

Abstract
Purpose
New therapies are urgently needed for patients with acute myeloid leukemia (AML). The novel NEDDylation inhibitor MLN4924 (pevonedistat) has demonstrated significant preclinical anti-leukemic activity and preliminary efficacy in patients with AML in a Phase I trial. Based on its anti-myeloid and DNA-damaging properties, we investigated the ability of MLN4924 to augment conventional cytarabine (ara-C) therapy.
Experimental Design
The effects of MLN4924/ara-C on viability, clonogenic survival, apoptosis, DNA damage and relevant pharmacodynamic targets were determined. The efficacy and pharmacodynamics of MLN4924/ara-C were assessed in an AML xenograft model.
Results
Co-treatment of AML cell lines and primary patient specimens with MLN4924 and ara-C led to diminished clonogenic survival, increased apoptosis, and synergistic levels of DNA damage. RNA interference demonstrated that stabilization of CDT-1, an event previously shown to mediate the DNA damaging effects of MLN4924, was not a key regulator of sensitivity to the MLN4924/ara-C combination. Global metabolic profiling revealed that MLN4924 disrupts nucleotide metabolism and depletes intracellular nucleotide pools in AML cells. Subsequent experiments showed that MLN4924 promoted increased incorporation of ara-C into the DNA of AML cells. This effect as well as the therapeutic benefit of the MLN4924/ara-C combination were antagonized by supplementation with the nucleotide building block ribose. Co-administration of MLN4924 and ara-C to mice bearing FLT3-ITD+ AML xenografts stably inhibited disease progression and increased DNA damage in vivo.
Conclusions
Our findings provide strong rationale for clinical investigation of the MLN4924/ara-C combination and establish a new link between therapeutic inhibition of NEDDylation and alterations in nucleotide metabolism.
Introduction
Acute myeloid leukemia (AML) is a multi-step, multi-pathway malignancy that is ultimately fatal in the majority of affected patients (1). Older adults with AML have an extremely poor prognosis and derive limited benefit from intensive chemotherapy due to their prevalence of unfavorable cytogenetics predicting chemotherapy refractoriness, pre-existing myelodysplastic syndromes (MDS) associated with a limited normal functioning stem cell compartment and serial genetic lesions evolving to disease manifestation, and expression of a multidrug resistant phenotype (2–4). No standard induction approach exists for older adults, due in part, to their poor representation in clinical studies. The last prospective trial comparing intensive chemotherapy to low-dose approaches was conducted over two decades ago (5). While induction with low dose cytarabine (LDAC) offers a survival advantage over supportive care for patients with good/intermediate prognosis cytogenetics, patients with poorer performance status and/or unfavorable cytogenetics have a low likelihood of achieving complete responses (CR) when treated with LDAC (1). Considering these issues, new therapeutic approaches that are effective and tolerated could be transformative particularly for older AML patients.
Protein turnover plays a critical regulatory role in many essential cellular processes including cell cycle progression, signal transduction, and cell death. Aberrant ubiquitin-proteasome system (UPS)-mediated protein degradation contributes to disease progression, metastasis, and drug resistance and is thus, an appealing anticancer strategy (6). The proteasome inhibitor, bortezomib (Velcade), was the first anticancer agent of this drug class to receive FDA approval (7). Bortezomib established proof of concept that targeting protein turnover is an effective anticancer strategy and is currently indicated for the treatment of multiple myeloma and mantle cell lymphoma and in combination with cytotoxic therapy, yielded high response rates in AML patients (8, 9). Following its success, a second proteasome inhibitor (carfilzomib) also earned FDA approval for the treatment of multiple myeloma patients who progressed following treatment with bortezomib and an immunomodulatory drug (10). More recent drug discovery efforts have focused on the development of novel agents that target key regulators of the UPS system. A major objective of this approach is to disrupt the turnover of selected subsets of proteins, which could potentially eliminate or reduce some of the off-target effects and toxicities that are associated with global proteasomal inhibition.
NEDD8 is a small ubiquitin-like (UBL) molecule that regulates the activity of the cullin-RING family of E3 ubiquitin ligases (CRLs) through covalent modification. The CRLs control the degradation of a number of proteins with essential roles in cell cycle progression, tumor suppression, signal transduction, and responses to DNA damage and other stress stimuli including p27, cyclin E, c-Myc, p53, phospho-IκBα, CDT-1, NRF-2, and HIF-1α (11). As degradation of these molecules could accelerate cancer progression and/or promote therapeutic resistance, disrupting the NEDDylation pathway has significant and logical therapeutic potential. MLN4924 (pevonedistat) is a first-in-class, selective small molecule inhibitor of NEDD8 activating enzyme (NAE), the proximal regulator of the NEDDylation cascade (12–14). We previously showed that MLN4924 overcomes multiple mechanisms of pro-survival signaling, triggers DNA damage associated with CDT1 stabilization and CHK1 activation, and induces stable disease regression in an AML mouse model (15). This was confirmed in a Phase I clinical trial in refractory MDS/AML patients, some of whom achieved complete responses following treatment with MLN4924 (16). The mechanistic basis for the anti-AML effects of MLN4924 remains to be fully elucidated. We conducted comprehensive metabolic profiling to further investigate the pharmacodynamic effects of MLN4924 in AML cells that contribute to its therapeutic activity and ability to augment the efficacy of ara-C.
Materials & Methods
Cells and cell culture
HL-60 and KG-1 cells were obtained from ATCC (Manassas, VA). MV4-11 and MOLM-13 cells were obtained from DSMZ (Braunschweig, Germany). Cell lines were authenticated by the source banks using DNA profiling techniques and were used for this study in accordance with AACR guidelines. Primary human AML cells were isolated from the bone marrow of AML patients after obtaining informed consent. All AML cells (primary and established cell lines) were cultured under identical conditions with RPMI 1640 medium supplemented with 10% FBS at 37°C with 5% CO2 as previously described (15).
Sample preparation, metabolic profiling, metabolite identification, and data analysis
The untargeted metabolic profiling platform employed for this analysis combined three independent platforms: ultra-high performance liquid chromatography/tandem mass spectrometry (UHLC/MS/MS2) optimized for basic species, UHLC/MS/MS2 optimized for acidic species, and gas chromatography/mass spectrometry (GC/MS). Detailed methods for the metabolomic analyses are available as an online supplement.
Chemicals and reagents
Reagents were obtained from: cytarabine (ara-C, Hospira, Inc., Lake Forest, IL), anti-active caspase-3, anti-p27, anti-CDT1 (Cell Signaling, Beverly, MA), anti-phospho-Ser317- and total CHK1, and γH2AX antibodies (Epitomics, Burlingame, CA), anti-β tubulin and ribose (Sigma, St. Louis, MO), anti-proliferating cell nuclear antigen (PCNA, Dako, Glostrup, Denmark), goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson Laboratories, West Grove, PA), rat anti-mouse IgG2a-HRP antibody (Serotec, Raleigh, NC), sheep anti-mouse-HRP and donkey anti-rabbit-HRP (Amersham, Pittsburgh, PA).
Cell viability assay
Cells were plated in triplicate and treated with defined concentrations of MLN4924, ara-C, or both drugs for 72 hours. Viable cells were quantified using ATPLite according to the manufacturer’s instructions (Perkin Elmer, Waltham, MA) as previously described (14, 15, 17).
Synergy analyses
The combination indices (CI) for MLN4924 and ara-C were calculated based on the effect of 72 h exposure to each drug on cell viability as determined by ATPLite assay. CompuSyn software (ComboSyn, Inc. Paramus, NJ) software was utilized to calculate CI values as previously described (18).
Analysis of drug-induced apoptosis
Drug-induced apoptosis was quantified by PI/FACS analysis of sub-G0/G1 DNA content as previously described (17, 19).
Colony assays
AML cells were treated with the indicated concentrations of MLN4924, ara-C or both drugs for 24h. Drugs were washed away and cells were seeded in Methocult methylcellulose-containing medium. Colonies were stained and scored as previously described (15, 20).
Immunoblotting
AML cells were incubated with MLN4924, ara-C, or the combination of both drugs for 24 h as indicated. Cells were then lysed for 1 h on ice in Triton X-100 lysis buffer (1% triton X-100, 150 mM NaCl, 25 mM Tris pH 7.5) with protease inhibitors. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blots were probed with the indicated antibodies and bands were detected by enhanced chemiluminescence (Alpha Innotech, San Leandro, CA) as previously described (21). β-tubulin documented equal loading.
Alkaline comet assay
AML cells were treated with MLN4924, ara-C, or both drugs for 24 h. Comet assays were performed using the CometAssay® kit (Trevigen, Gaithersburg, MD) according to the manufacturer’s instructions as previously described (17). Cells were imaged using fluorescent microscopy and tail moments (product of DNA amount in tail and distance of tail migration) from 50 cells per slide were calculated.
shRNA knockdown of CDT1
MOLM-13 human AML cells were infected with lentiviral particles containing non-targeted (control) or targeted shRNA directed at CDT1 (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer’s instructions. Positively infected cells were selected with puromycin treatment. Infected cells were treated with the indicated concentrations of MLN4924 and ara-C for 48 h. Drug-induced apoptosis was quantified by PI/FACS. Knockdown efficiency was assessed by immunoblotting.
Quantification of [3H]ara-C DNA incorporation
[3H]ara-C was purchased from Moravek Biochemicals, Brea, CA. The incorporation of [3H]ara-C into AML cells was evaluated using the method developed by Kufe et al (22). Briefly, cells were incubated with non-radiolabeled MLN4924, ribose, or the combination of MLN4924 and ribose in the presence and absence [3H]ara-C for 24 h. Cells were washed twice in PBS and total DNA was isolated from cells in all experimental groups using the DNeasy kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). DNA concentrations of each sample were quantified by spectrophotometry and normalized. An equivalent amount of DNA from each sample was diluted in 1 mL of scintillation fluid and 3H counts were determined using a standard liquid scintillation method (Beckman-Coulter, Brea, CA).
In vivo evaluation of MLN4924 and ara-C
MOLM-13 FLT3-ITD+ human leukemia cells were injected into the flanks of nude mice as previously described (23). After tumor growth reached 150 mm3, mice were randomly assigned (n = 10 per group for all regimens) to receive vehicle control, 60 mg/kg MLN4924 BID, 50 mg/kg ara-C QD, or the combination of both drugs for 21 days. Tumor growth and animal toxicity was assessed as previously described (24).
Immunohistochemistry
Paraffin-embedded tumor sections were deparaffinized in xylene, exposed to a graded series of alcohol, and rehydrated in PBS (pH 7.5). Heat-induced epitope retrieval on paraffin-embedded sections and probing with specific antibodies was conducted as previously described (17). Positive reactions were visualized using 3,3′-diaminobenzidine (Dako, Glostrup, Denmark). Images were captured using an Olympus fluorescent microscope (Center Valley, PA) with a DP71 camera and a 20X objective. Image-Pro Plus software Version 6.2.1 (MediaCybernetics, Bethesda, MD) was used for image acquisition. ImageJ software was used for quantification of γH2AX and p27 levels by densitometric analysis of five random fields containing viable tumor cells. Quantification of PCNA and cleaved caspase-3 was conducted by counting the number of positive cells in five random fields as previously described (25).
Statistical analyses
Statistical significance of differences observed between samples was determined using the Tukey-Kramer Comparison Test or the Student’s t test. Differences were considered significant in all experiments at p < 0.05 with two-sided comparisons.
Results
MLN4924 and ara-C cooperate to antagonize clonogenic survival and trigger apoptosis
To investigate inhibiting NEDDylation with MLN4924 as a novel approach to increase the anti-AML activity of ara-C, we first treated 4 human AML cell lines (MV4-11, MOLM-13, HL-60, KG-1) with MLN4924, ara-C, or both drugs for 72 h and quantified the resulting impact of drug exposure on cell viability by ATPLite assay. Pilot drug sequencing studies showed that pre-treatment with one drug first for 24 h followed by the other agent for the remainder of the assay period did not yield significantly different anti-leukemic effects as compared with simultaneous drug treatment (not shown). We therefore used simultaneous treatment conditions for all in vitro combination assays in this study. Combined treatment with MLN4924 and ara-C reduced cell viability significantly more effectively than either single agent (Fig. 1A). Formal synergy analyses demonstrated a combination index (CI) range of 0.5 – 0.8 across all concentrations tested. CI values less than 1.0 indicate synergy. Notably, FLT3-ITD status did not appear to affect cellular sensitivity to this combination as cells with (MV4-11, MOLM-13) and without (HL-60, KG-1) FLT3-ITD expression benefited similarly from treatment with MLN4924 and ara-C. Treatment of primary AML blasts from patients (n = 10) with MLN4924 and ara-C resulted in a significantly greater reduction in viability than treatment with single agents. Moreover, primary cells from chemo-naïve newly diagnosed (n = 4) and previously treated patients (n = 6) with AML did not differ in their sensitivity to the MLN4924/ara-c combination (Fig. 1B). Patient characteristics are detailed in Supplemental Table 1. Consistent with the results of our cell viability analyses, co-treatment with MLN4924 and ara-C resulted in significantly greater diminished clonogenic survival (Fig. 1C) and levels of drug-induced apoptosis (Fig. 1D) than exposure to either drug alone.
Fig. 1. MLN4924 and ara-C cooperate to diminish clonogenic survival and trigger apoptosis.
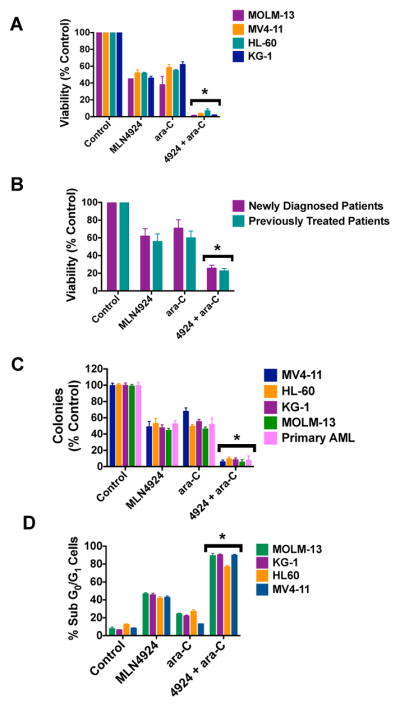
A, The combination of MLN4924 and ara-C potently reduces the viability of AML cells. MOLM-13, MV4-11, HL-60, and KG-1 cell lines were treated with 100 nM MLN4924, 100 nM ara-C, or both drugs for 72 hours. Viability was assessed by the ATPLite assay. n = 3 ± SD. B, The MLN4924/ara-C combination has activity in primary AML cells. Primary cells were obtained from 10 AML patients (n = 4 newly diagnosed, n = 6 previously treated). Patient characteristics are detailed in Supplemental Table 1. Primary cells were treated with MLN4924, ara-C, or both drugs for 72 hours. Viability was assessed by the ATPLite assay. C, Inhibition of clonogenic survival following treatment with MLN4924 and ara-C. MV4-11, HL-60, KG-1, MOLM-13, and primary AML cells were treated with MLN4924, ara-C, or both drugs for 24h. Drug was washed away, cells were seeded in Methocult and colonies were scored on Day 10. n = 3, bars represent the mean ± SD. D, Quantification of drug-induced apoptosis. MOLM-13, KG-1, HL-60, and MV4-11 cells were treated with 100 nM MLN4924, 100 nM ara-C, or both drugs for 48h. Percentages of cells with sub-diploid DNA were determined by PI/FACS. n = 3 ± SD, *P < 0.05, indicates a significant difference from either single agent treatment.
The combination of MLN4924 and ara-C promotes increased levels of DNA damage
The induction of DNA damage is one of the hallmark features of the anticancer mechanism of action of ara-C and other nucleoside analogs. We and others have also demonstrated that MLN4924 triggers DNA damage by promoting CDT-1 dependent DNA re-replication (15, 17, 26). We hypothesized that co-treatment with MLN4924 and ara-C would yield significantly greater levels of DNA damage. Accordingly, MLN924 and ara-C cooperated to stimulate increased levels of the phosphorylated (active) form of the DNA damage sensor CHK1 (Ser317), and the DNA damage marker γH2AX (Fig. 2A). These effects coincided with an elevation in the expression of the cyclin-dependent kinase inhibitor p27 and the active form of caspase-3, which would be expected based upon the combined effects of these agents on cell viability and apoptosis shown in Fig. 1. We next conducted alkaline comet assays to generate another readout of drug-induced DNA damage. These experiments demonstrated that the MLN4924/ara-C combination significantly increased the DNA tail moment, indicating heightened levels of DNA damage (Fig. 2B–C).
Fig. 2. Induction of DNA damage by MLN4924 and ara-C.

A, MLN4924 and ara-C activate the DNA damage response. Cells were treated with 100 nM MLN4924, 100 nM ara-C, or both drugs for 24h, lysed, subjected to SDS-PAGE, and probed with p27, pCHK1 (Ser317), total CHK1, γH2AX, and cleaved (active) caspase-3 -specific antibodies. Tubulin documented equal loading. B-C, Induction of DNA damage by MLN4924 and ara-C. Cells were treated with 100 nM MLN4924, 100 nM ara-C, or both drugs for 16 h. The DNA tail moment for each experimental condition was quantified by alkaline comet assay. Representative images are shown. n = 3 ± SD. *P < 0.05, indicates a significant difference from either single agent treatment.
CDT1 stabilization is not essential for the pro-apoptotic effects of the MLN4924/ara-C combination
As mentioned earlier, CDT1 stabilization has been previously suggested to be an important pharmacodynamic effect that facilitates DNA damage and contributes to the anticancer activity of MLN4924. In order to investigate whether this specific effect may be responsible, at least in part, for the benefit we observed with the MLN4924/ara-C combination, we utilized shRNA to target is expression in MOLM-13 cells. Knockdown efficiency was assessed by immunoblotting (Fig. 3A). A comparison of the pro-apoptotic effects of the MLN4924/ara-C combination in the presence and absence of CDT1 shRNA showed that CDT1 expression levels had a modest impact on single agent MLN4924 treatment, but had a negligible effect on the combination of both drugs (Fig. 3B). These results suggest that other aspects of the mechanism of action of MLN4924 may be mediating its ability to augment ara-C activity.
Fig. 3. CDT1 stabilization is not required for MLN4924 to augment ara-C activity.
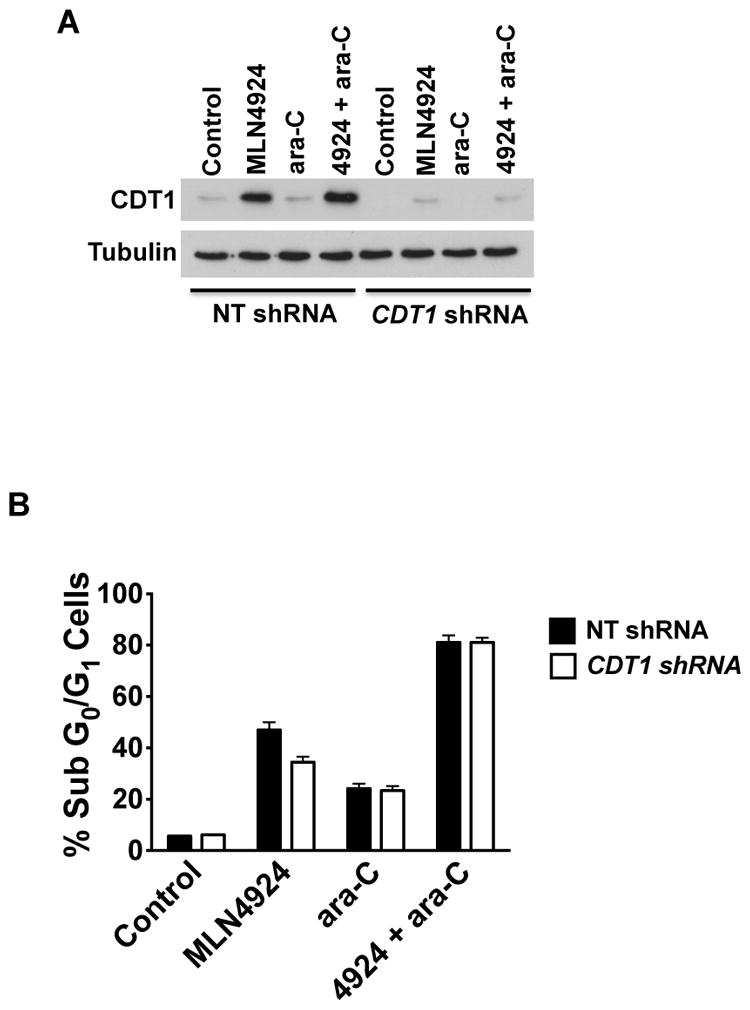
A, Knockdown of CDT1. Lentiviral shRNA was utilized to knockdown CDT1 expression. MOLM-13 cells infected with CDT1-directed or non-targeted control shRNA were treated with 100 nM MLN4924, 100 nM ara-C, or both drugs for 24 h. Knockdown efficiency was evaluated by immunoblotting. Tubulin documented equal protein loading. B, CDT1 impairment does not significantly impact the pro-apoptotic effects of the MLN4924/ara-C combination. Cells infected with non-targeted control or CDT1 directed shRNA were treated with 100 nM MLN4924, 100 nM ara-C, or both drugs for 48 h. Drug-induced apoptosis was quantified by PI/FACS analysis. n = 3 ± SD.
MLN4924 disrupts nucleotide metabolism and diminishes endogenous nucleotide pools
We next conducted global metabolic profiling to obtain a more comprehensive assessment of the pharmacodynamic effects of MLN4924. MV4-11 cells were treated with 100 nM MLN4924 for 12 h and 24 h. A total of 268 distinct biochemicals were specifically identified and quantified in experimental samples. Heatmap analysis revealed that exposure to MLN4924 caused time-dependent alterations in multiple metabolic superpathways (Fig. 4A). Quantification of individual biochemicals revealed that the levels of 112 biochemicals (19 increased, 93 decreased) were significantly altered (P<0.05) following 12 h treatment with MLN4924 compared to controls. Additional exposure to MLN4924 (24 h) resulted in 199 significant alterations (53 increased, 146 decreased, P<0.05). The overall trend at both timepoints indicated impairment rather than activation of the majority of the affected superpathways.
Fig. 4. MLN4924 disrupts nucleotide metabolism in AML cells.

A, Cells were treated with 100 nM MLN4924 for 12 h and 24 h. Cells were processed for global metabolic analyses as described in the Methods. Heatmap depicts superpathway analysis for both timepoints. Changes for each timepoint are quantified relative to control, n = 4 for each experimental condition. B, Cells were treated with 100 nM MLN4924 for 12 h and 24 h. Cells were processed for global metabolic analyses as described in the Methods. Heatmap specifically depicts nucleotide metabolism pathway analysis for both timepoints. Changes for each timepoint are quantified relative to control, n = 4 for each experimental condition.
Additional statistical analyses revealed that the nucleotide biosynthesis superpathway was one of the most altered overall pathways following treatment with MLN4924. Heatmap analysis (Fig. 4B) and quantification of the effects of drug treatment on the levels of individual nucleotide pathway components (Suppl. Fig. 1) demonstrated the time-dependent nature of the pathway alterations. Detailed schematics illustrating the key elements of pyrimidine and purine metabolism are shown in Supplemental Figures 2–3. The combined effect of drug-induced decreases in the levels of multiple key elements of nucleotide biosynthesis indicated severe pathway impairment. In particular, the dramatic diminishment of intracellular nucleotide pools following exposure to MLN4924 suggested that cells treated with MLN4924 may increase the cytotoxicity of conventional chemotherapy with nucleoside analogs such as ara-C by reducing endogenous competition for incorporation into DNA. The findings indicate that the effects of MLN4924 on nucleotide metabolism could contribute significantly to its ability to augment the efficacy of ara-C treatment.
MLN4924 facilitates increased incorporation of ara-C into the DNA of AML cells
We hypothesized that the therapeutic benefit derived from combining MLN4924 with ara-C could be due, in part, to increased DNA incorporation of ara-C facilitated by reduced competition from endogenous nucleotides/nucleosides. To test this possibility, we quantified the inclusion of radiolabeled [3H]ara-C into the DNA of MOLM-13 and primary AML cells in the presence and absence of MLN4924. Notably, the quantity of [3H]ara-C was significantly higher in cells co-treated with MLN4924 in both established and primary AML cells (Fig. 5A). We next sought to determine if this effect could be overcome by exogenous supplementation with the nucleotide building block ribose to antagonize the nucleotide depletion caused by MLN4924 treatment. As shown in Fig. 5A, the addition of ribose significantly blunted the enhancement of [3H]ara-C incorporation that we observed in the presence of MLN4924. Accordingly, quantification of drug-induced apoptosis triggered by the MLN4924/ara-C combination in the presence and absence of ribose supplementation showed that in addition to diminishing the integration of [3H]ara-C into DNA, exogenous ribose also tempered MLN4924/ara-C induced cell death (Fig. 5B). Our collective results demonstrate that the combination benefit of MLN4924 and ara-C is at least partially due to elevated incorporation of ara-C into the DNA of AML cells.
Fig. 5. MLN4924 promotes increased incorporation of ara-C into the DNA of AML cells.

A, Quantification of [3H]ara-C DNA incorporation. MOLM-13 and primary AML cells were treated with MLN4924, ribose, [3H]ara-C, and the indicated combinations for 24 h. The inclusion of [3H]ara-C under each experimental condition was determined as described in the Methods. n = 3 ± SD. *P < 0.05 for the comparison between [3H]ara-C versus [3H]ara-C + ribose, **P < 0.05 for the comparison between [3H]ara-C + MLN4924 versus [3H]ara-C + MLN4924 + ribose. B, Quantification of drug-induced apoptosis. MOLM-13 and primary AML cells were treated with 100 nM MLN4924, 100 nM ara-C, 10 mM ribose or the indicated combinations for 48 h. Percentages of cells with sub-diploid DNA were determined by PI/FACS. n = 3 ± SD, *P < 0.05 for the comparison between ara-C versus ara-C + ribose, **P < 0.05 for the comparison between ara-C + MLN4924 versus ara-C + MLN4924 + ribose.
The combination of MLN4924 and ara-C effectively inhibits the progression of FLT3-ITD+ AML xenografts in vivo
The in vivo anticancer activity of the MLN4924/ara-C combination was evaluated by administering vehicle control, MLN4924 (60 mg/kg BID), ara-C (50 mg/kg QD), or both drugs to mice implanted with MOLM-13 FLT3-ITD+ xenografts. Both MLN4924 and ara-C monotherapies significantly inhibited disease progression (Fig. 6A). Co-administration of MLN4924 and ara-C provided a significantly greater therapeutic benefit than either drug alone and resulted in stable inhibition of disease progression throughout our study. The combination was well tolerated and no notable toxicities were observed other than a very modest reduction in mean body weight. Immunohistochemical analysis of specimens collected from animals in all experimental groups demonstrated superior in vivo growth inhibition (elevated p27 and reduced PCNA levels), augmented DNA damage (γH2AX), and increased apoptosis (cleaved caspase-3) following treatment with the MLN4924/ara-C combination compared to either single agent (Fig. 6B, Suppl. Fig. 4).
Fig. 6. Combined administration of MLN4924 and ara-C suppresses disease progression in a FLT3-ITD+ AML xenograft model.

A, Administration of MLN4924 and ara-C to mice bearing MOLM-13 xenografts leads to stable suppression of disease progression. Mice received vehicle control, 60mg/kg MLN4924 BID, 50 mg/kg ara-C QD, or both drugs for 21 days. Tumor volume was measured by calipers. n = 10 per group, bars represent the mean ± SEM. *P<0.05, single agents versus control, **P<0.05, combination versus single agents. B, In vivo pharmacodynamic effects of MLN4924 and ara-C. Immunohistochemistry was utilized to measure the levels of p27, PCNA, γH2AX, and cleaved (active) caspase-3 in tumor specimens collected from mice in each treatment group. Representative images are shown.
Discussion
Cytotoxic-based therapy remains the standard for the treatment of AML, and has changed little in the past four decades. New agents/strategies for AML therapy are clearly needed in general, but are urgently required for patients that have unfavorable cytogenetics and/or are older and unfit for cytotoxic induction chemotherapy as they have the lowest probability of achieving long-term survival (<20%) with existing therapies (4, 27, 28). Inhibition of the NEDDylation pathway with the investigational drug MLN4924 is a novel therapeutic approach that has demonstrated encouraging anti-AML activity in the preclinical and clinical setting (15, 16).
However, our understanding of the pharmacodynamic effects of MLN4924 that are responsible for its therapeutic activity in AML remains incomplete. We hypothesized that MLN4924 may augment the efficacy of ara-C based on its DNA damaging properties that we and others have reported. Our initial data supported this hypothesis. Yet, targeted knockdown of CDT1, a chromatin licensing factor and NEDD8 substrate that has been suggested to be a key mediator of MLN4924’s DNA damaging properties and overall anticancer activity, did not indicate that it was an important regulator of sensitivity to the MLN4924/ara-C combination. We subsequently conducted global metabolic profiling of AML cells treated with MLN4924 to identify additional pharmacodynamic effects that may promote sensitization to ara-C. Our results showed that inhibition of NEDD8-mediated protein turnover has effects on multiple metabolic superpathways, but was particularly effective at antagonizing both purine and pyrimidine nucleotide metabolism. Although several preclinical studies have reported on the pharmacodynamic characteristics of MLN4924 (29), our investigation is the first to report the specific drug-related effects on nucleotide metabolism. In addition to establishing a new link between NEDD8-mediated protein turnover and nucleotide homeostasis, our findings demonstrate the value of global metabolic profiling in the pharmacodynamic analysis of investigational anticancer agents.
The MLN4924-induced disruption of nucleotide homeostasis that we observed provided a potential mechanistic explanation underlying its ability to improve the activity of the nucleoside analog ara-C. We hypothesized that the depletion of endogenous nucleotide pools by MLN4924 would yield more efficient incorporation of ara-C into the DNA of AML cells. A series of experiments demonstrated the benefit of the MLN4924/ara-C combination, which was due, at least in part, to MLN4924-mediated heightened incorporation of ara-C into DNA. This newly discovered DNA-related pharmacodynamic effect of MLN4924 builds upon several earlier investigations, which reported that the DNA damaging properties of MLN4924 are linked to the stabilization of the chromatin licensing factor CDT1 and subsequent DNA re-replication stress (14, 26, 30–32).
Another interesting aspect of our study is related to the similar sensitivity of the primary AML cells from both newly diagnosed and previously treated AML patients to the MLN4924/ara-C combination. Although the number of specimens we analyzed is relatively small (n = 4 newly diagnosed, n = 6 previously treated), the benefit we observed in cells that were clinically refractory to the ara-C based “7+3” regimen could have important translational implications. It is possible that NEDDylation may play a previously unstudied role in ara-C resistance and that inhibiting the pathway with MLN4924 may be sufficient to counter resistance to ara-C and other anticancer agents. Further investigation is required to fully evaluate this possibility, but the complete responses of refractory AML patients that were observed in the Phase I clinical trial of MLN4924 are encouraging (16). Moreover, the ability of MLN4924 to antagonize drug resistance does not appear to be restricted to AML and/or the conventional agents that are used to treat this disease. We recently showed that MLN4924 re-sensitizes cisplatin-resistant ovarian cancer cells to platinum chemotherapy (17). Our findings regarding the ability of MLN4924 to overcome platinum resistance were confirmed shortly after our initial report by an independent research team (33). These observations suggest that the mechanisms of resistance to MLN4924 do not directly overlap with those of cytotoxic chemotherapeutic agents and that NEDD8-mediated protein turnover may play a more significant role in controlling chemosensitivity than previously known.
At a minimum, these observations provide rationales to further explore potential applications for MLN4924 in the treatment of malignancies that are resistant to cytotoxic chemotherapy and to more rigorously study the mechanisms by which aberrant NEDDylation promotes malignant progression and drug resistance. The identification of a predictive biomarker of sensitivity to MLN4924 would also be a significant catalyst to its successful clinical development. Despite extensive ongoing preclinical and clinical research efforts with MLN4924, no specific predictive biomarker has been identified to date. The increasingly more powerful dataset that is developing as additional patients are treated with MLN4924 on clinical studies may help to address this issue.
In conclusion, our data establish that inhibition of NAE with MLN4924 is a novel strategy to augment the efficacy of ara-C. A clinical trial further investigating the safety and efficacy of MLN4924 in combination with ara-C therapy is warranted with ultimate comparison to LDAC monotherapy in older adults with AML. Considering the dramatic MLN4924-induced disruption of both purine and pyrimidine nucleotide metabolism we observed in this study, this approach may also have potential applications for other malignancies that use nucleoside analog-based therapy.
Supplementary Material
Translational Relevance.
Novel therapeutic strategies are urgently needed to improve the survivorship of patients with AML. MLN4924 (pevonedistat) is a first-in-class NEDDylation inhibitor currently under investigation in multiple clinical trials. Here we report that MLN4924 significantly augments the efficacy of conventional cytarabine (ara-C) therapy in preclinical models of AML and primary patient specimens. Global metabolic profiling analyses demonstrated that MLN4924 disrupted nucleotide metabolism and diminished intracellular nucleotide pools. These effects were associated with increased DNA damage and augmented incorporation of ara-C into the DNA of AML cells. Co-administration of MLN4924 and ara-C to mice bearing FLT3-ITD+ AML xenografts stably inhibited disease progression and increased DNA damage in vivo. Our findings provide strong rationale for clinical investigation of the MLN4924/ara-C combination.
Acknowledgments
This work was supported by a grant from the National Cancer Institute (R01CA172443, JSC)
Footnotes
Disclosure of conflicts of interest: PGS is a former employee of Millennium Pharmaceuticals, Inc.
References
- 1.Burnett AK, Milligan D, Prentice AG, Goldstone AH, McMullin MF, Hills RK, Wheatley K. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007;109:1114–24. doi: 10.1002/cncr.22496. [DOI] [PubMed] [Google Scholar]
- 2.Appelbaum FR, Gundacker H, Head DR, Slovak ML, Willman CL, Godwin JE, Anderson JE, Petersdorf SH. Age and acute myeloid leukemia. Blood. 2006;107:3481–5. doi: 10.1182/blood-2005-09-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kantarjian H, Ravandi F, O’Brien S, Cortes J, Faderl S, Garcia-Manero G, Jabbour E, Wierda W, Kadia T, Pierce S, Shan J, Keating M, Freireich EJ. Intensive chemotherapy does not benefit most older patients (age 70 years or older) with acute myeloid leukemia. Blood. 2010;116:4422–9. doi: 10.1182/blood-2010-03-276485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekeres MA, Stone R. Older adults with acute myeloid leukemia. Curr Oncol Rep. 2002;4:403–9. doi: 10.1007/s11912-002-0034-y. [DOI] [PubMed] [Google Scholar]
- 5.Lowenberg B, Zittoun R, Kerkhofs H, Jehn U, Abels J, Debusscher L, Cauchie C, Peetermans M, Solbu G, Suciu S, et al. On the value of intensive remission-induction chemotherapy in elderly patients of 65+ years with acute myeloid leukemia: a randomized phase III study of the European Organization for Research and Treatment of Cancer Leukemia Group. J Clin Oncol. 1989;7:1268–74. doi: 10.1200/JCO.1989.7.9.1268. [DOI] [PubMed] [Google Scholar]
- 6.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 7.Roccaro AM, Hideshima T, Richardson PG, Russo D, Ribatti D, Vacca A, Dammacco F, Anderson KC. Bortezomib as an antitumor agent. Curr Pharm Biotechnol. 2006;7:441–8. doi: 10.2174/138920106779116865. [DOI] [PubMed] [Google Scholar]
- 8.Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, Kolitz JE, Powell BL, Voorhees P, Wang ES, Blum W, Stone RM, Marcucci G, Bloomfield CD, Moser B, Larson RA. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013;31:923–9. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Advani A, Elson P, HSiED, Davis R, Kalaycio M, Sobecks R, Copelan E, Duong HK, Foster B, Rush ML, Farhat L, Sekeres MA. A phase I trial of MEC (mitoxantrone, etoposide, cytarabine) in combination with bortezomib for relapsed/refractory acute myeloid leukemia. Blood. 2012;120:3595. [Google Scholar]
- 10.Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M, Chanan-Khan A, Buadi F, Reu FJ, Somlo G, Zonder J, Song K, Stewart AK, Stadtmauer E, Kunkel L, Wear S, Wong AF, Orlowski RZ, Jagannath S. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120:2817–25. doi: 10.1182/blood-2012-05-425934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petroski MD, Deshaies RJ. In vitro reconstitution of SCF substrate ubiquitination with purified proteins. Methods Enzymol. 2005;398:143–58. doi: 10.1016/S0076-6879(05)98013-0. [DOI] [PubMed] [Google Scholar]
- 12.Podust VN, Brownell JE, Gladysheva TB, Luo RS, Wang C, Coggins MB, Pierce JW, Lightcap ES, Chau V. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci U S A. 2000;97:4579–84. doi: 10.1073/pnas.090465597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, Pierce JW, Podust VN, Luo RS, Chau V, Palombella VJ. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–33. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 15.Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, Oberheu K, Padmanabhan S, O’Dwyer M, Nawrocki ST, Giles FJ, Carew JS. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 16.De Angelo D, Erba HP, Maris MB, Swords RT, Anwer F, Alrman J, Hua Z, Blakemore S, Faessel H, Dezube BJ, Medeiros BC. MLN4924, a novel investigational inhibitor of NEDD8-activating enzyme (NAE), in adult patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS): results from multiple dosing schedules in a phase I study. Blood. 2013;122:1443. [Google Scholar]
- 17.Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA, Milhollen M, Blakemore S, Thomas M, Berger A, Carew JS. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res. 2013;19:3577–90. doi: 10.1158/1078-0432.CCR-12-3212. [DOI] [PubMed] [Google Scholar]
- 18.Mahalingam D, Medina EC, Esquivel JA, 2nd, Espitia CM, Smith S, Oberheu K, Swords R, Kelly KR, Mita MM, Mita AC, Carew JS, Giles FJ, Nawrocki ST. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res. 2010;16:141–53. doi: 10.1158/1078-0432.CCR-09-1385. [DOI] [PubMed] [Google Scholar]
- 19.Carew JS, Nawrocki ST, Krupnik YV, Dunner K, Jr, McConkey DJ, Keating MJ, Huang P. Targeting endoplasmic reticulum protein transport: a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood. 2006;107:222–31. doi: 10.1182/blood-2005-05-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles FJ, Cleveland JL. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–22. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nawrocki ST, Carew JS, Maclean KH, Courage JF, Huang P, Houghton JA, Cleveland JL, Giles FJ, McConkey DJ. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood. 2008;112:2917–26. doi: 10.1182/blood-2007-12-130823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kufe DW, Major PP, Egan EM, Beardsley GP. Correlation of cytotoxicity with incorporation of ara-C into DNA. J Biol Chem. 1980;255:8997–900. [PubMed] [Google Scholar]
- 23.Kelly KR, Nawrocki ST, Espitia CM, Zhang M, Yang JJ, Padmanabhan S, et al. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int J Cancer. 2012;131:2693–703. doi: 10.1002/ijc.27579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gontarewicz A, Balabanov S, Keller G, Colombo R, Graziano A, Pesenti E, Benten D, Bokemeyer C, Fiedler W, Moll J, Brummendorf TH. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood. 2008;111:4355–64. doi: 10.1182/blood-2007-09-113175. [DOI] [PubMed] [Google Scholar]
- 25.Carew JS, Nawrocki ST, Reddy VK, Bush D, Rehg JE, Goodwin A, Houghton JA, Casero RA, Jr, Marton LJ, Cleveland JL. The novel polyamine analogue CGC-11093 enhances the antimyeloma activity of bortezomib. Cancer Res. 2008;68:4783–90. doi: 10.1158/0008-5472.CAN-07-6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71:3042–51. doi: 10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- 27.Nazha A, Ravandi F. Acute myeloid leukemia in the elderly: do we know who should be treated and how? Leuk Lymphoma. 2013 doi: 10.3109/10428194.2013.828348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. American journal of hematology. 2013;88:318–27. doi: 10.1002/ajh.23404. [DOI] [PubMed] [Google Scholar]
- 29.Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21:1563–73. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- 30.Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, Smith PG, Morgan MA, Sun Y. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282–93. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang D, Tan M, Wang G, Sun Y. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS One. 2012;7:e34079. doi: 10.1371/journal.pone.0034079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kee Y, Huang M, Chang S, Moreau LA, Park E, Smith PG, D’Andrea AD. Inhibition of the Nedd8 system sensitizes cells to DNA interstrand cross-linking agents. Mol Cancer Res. 2012;10:369–77. doi: 10.1158/1541-7786.MCR-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jazaeri AA, Shibata E, Park J, Bryant JL, Conaway MR, Modesitt SC, Smith PG, Milhollen MA, Berger AJ, Dutta A. Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor MLN4924. Mol Cancer Ther. 2013;12:1958–67. doi: 10.1158/1535-7163.MCT-12-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.