Jensen RT. Neuropeptides as autocrine growth factors in cancer cells. Curr Pharm Des. 2003;9:495–509. doi: 10.2174/1381612033391621. [DOI] [PubMed] [Google Scholar]
- 11.Harmar AJ, Fahrenkrug J, Gozes I, Laburthe M, May V, Pisegna JR, Vaudry D, Vaudry H, Waschek JA, Said SI. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: Iuphar review 1. Br J Pharmacol. 2012;166:4–17. doi: 10.1111/j.1476-5381.2012.01871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- 13.Abad C, Martinez C, Juarranz MG, Arranz A, Leceta J, Delgado M, Gomariz RP. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of crohn's disease. Gastroenterology. 2003;124:961–971. doi: 10.1053/gast.2003.50141. [DOI] [PubMed] [Google Scholar]
- 14.Abad C, Juarranz Y, Martinez C, Arranz A, Rosignoli F, Garcia-Gomez M, Leceta J, Gomariz RP. Cdna array analysis of cytokines, chemokines, and receptors involved in the development of tnbs-induced colitis: Homeostatic role of vip. Inflamm Bowel Dis. 2005;11:674–684. doi: 10.1097/01.mib.0000171872.70738.58. [DOI] [PubMed] [Google Scholar]
- 15.Arranz A, Abad C, Juarranz Y, Torroba M, Rosignoli F, Leceta J, Gomariz RP, Martinez C. Effect of vip on tlr2 and tlr4 expression in lymph node immune cells during tnbs-induced colitis. Ann N Y Acad Sci. 2006;1070:129–134. doi: 10.1196/annals.1317.001. [DOI] [PubMed] [Google Scholar]
- 16.Gomariz RP, Arranz A, Abad C, Torroba M, Martinez C, Rosignoli F, Garcia-Gomez M, Leceta J, Juarranz Y. Time-course expression of toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of vip. J Leukoc Biol. 2005;78:491–502. doi: 10.1189/jlb.1004564. [DOI] [PubMed] [Google Scholar]
- 17.Conlin VS, Wu X, Nguyen C, Dai C, Vallance BA, Buchan AM, Boyer L, Jacobson K. Vasoactive intestinal peptide ameliorates intestinal barrier disruption associated with citrobacter rodentium-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G735–750. doi: 10.1152/ajpgi.90551.2008. [DOI] [PubMed] [Google Scholar]
- 18.Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA. Disrupted circadian rhythms in vip- and phi-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R939–949. doi: 10.1152/ajpregu.00200.2003. [DOI] [PubMed] [Google Scholar]
- 19.Gay J, Kokkotou E, O'Brien M, Pothoulakis C, Karalis KP. Interleukin-6 genetic ablation protects from trinitrobenzene sulfonic acid-induced colitis in mice. Putative effect of antiinflammatory cytokines Neuroimmunomodulation. 2006;13:114–121. doi: 10.1159/000096656. [DOI] [PubMed] [Google Scholar]
- 20.Delgado M. Vip: A very important peptide in t helper differentiation. Trends Immunol. 2003;24:221–224. doi: 10.1016/s1471-4906(03)00069-3. [DOI] [PubMed] [Google Scholar]
- 21.van Lierop PP, de Haar C, Lindenbergh-Kortleve DJ, Simons-Oosterhuis Y, van Rijt LS, Lambrecht BN, Escher JC, Samsom JN, Nieuwenhuis EE. T-cell regulation of neutrophil infiltrate at the early stages of a murine colitis model. Inflamm Bowel Dis. 2010;16:442–451. doi: 10.1002/ibd.21073. [DOI] [PubMed] [Google Scholar]
- 22.Martinez C, Delgado M, Abad C, Gomariz RP, Ganea D, Leceta J. Regulation of vip production and secretion by murine lymphocytes. J Neuroimmunol. 1999;93:126–138. doi: 10.1016/s0165-5728(98)00216-1. [DOI] [PubMed] [Google Scholar]
- 23.Delgado M, Martinez C, Pozo D, Calvo JR, Leceta J, Ganea D, Gomariz RP. Vasoactive intestinal peptide (vip) and pituitary adenylate cyclase-activation polypeptide (pacap) protect mice from lethal endotoxemia through the inhibition of tnf-alpha and il-6. J Immunol. 1999;162:1200–1205. [PubMed] [Google Scholar]
- 24.Delgado M, Abad C, Martinez C, Juarranz MG, Arranz A, Gomariz RP, Leceta J. Vasoactive intestinal peptide in the immune system: Potential therapeutic role in inflammatory and autoimmune diseases. J Mol Med. 2002;80:16–24. doi: 10.1007/s00109-001-0291-5. [DOI] [PubMed] [Google Scholar]
- 25.Duffy LC, Zielezny MA, Riepenhoff-Talty M, Byers TE, Marshall J, Weiser MM, Graham S, Ogra PL. Vasoactive intestinal peptide as a laboratory supplement to clinical activity index in inflammatory bowel disease. Dig Dis Sci. 1989;34:1528–1535. doi: 10.1007/BF01537105. [DOI] [PubMed] [Google Scholar]
- 26.O'Morain C, Bishop AE, McGregor GP, Levi AJ, Bloom SR, Polak JM, Peters TJ. Vasoactive intestinal peptide concentrations and immunocytochemical studies in rectal biopsies from patients with inflammatory bowel disease. Gut. 1984;25:57–61. doi: 10.1136/gut.25.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider J, Jehle EC, Starlinger MJ, Neunlist M, Michel K, Hoppe S, Schemann M. Neurotransmitter coding of enteric neurones in the submucous plexus is changed in non-inflamed rectum of patients with crohn's disease. Neurogastroenterol Motil. 2001;13:255–264. doi: 10.1046/j.1365-2982.2001.00265.x. [DOI] [PubMed] [Google Scholar]
- 28.Bishop AE, Polak JM, Bryant MG, Bloom SR, Hamilton S. Abnormalities of vasoactive intestinal polypeptide-containing nerves in crohn's disease. Gastroenterology. 1980;79:853–860. [PubMed] [Google Scholar]
- 29.Kubota Y, Petras RE, Ottaway CA, Tubbs RR, Farmer RG, Fiocchi C. Colonic vasoactive intestinal peptide nerves in inflammatory bowel disease. Gastroenterology. 1992;102:1242–1251. [PubMed] [Google Scholar]
- 30.Baticic L, Detel D, Kucic N, Buljevic S, Pugel EP, Varljen J. Neuroimmunomodulative properties of dipeptidyl peptidase iv/cd26 in a tnbs-induced model of colitis in mice. J Cell Biochem. 2011;112:3322–3333. doi: 10.1002/jcb.23261. [DOI] [PubMed] [Google Scholar]
- 31.Sigalet DL, Wallace L, De Heuval E, Sharkey KA. The effects of glucagon-like peptide 2 on enteric neurons in intestinal inflammation. Neurogastroenterol Motil. 2010;22:1318–e1350. doi: 10.1111/j.1365-2982.2010.01585.x. [DOI] [PubMed] [Google Scholar]
- 32.Linden DR, Couvrette JM, Ciolino A, McQuoid C, Blaszyk H, Sharkey KA, Mawe GM. Indiscriminate loss of myenteric neurones in the tnbs-inflamed guinea-pig distal colon. Neurogastroenterol Motil. 2005;17:751–760. doi: 10.1111/j.1365-2982.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- 33.Abad C, Tan YV, Lopez R, Nobuta H, Dong H, Phan P, Feng JM, Campagnoni AT, Waschek JA. Vasoactive intestinal peptide loss leads to impaired cns parenchymal t-cell infiltration and resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:19555–19560. doi: 10.1073/pnas.1007622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abad C, Tan YV, Cheung-Lau G, Nobuta H, Waschek JA. Vip deficient mice exhibit resistance to lipopolysaccharide induced endotoxemia with an intrinsic defect in proinflammatory cellular responses. PLoS One. 2012;7:e36922. doi: 10.1371/journal.pone.0036922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yusta B, Holland D, Waschek JA, Drucker DJ. Intestinotrophic glucagon-like peptide-2 (glp-2) activates intestinal gene expression and growth factor-dependent pathways independent of the vasoactive intestinal peptide gene in mice. Endocrinology. 2012;153:2623–2632. doi: 10.1210/en.2012-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lelievre V, Favrais G, Abad C, Adle-Biassette H, Lu Y, Germano PM, Cheung-Lau G, Pisegna JR, Gressens P, Lawson G, Waschek JA. Gastrointestinal dysfunction in mice with a targeted mutation in the gene encoding vasoactive intestinal polypeptide: A model for the study of intestinal ileus and hirschsprung's disease. Peptides. 2007;28:1688–1699. doi: 10.1016/j.peptides.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeiffer CJ, Qiu B, Lam SK. Reduction of colonic mucus by repeated short-term stress enhances experimental colitis in rats. J Physiol Paris. 2001;95:81–87. doi: 10.1016/s0928-4257(01)00012-2. [DOI] [PubMed] [Google Scholar]
- 38.Krimi RB, Kotelevets L, Dubuquoy L, Plaisancie P, Walker F, Lehy T, Desreumaux P, Van Seuningen I, Chastre E, Forgue-Lafitte ME, Marie JC. Resistin-like molecule beta regulates intestinal mucous secretion and curtails tnbs-induced colitis in mice. Inflamm Bowel Dis. 2008;14:931–941. doi: 10.1002/ibd.20420. [DOI] [PubMed] [Google Scholar]
- 39.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. Muc2-deficient mice spontaneously develop colitis, indicating that muc2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 40.Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in ibd. Nat Rev Gastroenterol Hepatol. 2012;9:599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 41.Yadav M, Huang MC, Goetzl EJ. Vpac1 (vasoactive intestinal peptide (vip) receptor type 1) g protein-coupled receptor mediation of vip enhancement of murine experimental colitis. Cell Immunol. 2011;267:124–132. doi: 10.1016/j.cellimm.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Delgado M, Munoz-Elias EJ, Kan Y, Gozes I, Fridkin M, Brenneman DE, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kb and camp response element-binding protein/c-jun. J Biol Chem. 1998;273:31427–31436. doi: 10.1074/jbc.273.47.31427. [DOI] [PubMed] [Google Scholar]
- 43.Delgado M, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit nuclear factor-kappa b-dependent gene activation at multiple levels in the human monocytic cell line thp-1. J Biol Chem. 2001;276:369–380. doi: 10.1074/jbc.M006923200. [DOI] [PubMed] [Google Scholar]
- 44.Delgado M, Martinez C, Johnson MC, Gomariz RP, Ganea D. Differential expression of vasoactive intestinal peptide receptors 1 and 2 (vip-r1 and vip-r2) mrna in murine lymphocytes. J Neuroimmunol. 1996;68:27–38. doi: 10.1016/0165-5728(96)00063-x. [DOI] [PubMed] [Google Scholar]
- 45.Lara-Marquez M, O'Dorisio M, O'Dorisio T, Shah M, Karacay B. Selective gene expression and activation-dependent regulation of vasoactive intestinal peptide receptor type 1 and type 2 in human t cells. J Immunol. 2001;166:2522–2530. doi: 10.4049/jimmunol.166.4.2522. [DOI] [PubMed] [Google Scholar]