

Abstract
When developed as targeting ligands for the in vivo delivery of biomaterials to biological systems, RNA aptamers immediately face numerous obstacles, in particular nuclease degradation and post-selection 2′ modification. This study aims to develop a novel class of highly stable, 2′-fully modified RNA aptamers that are ideal for the targeted delivery of biomaterials. We demonstrated the facile transcription of a fGmH (2′-F-dG, 2′-OMe-dA/dC/dU) RNA library with unexpected hydrophobicity, the direct selection of aptamers from a fGmH RNA library that bind Staphylococcus aureus Protein A (SpA) as a model target, and the superior nuclease and serum stability of these aptamers compared to 2′-partially modified RNA variants. Characterizations of fGmH RNA aptamers binding to purified SpA and to endogenous SpA present on the surface of S. aureus cells demonstrate fGmH RNA aptamer selectivity and stability. Significantly, fGmH RNA aptamers were able to functionalize, stabilize, and further deliver aggregation-prone silver nanoparticles (AgNPs) to S. aureus with SpA-dependent antimicrobial effects. This study describes a novel aptamer class with considerable potential to improve the in vivo applicability of nucleic acid-based affinity molecules to biomaterials.
INTRODUCTION
Currently, there is a significant dearth of targeting ligands that are suitable for conferring ‘smartness’ to biomaterials. Although different classes of targeting ligands, such as small molecules, polypeptide-based peptides/proteins, and nucleic acid-based aptamers, can be used for the targeted delivery of biomaterials, aptamers possess unique advantages. Developed in the 1990s by the Szostak, Gold, and Joyce groups through an in vitro selection process, aptamers are short single-stranded nucleic acids (RNA or DNA) capable of diverse structures with the potential for binding many biochemical and non-biochemical targets, from small molecules to large proteins [1–4]. This ability derives from a nucleic acid library in which every sequence contains 20 to 50 random residues, which determines the diversity of the potential aptamer pool. Although the theoretical diversity is an astronomical figure, the realistic diversity that can be achieved experimentally is typically in the range of 1 × 1013 to 1 × 1015 unique sequences. The high sequence and conformational diversity of the initial nucleic acid pools ensures a high probability of discovering aptamers that bind to numerous targets of interest [5, 6]. Aptamer selection involves iterative rounds of binding to a target of interest, partitioning between binding versus non-binding sequences, and amplification of the enriched target binding aptamers for the next round of selection.
The attractiveness of aptamers as biopolymers for “smart” biomaterial targeting, particularly for in vivo applications, derives from multiple properties that allow aptamers to outperform affinity ligands from other classes. Aptamers are typically non-toxic, non-immunogenic, functionalizable, and chemically synthesizable with little batch variation [7]. Diminutive compared to most biologics and able to form compact structures, aptamers can often bind epitopes, clefts, and enzymatic active sites that are relatively inaccessible to antibodies [8]. Indeed, the selection of aptamers with target-binding affinities higher than natural ligands, typically in the low nanomolar to picomolar range, is routine [9–11]. This is due to the imposition of specific evolutionary pressures on a simplified experimental system by the researcher, instead of natural selection on complex biological systems. Affinities of selected aptamers can be improved further via well-established procedures that reintroduce diversity to the selected aptamer pool, such as via additional selection of aptamer pools generated via doped synthesis of a nucleic acid sequence with demonstrable affinity for the target [12]. Importantly, it is possible to synthesize aptamers with a specific functional moiety, such as a carboxylate, amino, sulfhydryl, or aldehyde, at only one end of the nucleic acid aptamer. This ensures, and greatly facilitates, site-specific conjugation with a wide variety of biomaterials and prevents the formation of heterogeneous mixtures. Development of any RNA aptamer as a potential in vivo biomaterial targeting ligand immediately faces major obstacles that include nuclease degradation, post-selection 2′ modification, and tedious laboratory-scale chemical synthesis and purification involving 2′-hydroxyl protection and deprotection (Figure 1A). Nucleases, highly abundant in the biological fluids of almost any organism, rapidly degrade aptamers derived from natural nucleic acids. Natural RNAs are rapidly and extensively degraded, whereas natural DNAs, although relatively more stable than RNAs, are also quickly degraded by deoxyribonucleases and many nonspecific nucleases, yielding half-lives in the range of 30 to 60 minutes [5]. In general, RNAs are more inclined to fold into stable and complicated secondary and tertiary structures for numerous biological functions [6]. RNA with 2′ modifications exhibits increased resistance to many nucleases by reducing hydrolysis of the phosphodiester backbone [13]. In vitro, natural RNA exhibits half-lives of seconds to a few minutes in various biological fluids including human serum, whereas RNAs partially modified at the 2′ position have extended half-lives of 5 to 15 hours [5, 13, 14]. 2′-O-methyl and 2′-fluoro modifications on pyrimidines and some purines have been commonly used [5, 13–16]. When aptamers intended for an in vivo application are first selected from natural or 2′-partially modified RNA libraries, further stabilization of these aptamers by installation of additional 2′-modified residues is desirable yet challenging. 2′-hydroxyls can participate in aptamer:target interactions or in the structural folding of functional aptamers via hydrogen bonding. Therefore, post-selection replacement of residues bearing 2′-hydroxyls with 2′-modified variants carries a high risk of disrupting the structure and target affinity of the aptamer.
Figure 1.


A) Diagram of the in vivo challenges that 2′-fully modified RNA aptamers can address; B) Diagram of the novel fGmH RNA described in this work.
Direct selection of aptamers from 2′-partially modified (2′-F-dC/dU, 2′-OH-rG/rA: fYrR) RNA libraries partially addresses this problem. For example, anti-VEGF aptamer NX1838, later developed into the only FDA-approved aptamer drug Macugen, was selected from an initial fYrR RNA library. Post-selection, every 2′-hydroxyl purine was replaced with a 2′-O-methyl purine analog one at a time. Each NX1838 variant bearing a new single 2′-O-methyl purine analog was chemically synthesized and tested for target binding. This systematic testing found that all but two purines bearing 2′-hydroxyls could be replaced with 2′-O-methyl purines and maintain target-binding affinity and inhibitory activity against VEGF165-mediated signaling [11, 17]. Though ultimately successful, the chemical synthesis and affinity testing performed via this strategy was exceptionally tedious, time-consuming, and still limited in addressing the problem. Direct selection of 2′-fully modified aptamers from 2′-fully modified RNA libraries provides a highly simplified approach for the development of aptamers intended as in vivo biomaterial targeting ligands.
Existing T7 RNA polymerase mutants do not efficiently generate 2′-fully modified RNAs, making direct selection of such libraries difficult [14, 15, 18, 19]. Work by Sousa and coworkers identified two T7 RNA polymerase mutants (Y639F and Sousa Variant Y639F/H784A) that allow for the efficient accommodation of 2′-F-pyrimidine ribonucleotides as substrates during in vitro transcription [20, 21]. Keefe and coworkers identified a reaction condition that allowed the Sousa Variant to incorporate significant amounts of 2′-OMe-dGTP into transcripts in the presence of 5.7% 2′-OH-rGTP, resulting in RNAs that were mostly fully modified for in vitro selection [14, 18]. To overcome the concern of 2′-OH-rGTP incorporation during transcription, they supplemented the transcribed initial library in the first round of selection with a library chemically synthesized with only 2′-OMe ribonucleotides. Whereas this strategy has a reasonable chance to work, the heterogeneous 2′-OMe-dG/2′-OH-rG nature of this system makes it difficult to rule out the possibility of 2′-OH-rG involvement in the selected aptamer structures. Because of this, time-consuming post-selection functional testing using chemically synthesized 2′-OMe modified aptamer variants should be performed. In addition, it is unknown whether this selection would have been successful if the chemically synthesized all 2′-OMe modified library, with a quality typically much lower than that of enzymatic synthesis, had not been included in the overall first round.
Due to the high electronegativity of fluorine, nucleosides bearing a 2′-F moiety adopt an RNA-type C3′-endo sugar conformation. RNA residues bearing a 2′-F moiety are known to potently increase the hybridization stability between oligonucleotides and RNA at approximately 1.8 °C per 2′-F residue, compared to about 1.3° and 1.0° per residue when 2′-OMe and 2′-OH residues are used, respectively [22]. However, the use of 2′-F residues in RNA does not greatly enhance nuclease resistance, which limited the desired number of 2′-F residues in the RNA composition pursued in this study. Therefore, fGmH RNA aptamers that are composed of one 2′-F and three 2′-OMe residues were hypothesized to have a higher probability of achieving greater conformational and nuclease stability for translational applications. Consequently, this study used a T7 RNA polymerase, described by Diener and colleagues, with the H784A mutation present in the Sousa Variant, as well as a K378R mutation, which was reported as potentially silent by Keefe and coworkers and that we confirmed had no discernible effect on catalysis [18, 23]. Unlike the Sousa Variant, however, this polymerase also bore the Y639L mutation, rather than Y639F. The “LAR” T7 RNA polymerase we used was an attempt to address problems of inefficient non-canonical ribonucleotide incorporation during in vitro transcription. LAR does not require doping of the 2′-X-dNTP pool with 2′-OH-rGTP to produce fGmH RNA with up to 30 to 40 copies and high quality homogeneous fGmH RNA libraries that are truly directly selectable. We then focused on the selection of fGmH RNA aptamers that would have the properties of enhanced hybridization and nuclease stability, discussed above, inherent in their functionality. We found that such fGmH RNAs have unexpected hydrophobicity compared to RNAs composed of all 2′-OMe-dGTP/dCTP/dATP/dUTP, which, therefore, represent a novel class of RNA molecules. The resulting fGmH RNA has some unique features that would greatly increase their ability to confer “smartness” to biomaterials: enhanced stability against many nucleases, increased hydrophobicity compared to other RNAs, and an inability of most endogenous polymerases to utilize the metabolic products of eventual fGmH RNA degradation (Figure 1B).
The following sections will describe a direct selection from an fGmH RNA library with a diversity of 7 × 1013 against a target of interest, S. aureus Protein A (SpA). SpA was chosen because its structure and interaction mode with IgG Fc have been well characterized, and because S. aureus provides a convenient and relevant biological system for biomaterial development. In addition, SpA has become increasingly recognized over the last ten years as a key determinant of S. aureus and, especially, MRSA pathogenesis. SpA has binding capabilities to a diverse number of host factors, such as many subclasses of IgG Fc, von Willebrand Factor (vWF), tumor necrosis factor receptor-1 (TNFR-1), epidermal growth factor receptor (EGFR), and the VH3 class Fabs [24]. Most well-known is the protective role that SpA confers to S. aureus by neutralizing the humoral response via IgG-Fc binding, thereby preventing opsonization. It is clear that highly stable fGmH RNA aptamer anti-SpA nanoparticle ligands are desirable as potential therapeutics and reagents for the study of various aspects of S. aureus biology.
The ability of LAR T7 RNA polymerase to transcribe a DNA library into fGmH RNA without doping of any 2′-OH-rNTPs abrogates the lengthy and limited post-selection optimization process of converting degradation-susceptible natural or 2′-partially modified RNA aptamers into homogeneous 2′-fully modified variants. We demonstrate that fGmH RNA aptamers are capable of stable folded structures that bind SpA with high specificity and affinity. These same aptamers have high stability in harsh, alkaline hydrolysis conditions, and against nucleases present in mouse serum. In addition, a selected aptamer, fmA12, was found capable of binding endogenous SpA present on the S. aureus cell walls, permitting fmA12 to specifically deliver silver nanoparticles (AgNPs) to S. aureus for SpA-dependent cell killing. The direct selection of fGmH RNA libraries, the high stability of fGmH RNA, and the potential for comparatively facile fGmH RNA chemical synthesis compared to conventional nucleic acids represents significant progress in the biomaterial targeting field and increases the potential of smart biomaterials in in vivo applications.
RESULTS
Expression, Purification, and Characterization of His-tagged LAR T7 RNA Polymerase
As discussed above, it is of great interest to develop a novel class of 2′-fully modified RNA aptamers as enhanced biomaterial targeting ligands consisting of a fGmH RNA composition. To generate a T7 RNA polymerase mutant that can efficiently accept 2′-F-dGTP and 2′-OMe-dATP/dCTP/dUTP as substrates, we performed protein engineering to test different combinations of mutant residues reported in the literature [14, 18, 20, 21, 25]. In so doing, we confirmed that T7 RNA polymerase mutant Y639L/H784A/K378R (LAR) was also able to efficiently accommodate a fGmH deoxynucleotide mixture as substrates, though this mutant was previously reported capable of generating RNA composed entirely of 2′-OMe-deoxynucleotides (MNA) [23]. Briefly, the cDNA coding the LAR T7 RNA polymerase with the Y639L, H784A, and K378R triple mutations was generated by site-directed mutagenesis from the vector coding wild-type T7 RNA polymerase (a kind gift from the Sousa lab). The resulting cDNA was cloned into a pDest17 vector downstream of the N-terminal 6 x histidine tag to facilitate affinity purification. As shown in Figure 2A, a recombinant protein of 99 kDa was expressed well and could be purified to homogeneity by using a Co2+-nitrilotriacetic acid (NTA) column. Potential contamination by nucleases and natural rNTPs was examined on a 20% denaturing PAGE gel. Incubation of 5 pmol aliquots of natural RNA with and without purified LAR T7 RNA polymerase did not indicate the presence of contaminating nucleases.
Figure 2.
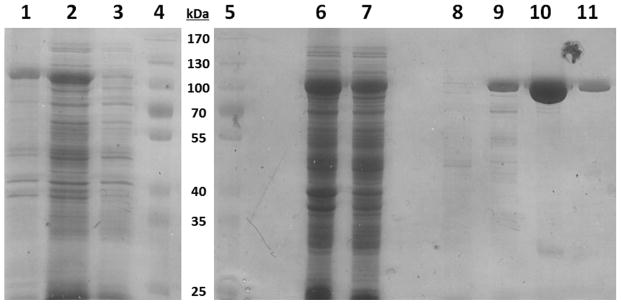
A) Electrophoresis (8%) PAGE gel of the expression and purification of His-LAR T7 RNA polymerase. Lanes 1) Induced insoluble fraction, 2) Induced soluble fraction, 3) Uninduced whole cell lysate, 4) Molecular weight marker, 5) Molecular weight marker, 6) Induced soluble fraction, 7) Flow-through of Talon metal affinity column, 8) Buffer A Wash (No imidazole), 9) Buffer B Wash (20 mM imidazole), 10) Buffer C Elution 1 (200 mM imidazole), 11) Buffer C Elution 2 (200 mM imidazole); B) Transcription efficiency of purified His-LAR T7 RNA polymerase using different 2′-X-dNTPs. Lane 1) fGmH RNA using 2′-F-dGTP and 2′-OMe-dA/U/CTP, 2) fGmH RNA repeat, 3) natural RNA using 2′-OH-rG/A/U/CTP, 4) 2′-OMe-dA/U/CTP, no 2′-F-dGTP, 5) 2′-OMe-dU/CTP, 2′-F-dGTP, no 2′-OMe-dATP, 6) 2′-OMe-dA/CTP, 2′-F-dGTP, no 2′-OMe-dUTP, 7) 2′-OMe-dA/UTP, 2′-F-dGTP, no 2′-OMe-dCTP.
As shown in Lanes 1 to 3 in Figure 2B, when primed with GMP in the presence of PEG-8000, a transcriptional initiator and molecular crowding adjuvant, respectively, His-LAR T7 RNA polymerase variant accepted 2′-F-dGTP and 2′-OMe-dATP/dCTP/dUTP very well, with a relative transcription efficiency of 96% compared to an LAR in vitro transcription reaction using all natural 2′-OH-rGTP/rATP/rCTP/rUTP. This contrasts with the various R425 T7 RNA polymerase mutants developed by Brakmann and coworkers, which demonstrated 4-fold lower activities in the presence of modified nucleotides compared to natural nucleotides [19]. We found that 30 to 40 copies of fGmH RNA were synthesized by His-LAR T7 RNA polymerase in the presence of 2′-F-dGTP and 2′-OMe-dATP/dCTP/dUTP (data not shown). When one of the four 2′-X-dNTPs was not included in the reaction mixture (Lanes 4 to 7), no full-length transcription products were obtained. With the availability of such a T7 RNA polymerase mutant, an fGmH RNA library with a diversity of 7 × 1013 unique sequences was enzymatically synthesized and purified for in vitro selection.
Direct in vitro Selection of fGmH RNA Aptamers that Bind S. aureus Protein A (SpA) from a Highly Diverse fGmH RNA Library
To investigate that 2′-fully modified aptamers can be directly selected from an fGmH RNA library enzymatically synthesized by LAR T7 RNA polymerase, we performed a proof-of-concept selection against S. aureus SpA. SpA was chosen as a model target because of its well-characterized structure, its role as a virulence factor for both antibiotic sensitive and resistant S. aureus, and because S. aureus offered a convenient and relevant biological system for later demonstrations of fGmH RNA aptamer-functionalized AgNPs [24]. Selection utilized magnetic sepharose beads immobilized with purified native Protein A via EDC:NHS conjugation with a density sufficient for a human IgG binding capacity of 27 mg/mL, as per manufacturer specifications. For each round of selection, the input fGmH RNA library, at ~0.7 μM, was incubated with 25 μL (1 column volume) of Protein A beads. Binding was performed in 6 column volumes of 1× Aptamer Selection Buffer for 1 h at 4 °C. Beads were then washed twice in 6 column volumes of 1× Aptamer Selection Buffer, each for 5 min at 4 °C. Presumably, the washing (12 column volumes total) removed sequences bound non-specifically, as well as target-binding sequences with rapid dissociation rates. Aptamers were eluted three times from the beads in a total of 18 column volumes of 1× Aptamer Selection Buffer, each for 20 min at 4 °C. This longer and extensive elution was designed to select aptamers with slower dissociation rates.
We used the Octet BLI method to monitor the evolution of desired binding characteristics within the selected pools during the selection. As shown in Figure 3A, enrichment of the SpA-binding aptamers was not detectable by this binding assay before Round 4. Binding was detected in Round 5, with a limited further increase in Round 6. Such phenomena are typical for SELEX. Cloning and sequencing, as described, were performed on Rounds 5, 6, 7, and 8 selected pools (N = 154) (Figure 3B). Despite the small sequencing sample size, the evolution of five distinct primary sequence groups was evident across these pools.
Figure 3.

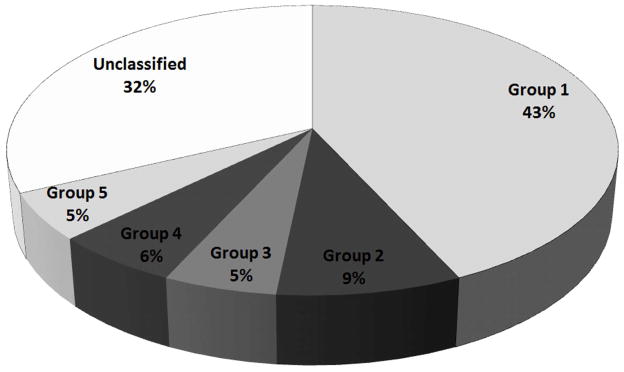
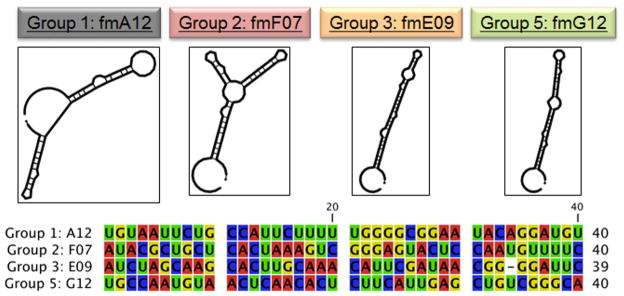
A) Real-time monitoring of the enrichment of fGmH RNA aptamers during the selection using Octet BLI; B) Percentages of sequences classified as one of the five primary sequence groups; C) The putative secondary structures and random region sequences of aptamers fmA12, fmF07, fmE09, and fmG12, representing Groups 1, 2, 3, and 5, respectively.
Sequences A12, F07, E09, E03, and G12, representing primary sequence groups 1–5 (Figure 3C), respectively, were prepared for kinetic characterization via PCR amplification followed by transcription using LAR T7 RNA polymerase. To measure the target-binding affinity, fGmH aptamers fmA12 and fmG12 were kinetically characterized initially via Biacore SPR using SpA immobilized on CM5 chips. It was found that the KDs for fmA12 and fmG12 were approximately 95 nM and 475 nM, respectively (data not shown). These results were confirmed via Octet BLI, and the binding curves measured for fmA12, the aptamer extensively used in the remainder of this study, are provided in Figure 4 and demonstrate saturable and monophasic binding. Due to the requirement of examining many different sequences and their variants, subsequent kinetic characterizations were performed using Octet BLI, which allowed the binding of 8 samples to SpA to be measured and analyzed in parallel. As listed in Table 1, fmA12 and fmG12 on Octet yielded KDs of 67.1 ± 6.1 nM and 220 ± 24 nM, respectively, which are comparable with those determined by Biacore. BLI analysis for fmF07 and fmE09 resulted in KDs of 38.9 ± 4.1 nM and 418 ± 112 nM, respectively, whereas fmE03 was found to be a non-binding sequence. These fGmH aptamers feature KDs that are reproducible in quadruplicate: a property that likely resulted from fGmH sequences forming stable structures.
Figure 4.

BLI kinetic characterization of fmA12 using biosensors immobilized with SpA.
Table 1.
Dissociation constants of fGmH RNA aptamers binding S. aureus SpA.
| Aptamer | Group | kON (Ms−1) | kOFF (μs−1) | KD (nM) |
|---|---|---|---|---|
| A12 | 1 | 12300 ± 207 | 823 ± 148 | 67.1 ± 6.1 |
| F07 | 2 | 8810 ± 901 | 333 ± 17 | 38.9 ± 4.1 |
| E09 | 3 | 3400 ± 292 | 1330 ± 276 | 418 ± 112 |
| G12 | 5 | 4930 ± 345 | 1080 ± 112 | 220 ± 24 |
Selected fGmH RNA SpA Aptamers are Specific for SpA, and the 2′ Modification States of the Selected Aptamers is Critical for SpA Affinity
To test the target-binding specificity of the selected fGmH RNA aptamers, we used Octet BLI binding assays to examine the affinity of these aptamers to S. aureus SpA and Protein G (SasG), and P. magnus Protein L (PpL), three bacterial proteins that have similar IgG binding features. As shown in Figure 5A, the previously characterized SpA-binding aptamer fmA12 did not exhibit measurable affinity for SasG or PpL. fmE09 and fmG12 yielded similar results (data not shown). fmF07, however, displayed minimal binding to both SasG and PpL, though with much lower affinity than SpA (2.76 ± 2.39 μM for SasG and 452 ± 92 nM for PpL, compared to 38.9 ± 4.1 nM for SpA). It is presumed that fmF07 is capable of binding a structurally conserved region present in all three proteins. However, the results for fmA12, fmE09, and fmG12 clearly indicate that selected fGmH RNA aptamers are capable of interacting specifically with the target used in selection.
Figure 5.


A) Binding specificity of fmA12 to Protein A, Protein G, and Protein L; B) Protein A (SpA) binding dependence of sequence A12 on the RNA 2′ modification state (WT A12 (wild-type; rN), bmA12 (fYrR), tmA12 (rGmH), fmA12 (fGmH)).
To assess whether the affinity of fGmH RNA aptamers for SpA is dependent on 2′ modifications, Octet BLI binding assays were performed comparing four A12 RNA compositions, each of a different 2′ modification state: rN RNA (every residue bears 2′-OH; WT A12), fYrR RNA (2′-F modifications on pyrimidines; bmA12), rGmH RNA (2′-OMe modifications on A/C/U; tmA12), and fGmH RNA (fmA12). As shown in Figure 5B, only 2′-fully modified fmA12 produced a binding curve matching those observed during kinetic characterization, whereas none of the unmodified (WT A12) or partially modified (bmA12 and tmA12) exhibited detectable SpA affinity. It is apparent that the introduction of 2′-hydroxyls (even when those hydroxyls are restricted to guanine residues) into A12 is sufficient to perturb and abolish aptamer binding to SpA. These results indicate that once selected, fGmH RNA aptamers must maintain their 2′ modification state to preserve target binding. As such, fGmH RNA aptamers constitute a distinct aptamer class. The lack of detectable tmA12 functionality also ruled out the concern that fmA12 guanine residues have heterogeneous 2′-hydroxyls, potentially from trace contamination of 2′-OH-rGTP in 2′-F-dGTP or rGMP used for transcription.
fmA12 can be Structurally Truncated while Preserving SpA Affinity
All identified SpA-binding aptamers have full-length sequences of 85 residues. To map the target-binding regions and facilitate future large-scale chemical synthesis using an oligonucleotide synthesizer, the high affinity SpA-binding aptamer fmA12 was shortened via comprehensive testing of nine truncation and stem stabilization variants (fmA12Δ1 through Δ9). Truncations fmA12Δ1/2/4/5/7/8 resulted in global disruption of the putative fmA12 secondary structure, as predicted by Mfold (data not shown). Truncations fmA12Δ3/6/9 successfully excised portions of full-length fmA12 without disrupting the putative secondary structure (Figure 6). Truncation fmA12Δ3 removed the last five residues at the 3′ end (red sequence in Figure 6), which appeared not to be involved in the stem or loop secondary structures as predicted by Mfold. Indeed, fmA12Δ3 affinity for SpA was preserved compared to full-length fmA12 with a KD of 69.7 ± 3.5 nM. Truncation fmA12Δ6 maintained Stem-loop Domain I (blue sequence in Figure 6) but completely removed Stem-loop Domain II (green sequence in Figure 6), resulting in more than a 1,000-fold reduction in binding affinity (KD of 63.1 ± 7.3 μM). Truncation fmA12Δ9 was designed with the fmA12Δ3 acaulescent Stem-loop Domain II and the 17-residue single-stranded region at the 5′ end removed (orange sequence in Figure 6). The resulting fmA12Δ9, with a length of 61 residues, exhibited preserved SpA binding with a KD of 69.1 ± 16.7 nM - almost identical to that of the full-length fmA12. Sequence truncation analysis indicated, in summary, that Domain I alone is insufficient for binding, and, presumably, that both domains are necessary to achieve high affinity SpA binding. Overall, sequence truncation found a version of full-length fmA12 (fmA12Δ9) that could be chemically synthesized if future need arose. For the purposes of this study, no further truncation was necessary though future applications may require different sequence length optimizations dependent on the features desired from the aptamer.
Figure 6.
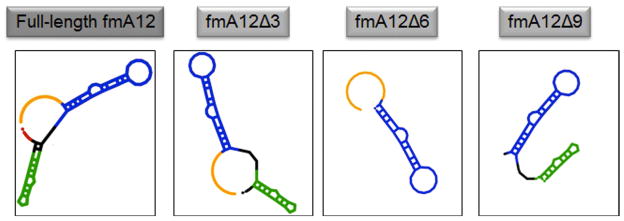
The predicted secondary structures of fmA12Δ3, fmA12Δ6, and fmA12Δ9 compared to that of full-length fmA12.
fGmH RNA Maintains High Stability Against In Vivo Degradation
One of the major challenges affecting the potential in vivo application of aptamers as biomaterial targeting ligands is the rapid degradation of natural nucleic acids by various nucleases present in almost every biological fluid. The enhanced stability of fYrR RNA bearing 2′-fluoro modifications on pyrimidines is well known with half-lives of 5 to 15 hours in serum [5, 13]. As mentioned in the Introduction, we pursued the fGmH RNA composition for the enhanced hybridization stability conferred by the 2′-F moiety on guanine. However, because this moiety does not greatly increase nuclease stability, the degradation susceptibility of fGmH RNA was investigated.
To compare fGmH RNA stability to the known 2′-fluoro-pyrimidine modified RNA composition, as well as other RNA compositions, A12 DNA template was transcribed into the same previously mentioned four RNA compositions of varying 2′ modification state: rN (WT A12), fYrR (bmA12), rGmH (tmA12), and fGmH (fmA12). These four A12 RNA variants were incubated in 10% mouse serum at 37 °C over 24 h (Figure 7A). As expected, the WT A12 variant proved unstable in serum within 5 minutes. fmA12 demonstrated much higher stability with 98% survival after five minutes. For bmA12, tmA12, and fmA12, gradual losses of the major bands were observed over the 24 hour incubation, with 31% bmA12, 59% tmA12, and 76% fmA12 surviving after 24 h. Linear regression curves fitted to these data indicate a T½ of ~15 h for bmA12, which agrees well with that reported in the literature [5]. Significantly, regression analysis predicts a T½ of ~29.5 h for tmA12 and, significantly, ~52 h for fmA12. Incubation of these RNAs in 50% serum produced similar results (Figure 7B). After 2 h, WT A12 is completely degraded (not detectable) and 23.7% bmA12, 71.6% tmA12, and 97.4% fmA12 survives. fmA12 exhibits a serum stability that was more than 4-fold greater than bmA12 after 2 h. After 5 h, WT A12 is completely degraded (not detectable), bmA12 is completely degraded (not detectable), and 40.7% tmA12 and 85.0% fmA12 survives. Because bmA12 of the canonical fYrR RNA composition was no longer detectable after 5 h, no fold comparison is possible between bmA12 and fmA12. However, after 5 h, fmA12 proved more than twice as stable as tmA12. These results indicate that despite some fmA12 degradation, the 2′-fully modified aptamer variant features superior pan-nuclease stability compared to the assayed RNA variants with lower 2′ modification states.
Figure 7.


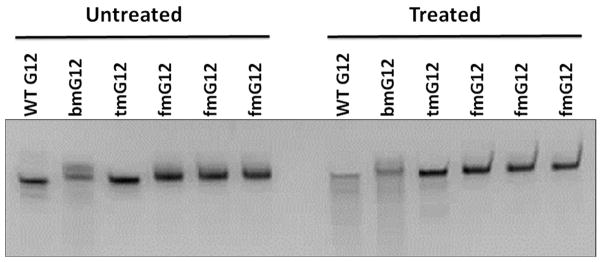
A) Percent survival of A12 RNAs of varying 2′ modification state in 10% serum (rN (WT A12), fYrR (bmA12), rGmH (tmA12), fGmH (fmA12)); B) Percent survival of A12 RNAs of varying 2′ modification state in 50% serum after 2 h and 5 h incubation at 37 °C; C) Degradation of G12 RNAs of varying 2′ modification state in alkaline hydrolysis conditions.
We further examined the in vitro stability of fGmH RNA aptamers against nucleases that degrade RNA via formation of a 2′, 3′-cyclic phosphate intermediate. To address this question, G12 DNA was in vitro transcribed into different 2′ modification states matching those described above for A12. The four G12 variants were incubated at 95 °C under alkaline conditions (pH 9.2) for 10 min. Degradation products were observed below the major bands for WT G12, bmG12, and tmG12 (Figure 7C). However, fmG12 did not yield any detectable degradation products under these harsh conditions. Percent survival was calculated by quantifying and comparing the treated and untreated fGmH RNA major bands, which indicated an fmG12 survival of 99.25 ± 4.86%. The results demonstrate that the substitution of all 2′-hydroxyls with a combination of fluoro and O-methyl groups effectively abrogates degradation by nucleases forming 2′, 3′-cyclic phosphate intermediates.
fmA12 Binds Specifically to S. aureus Cells with High SpA Expression
To demonstrate the biological application of fmA12 as a targeting ligand for biomaterials such as AgNPs, confocal microscopy was performed to visualize fmA12 binding to S. aureus cells with both high and low SpA expression [26, 27]. Full-length fmA12 and tmA12, both intercalated with SYBR Green I for signal detection, were incubated with S. aureus strains 12598 (high SpA expression) and 10832 (low SpA expression). Nonfunctional tmA12 incubation with strain 12598, and functional fmA12 aptamer incubation with strain 10832 served as negative controls. Incubation of strains 12598 and 10832 with 10 nM SYBR Green I showed no detectable fluorescent signal, indicating that the signal observed later from fmA12 samples was not due to background fluorescence [28]. As shown in Figure 8, 500 nM fmA12 produced a strong fluorescent signal upon incubation with high SpA expressing strain 12598. Compared to fmA12, 500 nM tmA12 yielded an approximately 90% reduced fluorescent signal upon binding to strain 12598. Neither fmA12 nor tmA12 exhibited detectable signal above background upon incubation with low SpA expressing strain 10832. These results indicate that fmA12 can bind endogenous S. aureus SpA expressed in situ, and fmA12 can be used for cellular applications.
Figure 8.

Confocal analysis of the binding of functional fmA12 and non-functional tmA12 to S. aureus cells with high (Strain 12598) and low (Strain 10832) SpA expression.
Functionalization of AgNPs in Preparation for S. aureus Antimicrobial Assays
As a further demonstration of the applicability of fGmH RNA to in vivo applications on biomaterials, fmA12 was used as a targeting ligand for delivery of antimicrobial AgNPs to S. aureus cells. AgNPs were chosen because of their diverse antimicrobial mechanisms: reactive oxygen species (ROS) generation, bacterial cell wall permeability and respiration perturbation via direct contact, and Ag+ shedding resulting from oxidation [29]. This study used AgNPs with 10 nm average diameter because of their superior Ag+ shedding rate compared to those with larger diameters [30]. AgNP-Aptamer buffer was formulated with acetate salts because AgNP antimicrobial activity decreases in buffers containing chloride and phosphate, presumably because silver acetate features superior aqueous solubility (Ksp = 1.94×10−3) compared to silver chloride and silver phosphate (Ksp = 1.77×10−10 and 8.89×10−17, respectively) [31].
To introduce fmA12 aptamers to AgNPs, either full-length fmA12 or fmA12Δ9 functionalized with a 5′ amino group would have been a strategy for direct functionalization. However, the presence of a 17 nt region at the 5′ terminus of fmA12 that was not involved in SpA binding prompted the use of a hybridization approach whereby a DNA oligo, antisense to the 5′ terminus of fmA12 and synthesized with a primary amine for later conjugation to AgNP surfaces, could serve as both a capture oligo and a flexible spacer to maintain fmA12 function upon immobilization on the AgNP surface. It was found, however, that replacement of GMP during in vitro transcription with GMP analogs bearing a 5′ biotin or amine resulted in lower yield (data not shown). Fortunately, the 5′ single-stranded region (17 residues) of full-length fmA12 is not necessary for aptamer structure or target binding, as demonstrated during sequence truncation analysis. Therefore, we pursued a strategy reliant on enzymatic synthesis for the precise and robust generation of both full-length fmA12 and tmA12. This strategy was compatible with an antisense oligo hybridization approach that functionalized AgNPs with a capture oligo complementary to the 5′ single-stranded region of full-length fmA12 and tmA12 (Figure 9A). This antisense capture oligo was synthesized with a 3′ amine. Reaction of the 3′ amine with carbon disulfide formed a dithiocarbamate, which are adsorbed much more strongly onto gold nanoparticles (AuNPs) than monothiols [32, 33]. We found that the capture oligo functionalized with dithiocarbamate was also adsorbed very well onto AgNPs. The reaction was characterized via measurement of the UV/Vis spectrum (Abs220–750) with a NanoDrop with subsequent analysis of peaks produced by AgNPs and capture oligo using the Beer-Lambert Law. Incubation of dithiocarbamate-functionalized capture oligos at a 1000:1 molar excess over AgNPs while gradually increasing the [Na+] of the conjugation buffer over 28 hours to a final concentration of 300 mM yielded a molar ratio of capture oligo:AgNP of ~40:1, a AgNP percent recovery of 88% was obtained after conjugation. Conjugation of the capture oligos to the AgNPs shifted the absorption maximum of the nanoparticles from 390 nm to 410 nm and decreased the height of the spectral peak (data not shown), which were consistent with observations reported by Tokareva and Hutter [34].
Figure 9.


A) Diagram of the binding of fmA12-functionalized AgNPs to SpA on the surface of BLI biosensors; B) BLI kinetic characterization of fmA12-functionalized AgNP SpA binding; C) S. aureus cell killing effect of fmA12-directed AgNPs based on CFU analysis: 1) AgNP-Aptamer \Buffer only, 2) Citrate-capped AgNPs alone, Unwashed, 3) AgNP-Capture:fmA12, 4) AgNP-Capture:tmA12, 5) fmA12 alone.
We found that the conjugated capture oligos stabilized the AgNPs in multiple physiological buffers, including LB broth, and allowed facile fmA12 docking by sense-antisense hybridization. This stabilization manifested as the maintenance of a characteristic AgNP peak between 390 nm and 410 nm, a visual brown color in solution (disperse AgNPs appear brown in solution, whereas aggregated AgNPs become gray), and the lack of an AgNP aggregation pellet after centrifugation at 3,000 x g. A fmA12:AgNP molar ratio of 10:1 yielded functional binding of the AgNPs to SpA with a 3 to 4-fold improvement as compared to fmA12 alone. BLI binding analysis showed that both ķON and ķOFF of the fmA12 functionalized AgNPs were slower (ķON of 2.68×103 Ms−1; ķOFF of 5.29×10−5 s−1) compared to free fmA12 alone (ķON of 1.23×104 M−1s−1; ķOFF of 8.23×10−4 s−1) (Figure 9B). The slower fmA12-AgNP association rate could be attributed to the slower diffusion rate of the approximately 60-fold greater molecular mass of the AgNPs (1.7 MDa) compared to fmA12 aptamer alone (28 kDa). However, the greater than 15-times slower dissociation rate could be due to the avidity effect from multiple fmA12 aptamers on the same nanoparticle interacting with SpA on the biosensor surface (Figure 9B). These results indicate that AgNPs functionalized with fmA12 can be used in physiological buffers for targeted delivery of antimicrobial nanoparticles.
fmA12 can Deliver AgNPs Specifically to S. aureus in a SpA-dependent Manner
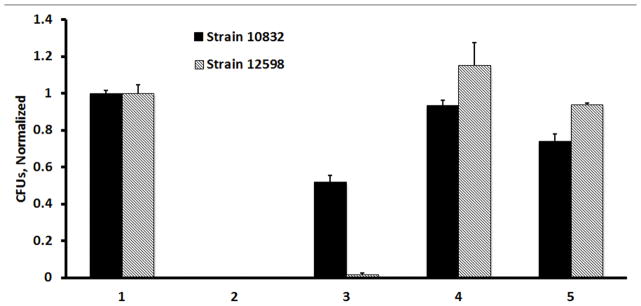
In a further demonstration of fGmH aptamer applicability to in vivo studies, fmA12 was used as a targeting ligand for specific delivery of antimicrobial AgNPs to S. aureus cells. S. aureus strains 10832 and 12598 were cultured overnight in LB broth as described. 1 × 106 CFUs of cells from each strain were incubated under the following conditions, in quadruplicate: AgNP-Aptamer buffer only (negative control), citrate-capped AgNPs alone - not removed from cells during the assay (positive killing control), fmA12-functionalized AgNPs, tmA12-functionalized AgNPs (negative control), and fmA12 alone (negative control). Each sample used the same concentration of AgNPs (except for fmA12 alone, which did not use AgNPs) and A12 RNA (either fmA12 or tmA12, except for positive control, which used non-functionalized, citrate-capped AgNPs).
The killing effect of each preparation above was evaluated by directly counting the CFUs and normalizing them against AgNP-Aptamer Buffer Only control. CFU counts for the tmA12-functionalized AgNPs negative control were indistinguishable from AgNP-Aptamer buffer only control, indicating that non-specific S. aureus cell killing from AgNPs was negligible. The same was observed for fmA12 aptamer alone, which gave a 93.5 ± 1.1% survival for strain 12598. However, fmA12 aptamer alone gave a 74.0 ± 2.2% survival for strain 10832 (Figure 9C). Though this result was unexpected, it is unlikely to be the result of non-specific binding or hydrophobicity. If either of these possibilities were real, then fmA12-functionalized AgNP cell killing would have been enhanced for Strain 10832 cells to levels equivalent to those observed for Strain 12598 cells. In addition, fmA12 alone would be expected to induce some cell killing in Strain 12598 cells. Neither of these results were observed. Instead, fmA12-functionalized AgNPs resulted in specific S. aureus antimicrobial activity with only a 1.6 ± 1.0% survival of 12598 cells and 51.9 ± 2.0% survival of 10832 cells. High inhibition of strain 12598 cell growth is likely due to high SpA expression, which would recruit and enhance fmA12-targeted AgNPs binding compared to strain 10832, resulting in greater antimicrobial action. Some growth inhibition of strain 10832 was not surprising because this strain still expresses SpA but at a much attenuated level [27]. Overall, fmA12 was able to target AgNPs specifically to S. aureus cells and inhibit cell growth in a SpA-dependent manner. Regarding the apparent reduction of survival for strain 10832 treated with fmA12 aptamer alone (74.0 ± 2.2%), this result may be explained by saturation of the low SpA levels present on the surfaces of these cells with fmA12. Schreiber and coworkers recently reported an effect where a SpA-deficient S. aureus strain demonstrated reduced adhesion to tested biomaterials due to saturation of available SpA by serum immunoglobulins [35]. Therefore, it is possible that fmA12 alone saturated the SpA available on strain 10832 cell surfaces and slightly affected the ability of this bacteria strain to adhere to LB agar media, leading to smaller colonies and fewer CFU counts. This effect, if real, would be even more pronounced for 10832 cells treated with fmA12-AgNPs due to the larger size of the nanoparticles. Therefore, the 51.9 ± 2.0% survival of 10832 cells treated with fmA12-AgNPs are due to either increased saturation of available SpA or reduced delivery of AgNPs to these cells compared to strain 12598 cells. For either of these explanations, the effect of fmA12 alone or fmA12-AgNPs on strain 10832 cells is negligible, as would be expected for SpA-deficient cells.
DISCUSSION
The 2′-fully modified RNA aptamers described in this work can be readily synthesized using an oligo-synthesizer to introduce a wide variety of functional groups at the 5′ or 3′ terminus, including carbonyl-reactive hydrazides or alkyoxyamines, amine-reactive activated carboxylates or imidoesters, sulfhydryl-reactive maleimides, haloacetyls, pyridyl disulfides, and clickable groups, such as azides, alkynes, or phosphines that can undergo bioorthogonal reactions [36]. Therefore, many other classes of biomaterials or nanomaterials can be targeted with fGmH aptamers, as far as they can be functionalized with a chemical group that is compatible with that on the aptamer. The chemistry available to aptamers also permits site-specific conjugation to biomaterials with correct orientation, a capability that is difficult to achieve for antibodies. This property, combined with the high target selectivity demonstrated by many aptamers, and the potential for improved bioavailability as suggested by the fGmH RNA stability data, indicates that fGmH aptamers would prove superior to peptides, protein domains, antibodies, and other targeting ligands in conferring smartness to biomaterials in vivo and ex vivo. As this study demonstrates, fGmH aptamers are well-suited to conferring smartness to silver nanoparticles. It would not be a large logical leap to assume that such aptamers would also outperform aptamers of lower 2′-modification aptamers and antibody-based affinity molecules that have already been used to functionalize various biosensors, including those on atomic force microscopy, graphene, BLI, and SPR chips [37–40]. fGmH aptamers, specific to a variety of cancer biomarkers, could be used to decorate microfluidic platforms or biomimetic hydrogels for the capture and potential culture of circulating tumor cells (CTCs) derived from a plethora of cancer types, similar to that reported by using EpCAM antibodies [41–43]. fGmH aptamers could also be logically applied to proteome profiling, particularly the highly sensitive single cell detection of cancer cells via a single-cell barcode chip (SCBC) [44]. In this instance, a single fGmH aptamer bearing a 5′ or 3′ tail can serve both docking and affinity functions, and with lower cost compared to antibodies. Such advantages could also improve the performance of lab-on-a-chip approaches, such as magnetophoretic digital circuits for single cell or molecular control, and for tuning the biological properties of nanofibrous scaffolds and hydrogels used in tissue engineering for regenerative medicine [45, 46].
The concept of direct in vitro selection of 2′-fully modified fGmH aptamers, intended for targeted biomaterial delivery, from in vitro transcribed fGmH RNA libraries was proven using S. aureus SpA: a structurally well characterized binding partner of human Fc, vWF, TNFR-1, EGFR, and VH3 class Fabs. The results obtained from target binding analysis clearly demonstrate that fGmH RNA aptamers with high affinity and specificity can be directly selected from an fGmH RNA library with high diversity. Prior to selection, we hypothesized that RNAs of this 2′-modified composition would have enhanced structural stability – a desirable property in an aptamer. The successful evolution of fGmH aptamers with high affinity and specificity to SpA proved that, at least, this composition did not disrupt the ability of RNA sequences to form stable structures. Octet BLI proved invaluable for assaying aptamer pool enrichment. Traditionally, the characterization of aptamer pool enrichment requires radio-labeling aptamers with either ATP [α-32P] (via in vitro transcription) or ATP [γ-32P] (via 5′ end labeling). Transcription using ATP [α-32P] is not desirable for fGmH aptamers because the replacement of 2′-OMe-dATP with 2′-OH-ATP [α-32P] would change the 2′ moiety of some adenosine residues that would likely disrupt aptamer function. 5′ end radiolabeling is compatible with fGmH aptamers, but it adds length to the binding assay procedure. Octet BLI, however, is a 96-well plate-based and label-free method of affinity testing that can be performed in parallel for instantaneous measurement and simultaneous comparison of samples from different selection rounds.
Interestingly, fGmH RNAs almost completely partitioned into the organic phase during phenol:chloroform extraction, whereas a synthesized MNA (all 2′-OMe) version of the same sequence did not (data not shown), suggesting a higher hydrophobicity compared to other 2′-partially modified RNAs, including fYrR, rGmH, and MNA variants. This unique property observed only in fGmH RNAs described in our work makes it impossible to separate fGmH RNAs from proteins via widely used phenol:chloroform extraction. To address the problem, we adopted a procedure to denature the proteins in the reaction mixture by heating. Importantly, the enhanced hydrophobicity did not abolish target specificity amongst the selected aptamers. Considering that a distinctive feature of conventional nucleic acid-based aptamers is their extreme hydrophilicity, the hydrophobicity of fGmH RNA reinforces these polynucleotides as a unique aptamer class. We hypothesize that the enhanced fGmH RNA hydrophobicity allows fGmH RNA aptamers to interact with their binding targets with more hydrophobic interactions, a capability that would expand the repertoire of interactions between nucleic acid aptamers and their targets. In the future, it would be of great interest to investigate the effect of this hydrophobicity on fGmH RNA aptamer structures and aptamer:target interactions.
Structurally, the four kinetically characterized aptamers resulting from SpA selection can be divided into two pairs: high affinity aptamers fmA12 and fmF07, and low affinity aptamers fmE09 and fmG12. Within the pairs, aptamers show considerable secondary structure similarity (Figure 3C), as predicted by MFold, despite significant primary sequence differences. fmA12 and fmF07 both have two stem-loop domains, whereas fmE09 and fmG12 have only one. Though the SpA epitopes interacting with these aptamers are unknown, fmA12 and fmF07 have dissociation constants 5- to 10-fold lower than fmE09 and fmG12, implying that the two predicted stem-loop domains could be enhancing target binding via avidity.
Binding of the selected aptamers to SpA does not appear tolerant of post-selection replacement of 2′-moieties (fluoro or O-methyl) with hydroxyl groups. When A12 DNA was transcribed into rN, fYrR, rGmH, and fGmH RNA, only 2′-fully modified fGmH fmA12 demonstrated SpA binding. It is presumed that functional dependence of fmA12 on the 2′ modification state of fGmH RNA is related to its structure. Specifically, post-selection replacement of fGmH RNA 2′-moieties with 2′-hydroxyls likely alters intra- and/or intermolecular hydrogen bonds (H-bonds), though intramolecular charge distributions may also be altered inductively. Unlike 2′-hydroxyls, 2′-fluoro and 2′-O-methyl groups can only accept H-bonds. Post-selection installation of 2′-hydroxyls, acting as both H-bond acceptors and donors, have the potential to rearrange the network of hydrogen bonds across the entire RNA sequence, thereby altering the aptamer original structure. It is likely, however, that short regions or individual residues within an fGmH RNA aptamer are tolerant of 2′-moiety changes. However, fGmH aptamer fmA12 lost affinity to SpA upon transcription into RNA variants other than fGmH. In the case of rGmH tmA12, where natural 2′-OH-rGTP is used instead of 2′-F-dGTP, every 2′-fluoro modified guanine residue was changed to natural guanines. Such substitution, even constrained only to guanine residues, would represent a global change to the hydrogen bonding network. It is assumed, therefore, that such a change almost always results in structural disruption and loss of target binding. It is assumed too that using mixed ribonucleotides during transcription (e.g. a mixture of 2′-OH-rGTP and 2′-F-dGTP) would yield aptamers with no or reduced affinities. Under such a situation, the incorporation of either 2′-OH-rGTP or 2′-F-dGTP at any given position would be stochastic or, more likely, that LAR T7 RNA polymerase would preferentially incorporate 2′-OH-rGTPs, making it difficult to attribute the loss of binding affinity.
The direct selection of fGmH RNA libraries nullifies the need for post-selection replacement of 2′-hydroxyls, but the selected fGmH RNA aptamers also hold other significant advantages over aptamers selected from natural or 2′-partially modified RNA libraries. One potential advantage involves chemical synthesis, which is tedious at the laboratory scale for natural and 2′-partially modified RNA because 2′-hydroxyls require protection and deprotection. In contrast, the 2′-F and 2′-OMe groups on fGmH RNA do not require protection. This would potentially greatly simplify chemical synthesis and post-synthesis deprotection of the selected fGmH RNAs using protocols as simple as those used for DNA.
Another advantage of fGmH RNA aptamers is that the 2′ modifications enhance aptamer resistance to nuclease degradation compared to natural and partially-modified RNA aptamers while introducing a 2′-F moiety for strong H-bond interactions that yield greater hybridization stabilization. A significant class of nucleases degrade natural RNA (and partially modified RNA to a lesser extent) via a SN2 reaction wherein the 2′-hydroxyl serves as a nucleophile to form a 2, ′ 3′-cyclic phosphate intermediate [47, 48]. Results from harsh alkaline hydrolysis at 95 °C, which mimics this cleavage, demonstrated the unusual high stability of selected fGmH RNA aptamers.
Alkaline hydrolysis, however, is not representative of the plethora of mechanisms used by the panoply of nucleases present in biological fluids. fGmH RNA still exhibited superior serum stability after 24 hours (76% survival) compared to rGmH RNA (59%), fYrR RNA (31%), and rN RNA (~0%), indicating a 17% and 45% increase in survival of fGmH RNA over rGmH RNA and fYrR RNA in serum, respectively. The significantly increased half-lives of fGmH RNA over the widely used fYrR RNA, in both alkaline hydrolysis and serum, clearly indicate that fGmH RNA would be a superior class of in vivo reagent compared to fYrR RNA. The minor amount of fGmH RNA degradation observed in serum is not surprising considering the presence of numerous nonspecific nucleases utilizing various mechanisms of action. Translationally, some nuclease sensitivity may be desired because intact fGmH RNA aptamers and their associated nanoparticles that persist may yield currently unknown deleterious effects in vivo.
After successfully proving fGmH RNA aptamer selection and examining the stability of fGmH RNA, the biological application of selected fGmH aptamer fmA12 was demonstrated. Historically, aptamer affinity for endogenous targets is not certain if in vitro selection was performed against a different version of the target protein. For example, Sullenger and coworkers found that aptamers selected against purified, recombinant EGFRvIII ectodomain expressed in Escherichia coli were incapable of binding endogenous EGFR on cancer cell surfaces due to differences in glycosylation [49]. Though SpA glycosylation was not a concern here, it was posited that SpA epitope structure and accessibility differences could exist between the SpA immobilized on the magnetic sepharose beads and endogenous S. aureus SpA on cell surfaces. Cell binding analysis using confocal microscopy addressed this concern, revealing that fmA12 could bind to endogenous SpA present on S. aureus. This experiment also indicated that fGmH RNA aptamers could function in a biologically-relevant environment.
fmA12 was then applied, as intended, as an AgNP targeting ligand for the selective killing of S. aureus cells. AgNPs were chosen for their antimicrobial characteristics, especially for the generation of Ag+ that disrupts multiple bacterial metabolic processes. Attempts to deliver AgNPs specifically to bacteria in biological fluids face a significant challenge: the maintenance of nanoparticle dispersity. Citrate-capped AgNPs alone aggregated irreversibly upon addition to LB broth or in buffers with NaCl as low as 2 mM, as described by the Derjaguin-Landau-Verwey-Overbeek (DLVO) theory [50]. A stabilizing agent, such as nucleic acids conjugated to the nanoparticle surface, is critical when using AgNPs under physiological conditions. To simultaneously stabilize and functionalize AgNPs, nanoparticles were first functionalized with a short DNA capture oligo. Optimization of capture oligo concentration during AgNP conjugation yielded approximately 40 capture oligos per AgNP. At the level of individual AgNPs, however, it is likely that functionalization with capture oligos follows a more Gaussian distribution, though this was not characterized in this study. Following this assumption, the stability of any given functionalized AgNP against [Na+] should also follow this Gaussian distribution. These potential AgNP stability differences, if real, however, did not have an effect of sufficient significance to be observed because overall, functionalized AgNPs were satisfactorily stabilized in physiological buffers, and featured tunable hybridization-mediated aptamer immobilization on AgNP surfaces.
AgNPs immobilized with fmA12 or tmA12 remained dispersed in buffer, and unbound AgNPs were easily separated from the bacterial pellet. Indeed, AgNPs docked with nonfunctional tmA12 demonstrated significantly lower bacterial killing compared to those docked with SpA-binding functional fmA12. This is because the AgNPs docked with tmA12 were easily removed during washing, whereas AgNPs docked with functional fmA12 aptamers, which tightly interacted with SpA on S. aureus cell surfaces, were not. These results also mitigated concerns that AgNP functionalization would interfere with Ag+ shedding. Obviously, either the effect of such interference was non-existent or not sufficiently strong to be evident in the obtained results. Instead, this study determined that the antimicrobial effect produced by functionalized AgNPs, unlike aggregated citrate-capped AgNPs, was SpA-dependent and specific.
In the future, it will be of great interest to study the secondary and tertiary structures of fGmH RNA aptamers in greater detail compared to lower 2′ modified variants. Such investigations, potentially involving DMS footprinting, x-ray crystallography, or NMR structure determination, may yield insights into the hydrogen bonding network driving fGmH RNA aptamer structure. In addition, the role, if any, that enhanced fGmH RNA hydrophobicity plays in the formation of intermolecular contacts should be elucidated. Future studies should focus on the development of fGmH RNA aptamers to other biologically-relevant targets for the expansion of the biomaterial targeting repertoire.
CONCLUSIONS
This study has demonstrated the successful direct selection of fGmH RNA aptamers with high target binding affinity and specificity from a 2′-fully modified RNA library transcribed by a T7 RNA polymerase mutant. Results from stability analysis revealed the high stability of fGmH RNA aptamers against alkaline hydrolysis, as well as many of the nucleases present in serum as compared to RNAs with different 2′ modification states. The selected fGmH RNA aptamers could have great potential in conferring smartness to biomaterials as demonstrated by their use as a targeting ligand for antimicrobial AgNP delivery to S. aureus cells in a SpA-dependent manner. The system described in this work permits the rapid selection of 2′-fully modified fGmH RNA aptamers that do not require lengthy and limited 2′ modification after selection. In addition, the protection and deprotection of 2′-hydroxyls is completely unnecessary, which would greatly facilitate the large-scale chemical synthesis of selected fGmH RNA aptamers for translational applications. Overall, fGmH RNA aptamer technology will allow the facile identification of highly stable fGmH aptamers that confer smartness to biomaterials via a multitude of potential applications.
MATERIAL AND METHODS
Reagents and Strains
Staphylococcus aureus strains 10832 (subsp. aureus Rosenbach, Wood 46, NRA105) and 12598 (subsp. aureus Rosenbach, NCTC 8530, Cowan I, NRS104) were obtained from ATCC (Manassas, VA) and cultured in Luria-Bertani (LB) broth for all experiments. Citrate BioPure™ silver nanoparticles (10 nm diameter) were purchased from NanoComposix (San Diego, CA). A capture oligo containing a 3′ terminal amine, used for docking full-length A12 sequences to AgNPs and bearing the sequence 5′-GTCATTGTTCTCTCCCA-NH2-3′, was synthesized by MWG Eurofins (Huntsville, AL).
His-LAR T7 RNA Polymerase Expression, Purification, and Characterization
The pDest17 expression vector bearing His-tagged LAR T7 RNA polymerase was transformed into E. coli BL21 (DE3) Rosseta cells (Novagen, Darmstadt, Germany). The positive clones were selected on LB plates containing carbenicillin (100 μg/mL) and chloramphenicol (34 μg/mL). A single colony was selected and grown in 5 mL of LB broth overnight at 37 °C. The resulting culture was added to a flask with 500 mL of LB broth containing carbenicillin (100 μg/mL) and chloramphenicol (34 μg/mL). The cells were grown at 37 °C until the optical density (Ab600) reached 0.5 to 1.0. IPTG with a final concentration of 1 mM was then added to the cell cultures, followed by incubation at 22 °C for 16 h. After induction, the cells were spun down at 3,000 × g for 10 min at 4 °C, and the pellet was stored at −20 °C until use. To purify Hi s-tagged LAR T7 RNA polymerase, the cell pellet was resuspended in buffer A (25 mM HEPES pH 7.4 and 300 mM NaCl) and lysed by sonication for 1 min for a total of 5 times. The soluble fraction was recovered by centrifugation at 12,000 × g for 10 min at 4 °C. The resulting fracti on was loaded onto a TALON metal affinity column (Clontech, Mountainview, CA) pre-equilibrated with buffer A. Approximately 20 column volumes of buffer A were used for initial washing followed by extensive washing (20 column volumes) with buffer B (buffer A with 20 mM imidazole). The protein of interest was eluted with buffer C (buffer A with 200 mM imidazole). The quality of the purified protein was examined by SDS-PAGE. The optimal amount of His-LAR T7 RNA polymerase to be used in each reaction was determined on a per batch basis via parallel fGmH RNA transcriptions using a serial dilution of the purified enzyme. Transcription products were separated on a 20% denaturing PAGE gel, visualized by ethidium bromide staining, and band intensities were quantified relative to each other. Purified LAR T7 RNA polymerase was quality controlled by checking for nuclease and natural ribonucleotide contamination. Nuclease contamination was tested by incubating each LAR T7 RNA polymerase dilution with 5 pmol of a control natural RNA sequence for 2 h at 37 °C. The presence of contaminating natural ribonucleotides in the purified protein was checked by performing transcription in the absence of externally added 2′-X-dNTPs. Transcription products were separated and visualized on 20% denaturing PAGE.
In vitro Selection
A N40 random fGmH RNA library was generated by transcription from a dsDNA library of the following primary sequence: 5′-TAATACGACTCACTATAGGGAGAGAACAATGACCTG-(N)40-GAGTGCATTGCATCACGTCAGTAG-3′. The DNA library was constructed previously using Klenow Fragment (3′ to 5′ exo-) (New England Biolabs, Ipswich, MA) in a reaction mixture of 1.2 mL containing 0.2 mM dNTPs, 0.1 μM single-stranded DNA (ssDNA) sequence ssEVS40LB (5′-CTACTGACGTGATGCAATGCACTC-(N)40-CAGGTCATTGTTCTCTCCCTA), and 0.15 μM ssDNA sequence PDAC-5P (5′-TAATACGACTCACTATAGGGAGAGAACAATGACCTG), at 37 °C for 1 h. The T7 promoter sequence is underlined. The initial pool, as well as reverse-transcribed enriched aptamer pools during selection, was PCR amplified using 0.5 μM of primers PDAC-5P and PDAC-3P (5′-CTACTGACGTGATGCAATGCACTC-3′) and cycling at 95 °C, 62 °C, and 72 °C for 15 seconds, respectively, for an appropriate number of cycles. In vitro transcription of the DNA library using His-LAR T7 RNA polymerase occurred at 37 °C for 20 h in an optimized transcription buffer containing 200 mM HEPES, pH 7.5, 2 mM spermidine, 2 mM spermine, 19.2 mM MgCl2, 5.8 mM MnCl2, 1.5 mM GMP, 1.5 mM 2′-F-dGTP, 1.5 mM 2′-OMe-dATP/dCTP/dUTP, 40 mM DTT, 10% w/v PEG-8000, 0.01% v/v Triton X-100, and 0.04 U/μL thermostable inorganic pyrophosphatase (New England Biolabs, Ipswich, MA). After transcription, the reaction mixture was treated with 0.017 U/μL RQ1 RNase-free DNase (Promega, Madison, WI) for 2 h at 37 °C. DNase was inactivated via addition of EDTA, pH 8.0 to a concentration 1.2× in excess of Mg2+, and heated at 70 °C for 10 min. The fGmH RNA reaction products were ethanol precipitated, desalted, and quantified via denaturing PAGE electrophoresis. The initial fGmH RNA library used for selection has a diversity of 7 × 1013 unique sequences.
Prior to target-binding, the initial fGmH RNA library, or regenerated aptamer pools, were folded by heating at 80°C for 3 min followed by incubation in 1× Aptamer Selection Buffer (20 mM HEPES, pH 7.4, 150 mM sodium chloride, 5 mM magnesium chloride, 1 mM calcium chloride, 1 mg/mL BSA, 0.1 mg/mL yeast tRNA) for 15 min at room temperature. Binding of the fGmH RNA library to Protein A Mag Sepharose Xtra beads (GE Healthcare, Fairfield, CT) occurred in 6 column volumes of 1× Aptamer Selection Buffer (1 column volume = 25 μL) for 1 h at 4 °C. Input fGmH RNA for each round of selection was approximately 100 pmol. After incubation, beads were washed twice, each with 6 column volumes of 1× Aptamer Selection Buffer for 5 min at 4 °C to remove sequences bound nonspecifically. Elution from the beads was performed three times, each using 6 column volumes of 1× Aptamer Selection Buffer incubated at 4 °C for 20 min to obtain aptamers with slower dissociation rates. Eluted aptamers were concentrated and desalted on a Vivaspin 500 5 kDa MWCO centrifugal concentrator (Sartorius Stedim Biotech, Bohemia, NY). fGmH RNA recovered from a selection round was prepared for reverse transcription by annealing to 2 μM PDAC-3P reverse primer by heating at 70 °C for 10 min and immediately cooling on ice for 5 min. After 2 min at 42 °C, reverse transcription occurred upon addition of 10 U/μL SuperScript II Reverse Transcriptase (Life Technologies, Grand Island, NY), in a buffer containing 50 mM Tris-HCl, pH 8.3, 0.5 mM dNTPs, 75 mM KCl, 3 mM MgCl2, and 10 mM DTT. cDNA products were amplified by PCR as described above for an optimized number of cycles. Aptamer pools from Rounds 5, 6, 7, and 8 were ligated into pJET1.2/blunt cloning vector (Thermo Scientific, Waltham, MA) and transformed into DH5α E.coli cells for sequencing (Eton Bioscience, Durham, NC). Dominant sequences, representing primary sequence groups, were selected for in vitro transcription and characterization. The secondary structures of sequences of interest were predicted by the online program Mfold.
Binding Analysis Using Surface Plasmon Resonance (SPR) and Biolayer Interferometry (BLI)
Aptamer affinity for S. aureus Protein A (SpA) was measured by using both a Biacore 3000 (GE Healthcare, Fairfield, CT) and Octet QK (Pall FortéBio, Menlo Park, CA). SPR assays were performed on a Biacore 3000 at room temperature. SpA from S. aureus Cowan (Sigma-Aldrich, St. Louis, MO) was pre-concentrated onto CM5 chips by injection of 300 μg/mL SpA in 5 mM sodium acetate, pH 4.2 at 10 μL/min. The acidic pH of this buffer was below the SpA isoelectric point (pI = 5.16) but above the pKa of the carboxyl groups of carboxymethyl dextran, permitting the net positively charged SpA to electrostatically interact, and pre-concentrate, with the negatively charged dextran on the CM5 chip. With the SpA pre-concentrated against CM5 dextran, covalent immobilization of SpA onto the chip occurred via reaction with 200 μL 0.8 M 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide (EDC) and 0.2 M N-hydroxysulfosuccinimide (NHS) at a 1:1 molar ratio at a flow rate of 10 μL/min at room temperature. The reaction was quenched with 1 M ethanolamine at pH 8.0 (10 μL/min and room temperature). SpA immobilization on CM5 chips typically produced ~2500 response units (RUs). Selected fGmH RNA aptamers fmA12 and fmG12, human IgG Fc fragment (Athens Research, Athens, GA)(positive control), the initial fGmH RNA library and fmE03 (negative controls) were analyzed in assay buffer (20 mM HEPES, pH 7.4, 150 mM sodium chloride, 5 mM magnesium chloride, 1 mM calcium chloride, 0.005% Tween 20). fGmH RNA aptamers were injected into the flow cell at 10 μL/min at 31.25 nM, 62.5 nM, 125 nM, and 250 nM, respectively. Binding parameters were calculated using BIAevaluation software by fitting data using a 1:1 Langmuir binding model.
The Octet QK is a relatively new, label-free method based on BLI technology that measures molecular interactions via fiber optic biosensors [51]. Due to its higher throughput, greater automation, and lower cost compared to other methods of affinity detection, BLI has been applied in the study of more and more molecular interactions [52–54]. BLI assays were run at 30 °C on an Octet QK using Protein A (ProA) biosensors (Pall ForteBio Corp., Menlo Park, CA) and black 96-well microplates. All aptamer samples were prepared in assay buffer (as above) and applied to a 96-well microplate in column arrangement. An aptamer dilution series tested the following concentrations: 157 nM, 313 nM, 625 nM, 1,000 nM, and 1,250 nM. The initial fGmH RNA library and non-binding fmE03 were used at 1,250 nM to evaluate nonspecific and background binding. Assays were run in quadruplicate. All data were acquired in fortéBio Data Acquisition 6.4 software and analyses were performed in fortéBIO Data Analysis 6.4 software. Data processing was performed by subtraction of the reference sensor signal (assay buffer only), application of Savitzky-Golay Filtering, and global curve fitting using a 1:1 model.
Other binding assays utilizing the Octet QK were performed under similar conditions as above. The binding specificity assay was performed using Protein A (ProA), Protein G (ProG), and Protein L (ProL) biosensors, with the major classes of the selected aptamers and human IgG Fc fragment positive control at 1.0 μM. The binding of A12 aptamer variants with different 2′ modification states was analyzed at 1.0 μM with ProA biosensors. Assays assessing the avidity of fmA12-functionalized AgNPs for ProA were performed in AgNP-Aptamer Buffer (100 mM HEPES, pH 7.4, 300 mM sodium acetate, 10 mM magnesium acetate).
Aptamer Stability Assays
fGmH RNA stability in biological fluids started with in vitro transcription of A12 DNA into four different 2′-modification states: natural 2′-unmodified RNA (rN RNA; WT A12), 2′-bi-modified 2′-F-dCTP/dUTP (fYrR RNA; bmA12), 2′-tri-modified 2′-OMe-dATP/dCTP/dUTP (rGmH RNA; tmA12), and 2′-fully modified 2′-F-dGTP/2′-OMe-dATP/dCTP/dUTP RNA (fGmH RNA; fmA12). LAR T7 RNA polymerase was used to transcribe the fYrR, rGmH, and fGmH A12 variants using the optimized buffer system described above, while wild-type T7 RNA polymerase (New England Biolabs, Ipswich, MA) was used to transcribe the natural A12 RNA. DNase-treatment, precipitation, desalting, and quantification procedures were followed as described above. 5 pmol of each A12 RNA variant was incubated in 10% mouse serum that was free of chelating agents, such as EDTA or citrate (Sigma-Aldrich, St. Louis, MO), for 0 min, 5 min, 2 h, 5 h, 8 h, 12 h, and 24 h, respectively. All samples after incubation were loaded onto a 20% denaturing PAGE for separation, followed by visualization and quantification. Percent survival for each time point was determined via relative quantification compared to 0 min time point (5 pmol A12 RNA input).
Alkaline hydrolysis was performed as a further test of fGmH RNA stability against nucleases that form 2′, 3′-cyclic phosphate intermediates. The cDNA template coding Aptamer G12 was in vitro transcribed into the same four 2′ modification states as described above. 5 pmol of each G12 RNA variant was subjected to incubation at 95 °C an d pH 9.2 for 10 min (in 50 mM sodium carbonate, pH 9.2, 1 mM EDTA), a condition that mimics nuclease degradation [55]. Major bands of each RNA composition, both treated and untreated, were separated, visualized and quantified via 20% denaturing PAGE electrophoresis. Percent survival was calculated by quantitative comparison of treated and untreated RNA samples.
Confocal Microscopy
S. aureus strains 12598 (high SpA expression) and 10832 (low SpA expression) were cultured in LB broth for 12 h at 37 °C. Culture optical density was adjusted to McFarland Standard No. 3 (Ab600 = 0.582) in 1 mL LB broth and distributed across sample tubes. Bacteria were pelleted via centrifugation at 3,000 × g for 5 min and washed with 1× Aptamer Selection Buffer. 500 nM fmA12 and tmA12 were folded as described above. SYBR Green I (Life Technologies, Grand Island, NY), a fluorophore that intercalates into double stranded nucleic acids for RNA detection and quantification, was incubated at 1.0 μM with 0.5 μM folded fmA12 or tmA12 in a total volume of 110 μL for 30 min at room temperature [28]. To minimize fluorescent background, excess free SYBR Green I was removed from the fmA12 and tmA12 preparations. fmA12 and tmA12 sample volumes were first adjusted to 500 μL in 1× Aptamer Selection Buffer and concentrated to 50 μL using a Vivaspin 500 5 kDa MWCO centrifugal concentrator. The samples were then diluted to 500 μL and concentrated to 50 μL a second time. These samples, after dilution to 110 μL, had a background SYBR Green I concentration of 10 nM. Incubation of fmA12 and tmA12 samples with 3,000 colony forming units (CFUs: viable bacteria capable of colony formation on LB agar) of S. aureus strain 12598 and 10832 cells, respectively, occurred at room temperature for 15 min. To remove unbound fmA12 and tmA12, the cells were pelleted and washed once with 30 μL of 1× Aptamer Selection Buffer at room temperature. Cells were resuspended in 1× Aptamer Selection Buffer, mounted with Fluoromount-G on slide glass, and examined using a Zeiss LSM 710 confocal microscope (Zeiss, Thornwood, NY).
Silver Nanoparticle Stabilization and Functionalization
The capture oligo with a 3′ amine was reacted with carbon disulfide to form a dithiocarbamate moiety. The reaction occurred at equimolar (50 μM) concentrations of capture oligo and carbon disulfide in 400 μL for 2 h at room temperature in 10 mM borate buffer, pH 9.0 according to the procedures published by Yan and coworkers [32]. Free carbon disulfide was removed from the capture oligos via passage through a Vivaspin 500 5 kDa MWCO centrifugal concentrator three times in 10 mM borate buffer. 20 nmol of the resulting capture oligo was incubated with 24 pmol of AgNPs in 95 μL of 10 mM borate buffer for 12 h at room temperature with constant shaking. The Na+ concentration was gradually adjusted to 300 mM by stepwise addition of AgNP Conjugation Buffer (100 mM HEPES, pH 7.4, 600 mM sodium acetate, 10 mM magnesium acetate) by first increasing the Na+ concentration to 75 mM and incubation for 4 h, then to 150 mM (4 h), and then to 300 mM (12 h), and finally storing at 4 °C. Excess capture oligos were removed from AgNPs via repeated dilution and concentration using a Vivaspin 500 30 kDa MWCO centrifugal concentrator. Docking and immobilization of A12 RNA (fmA12 and tmA12) to AgNPs conjugated with capture oligo (AgNP-Capture) occurred at a molar ratio of 10:1 in AgNP-Aptamer Buffer (20 mM HEPES, pH 7.4, 150 mM sodium acetate, 5 mM magnesium chloride, 1 mM calcium chloride) at room temperature for 5 min. AgNPs functionalized with fmA12 and tmA12 remained dispersed in multiple physiologically relevant buffers, including 1× Aptamer Selection Buffer, 1× Aptamer Selection Buffer-Phosphate (chloride ions replaced with phosphates), AgNP-Aptamer Buffer, and LB broth.
Targeted Killing of S. aureus Using SpA-targeted Silver Nanoparticles
S. aureus strains 12598 and 10832 were cultured and adjusted as above. Prior to assay execution, the dispersity and concentration of AgNPs docked with fmA12 or tmA12 was confirmed via spectral measurement of the 410 nm peak, with subsequent nanoparticle quantification using the molar extinction coefficient reported by Navarro and Werts [56]. 1 × 106 CFUs of each S. aureus strain were incubated in quadruplicate for 2 h at 37 °C (total sample volume of 10 μL) with: AgNP-Aptamer Buffer only (negative control), 280 nM fmA12 only (negative control), 91 μg/mL citrate-capped AgNPs (positive killing control), 91 μg/mL fmA12-functionalized AgNP-Capture (AgNP-Capture:fmA12), and 91 μg/mL AgNP-Capture:tmA12 (negative control), respectively. After 2 h incubation, all samples, except citrate-capped AgNP control, were pelleted and washed with 10 μL AgNP-Aptamer Buffer. Samples were incubated in LB broth for 4 h at 37 °C, followed by 20 h at 25 °C. These growth conditions permitted the S. aureus cells in the negative controls to reach McFarland Standard No. 3 (~1 × 109 CFUs/mL). Dilution of the cultures 1:10,000 ensured proportional plating of approximately 100 CFUs per sample if no killing occurred. OD600 values were measured for each sample immediately prior to plating. CFUs were counted on each plate after overnight incubation at 37 °C. Counted CFUs f or each sample were normalized to AgNP-Aptamer Buffer Only negative control to estimate cell-killing efficiency.
Acknowledgments
The authors would like to acknowledge Chuan Fu for his generation of the vector containing the LAR T7 RNA polymerase. The UNC Macromolecular Interactions Facility provided the Biacore 3000 used in this study.
FUNDING
This work was supported by National Institutes of Health Grants CA151652 and CA157738 (to R.L.), and by an American Foundation for Pharmaceutical Education (AFPE) Pre-doctoral Fellowship (to A.D.F.). A.D.F. is a Howard Hughes Medical Institute (HHMI) Med into Grad Scholar supported in part by a grant to the University of North Carolina at Chapel Hill from HHMI through the Med into Grad Initiative.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346(6287):818–22. doi: 10.1038/346818a0. Epub 1990/08/30. [DOI] [PubMed] [Google Scholar]
- 2.Gold L, Polisky B, Uhlenbeck O, Yarus M. Diversity of oligonucleotide functions. Annual review of biochemistry. 1995;64:763–97. doi: 10.1146/annurev.bi.64.070195.003555. Epub 1995/01/01. [DOI] [PubMed] [Google Scholar]
- 3.Robertson DL, Joyce GF. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature. 1990;344(6265):467–8. doi: 10.1038/344467a0. Epub 1990/03/29. [DOI] [PubMed] [Google Scholar]
- 4.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249(4968):505–10. doi: 10.1126/science.2200121. Epub 1990/08/03. [DOI] [PubMed] [Google Scholar]
- 5.White RR, Sullenger BA, Rusconi CP. Developing aptamers into therapeutics. The Journal of clinical investigation. 2000;106(8):929–34. doi: 10.1172/JCI11325. Epub 2000/10/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson DS, Szostak JW. In vitro selection of functional nucleic acids. Annual review of biochemistry. 1999;68:611–47. doi: 10.1146/annurev.biochem.68.1.611. Epub 2000/06/29. [DOI] [PubMed] [Google Scholar]
- 7.Jayasena SD. Aptamers: An Emerging Class of Molecules That Rival Antibodies in Diagnostics. Clin Chem. 1999;45(9):1628–50. [PubMed] [Google Scholar]
- 8.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nature reviews Drug discovery. 2010;9(7):537–50. doi: 10.1038/nrd3141. Epub 2010/07/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355(6360):564–6. doi: 10.1038/355564a0. Epub 1992/02/06. [DOI] [PubMed] [Google Scholar]
- 10.Li N, Nguyen HH, Byrom M, Ellington AD. Inhibition of cell proliferation by an anti-EGFR aptamer. PloS one. 2011;6(6):e20299. doi: 10.1371/journal.pone.0020299. Epub 2011/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruckman J, Green LS, Beeson J, Waugh S, Gillette WL, Henninger DD, et al. 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. The Journal of biological chemistry. 1998;273(32):20556–67. doi: 10.1074/jbc.273.32.20556. Epub 1998/08/01. [DOI] [PubMed] [Google Scholar]
- 12.Famulok M. Molecular recognition of amino acids by RNA-aptamers: an L-citrulline binding RNA motif and its evolution into an L-arginine binder. Journal of the American Chemical Society. 1994;116:1698–706. [Google Scholar]
- 13.Pieken WA, Olsen DB, Benseler F, Aurup H, Eckstein F. Kinetic characterization of ribonuclease-resistant 2′-modified hammerhead ribozymes. Science. 1991;253(5017):314–7. doi: 10.1126/science.1857967. Epub 1991/07/19. [DOI] [PubMed] [Google Scholar]
- 14.Burmeister PE, Lewis SD, Silva RF, Preiss JR, Horwitz LR, Pendergrast PS, et al. Direct in vitro selection of a 2′-O-methyl aptamer to VEGF. Chemistry & biology. 2005;12(1):25–33. doi: 10.1016/j.chembiol.2004.10.017. Epub 2005/01/25. [DOI] [PubMed] [Google Scholar]
- 15.Chelliserrykattil J, Ellington AD. Evolution of a T7 RNA polymerase variant that transcribes 2′-Omethyl RNA. Nature biotechnology. 2004;22(9):1155–60. doi: 10.1038/nbt1001. Epub 2004/08/10. [DOI] [PubMed] [Google Scholar]
- 16.Layzer JM, Sullenger BA. Simultaneous generation of aptamers to multiple gamma-carboxyglutamic acid proteins from a focused aptamer library using DeSELEX and convergent selection. Oligonucleotides. 2007;17(1):1–11. doi: 10.1089/oli.2006.0059. Epub 2007/04/28. [DOI] [PubMed] [Google Scholar]
- 17.Bell C, Lynam E, Landfair DJ, Janjic N, Wiles ME. Oligonucleotide NX1838 inhibits VEGF165- mediated cellular responses in vitro. In vitro cellular & developmental biology Animal. 1999;35(9):533– 42. doi: 10.1007/s11626-999-0064-y. Epub 1999/11/05. [DOI] [PubMed] [Google Scholar]
- 18.Burmeister PE, Wang C, Killough JR, Lewis SD, Horwitz LR, Ferguson A, et al. 2′-Deoxy purine, 2′-O-methyl pyrimidine (dRmY) aptamers as candidate therapeutics. Oligonucleotides. 2006;16(4):337–51. doi: 10.1089/oli.2006.16.337. Epub 2006/12/13. [DOI] [PubMed] [Google Scholar]
- 19.Ibach J, Dietrich L, Koopmans KR, Nobel N, Skoupi M, Brakmann S. Identification of a T7 RNA polymerase variant that permits the enzymatic synthesis of fully 2′-O-methyl-modified RNA. Journal of biotechnology. 2013;167(3):287–95. doi: 10.1016/j.jbiotec.2013.07.005. Epub 2013/07/23. [DOI] [PubMed] [Google Scholar]
- 20.Padilla R, Sousa R. Efficient synthesis of nucleic acids heavily modified with non-canonical ribose 2′-groups using a mutantT7 RNA polymerase (RNAP) Nucleic acids research. 1999;27(6):1561–3. doi: 10.1093/nar/27.6.1561. Epub 1999/02/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padilla R, Sousa R. A Y639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucleic acids research. 2002;30(24):e138. doi: 10.1093/nar/gnf138. Epub 2002/12/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki AM, Casper MD, Freier SM, Lesnik EA, Zounes MC, Cummins LL, et al. Uniformly modified 2′-deoxy-2′-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. Journal of medicinal chemistry. 1993;36(7):831–41. doi: 10.1021/jm00059a007. [DOI] [PubMed] [Google Scholar]
- 23.Diener JL, Keefe AD, Thompson K, Wang C, Zhu S. Materials and methods for the generation of fully 2′-modified nucleic acid transcripts. 8105813 B2. Google Patents; US. 2012
- 24.Gomez MI, Seaghdha MO, Prince AS. Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. The EMBO journal. 2007;26(3):701–9. doi: 10.1038/sj.emboj.7601554. Epub 2007/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu C, Friedman A, Liu R. A Systematic Examination of T7 RNA Polymerase Variants and 2′-Modified NTPs: A Guideline for SELEX Using Non-canonical Substrates. To be published. [Google Scholar]
- 26.Kernodle DS, McGraw PA, Barg NL, Menzies BE, Voladri RK, Harshman S. Growth of Staphylococcus aureus with nafcillin in vitro induces alpha-toxin production and increases the lethal activity of sterile broth filtrates in a murine model. The Journal of infectious diseases. 1995;172(2):410– 9. doi: 10.1093/infdis/172.2.410. Epub 1995/08/01. [DOI] [PubMed] [Google Scholar]
- 27.Suci PA, Berglund DL, Liepold L, Brumfield S, Pitts B, Davison W, et al. High-density targeting of a viral multifunctional nanoplatform to a pathogenic, biofilm-forming bacterium. Chemistry & biology. 2007;14(4):387–98. doi: 10.1016/j.chembiol.2007.02.006. Epub 2007/04/28. [DOI] [PubMed] [Google Scholar]
- 28.Zipper H, Brunner H, Bernhagen J, Vitzthum F. Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications. Nucleic acids research. 2004;32(12):e103. doi: 10.1093/nar/gnh101. Epub 2004/07/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Castanon G, Nino-Martinez N, Martinez-Gutierrez F, Martinez-Mendoza J, Facundo R. Synthesis and antibacterial activity of silver nanoparticles with different sizes. J Nanopart Res. 2008;10:1343–8. [Google Scholar]
- 30.Liu J, Hurt RH. Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environmental science & technology. 2010;44(6):2169–75. doi: 10.1021/es9035557. Epub 2010/02/24. [DOI] [PubMed] [Google Scholar]
- 31.Choi O, Clevenger TE, Deng B, Surampalli RY, Ross L, Jr, Hu Z. Role of sulfide and ligand strength in controlling nanosilver toxicity. Water research. 2009;43(7):1879–86. doi: 10.1016/j.watres.2009.01.029. Epub 2009/03/03. [DOI] [PubMed] [Google Scholar]
- 32.Sharma J, Chhabra R, Yan H, Liu Y. A facile in situ generation of dithiocarbamate ligands for stable gold nanoparticle-oligonucleotide conjugates. Chem Commun (Camb) 2008;(18):2140–2. doi: 10.1039/b800109j. Epub 2008/04/29. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Perez-Segarra W, Shi Q, Wei A. Dithiocarbamate assembly on gold. Journal of the American Chemical Society. 2005;127(20):7328–9. doi: 10.1021/ja050432f. Epub 2005/05/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tokareva I, Hutter E. Hybridization of oligonucleotide-modified silver and gold nanoparticles in aqueous dispersions and on gold films. Journal of the American Chemical Society. 2004;126(48):15784– 9. doi: 10.1021/ja046779k. Epub 2004/12/02. [DOI] [PubMed] [Google Scholar]
- 35.Schuster S, Yu W, Nega M, Chu YY, Zorn S, Zhang F, et al. The role of serum proteins in Staphylococcus aureus adhesion to ethylene glycol coated surfaces. International journal of medical microbiology : IJMM. 2014 doi: 10.1016/j.ijmm.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 36.Friedman AD, Claypool SE, Liu R. The smart targeting of nanoparticles. Current pharmaceutical design. 2013;19(35):6315–29. doi: 10.2174/13816128113199990375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu CH, Yang HH, Zhu CL, Chen X, Chen GN. A graphene platform for sensing biomolecules. Angewandte Chemie. 2009;48(26):4785–7. doi: 10.1002/anie.200901479. [DOI] [PubMed] [Google Scholar]
- 38.Miyachi Y, Shimizu N, Ogino C, Kondo A. Selection of DNA aptamers using atomic force microscopy. Nucleic acids research. 2010;38(4):e21. doi: 10.1093/nar/gkp1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tombelli S, Minunni M, Luzi E, Mascini M. Aptamer-based biosensors for the detection of HIV-1 Tat protein. Bioelectrochemistry. 2005;67(2):135–41. doi: 10.1016/j.bioelechem.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Zichel R, Chearwae W, Pandey GS, Golding B, Sauna ZE. Aptamers as a sensitive tool to detect subtle modifications in therapeutic proteins. PloS one. 2012;7(2):e31948. doi: 10.1371/journal.pone.0031948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malinen MM, Kanninen LK, Corlu A, Isoniemi HM, Lou YR, Yliperttula ML, et al. Differentiation of liver progenitor cell line to functional organotypic cultures in 3D nanofibrillar cellulose and hyaluronan-gelatin hydrogels. Biomaterials. 2014;35(19):5110–21. doi: 10.1016/j.biomaterials.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Picollet-D’hahan N, Gerbaud S, Kermarrec F, Alcaraz JP, Obeid P, Bhajun R, et al. The modulation of attachment, growth and morphology of cancerous prostate cells by polyelectrolyte nanofilms. Biomaterials. 2013;34(38):10099–108. doi: 10.1016/j.biomaterials.2013.08.093. [DOI] [PubMed] [Google Scholar]
- 43.Yoon HJ, Kim TH, Zhang Z, Azizi E, Pham TM, Paoletti C, et al. Sensitive capture of circulating tumour cells by functionalized graphene oxide nanosheets. Nature nanotechnology. 2013;8(10):735–41. doi: 10.1038/nnano.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi Q, Qin L, Wei W, Geng F, Fan R, Shin YS, et al. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(2):419–24. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng M, James R, Laurencin CT, Kumbar SG. Nanostructured polymeric scaffolds for orthopaedic regenerative engineering. IEEE transactions on nanobioscience. 2012;11(1):3–14. doi: 10.1109/TNB.2011.2179554. [DOI] [PubMed] [Google Scholar]
- 46.Lim B, Reddy V, Hu X, Kim K, Jadhav M, Abedini-Nassab R, et al. Magnetophoretic circuits for digital control of single particles and cells. Nature communications. 2014;5:3846. doi: 10.1038/ncomms4846. [DOI] [PubMed] [Google Scholar]
- 47.Abelson J, Trotta CR, Li H. tRNA splicing. The Journal of biological chemistry. 1998;273(21):12685–8. doi: 10.1074/jbc.273.21.12685. Epub 1998/05/28. [DOI] [PubMed] [Google Scholar]
- 48.Raines RT. Ribonuclease A. Chemical reviews. 1998;98(3):1045–66. doi: 10.1021/cr960427h. Epub 2002/02/19. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Kuan CT, Mi J, Zhang X, Clary BM, Bigner DD, et al. Aptamers selected against the unglycosylated EGFRvIII ectodomain and delivered intracellularly reduce membrane-bound EGFRvIII and induce apoptosis. Biological chemistry. 2009;390(2):137–44. doi: 10.1515/BC.2009.022. Epub 2008/12/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, Lenhart JJ, Walker HW. Dissolution-accompanied aggregation kinetics of silver nanoparticles. Langmuir : the ACS journal of surfaces and colloids. 2010;26(22):16690–8. doi: 10.1021/la101768n. Epub 2010/10/01. [DOI] [PubMed] [Google Scholar]
- 51.Abdiche Y, Malashock D, Pinkerton A, Pons J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Analytical biochemistry. 2008;377(2):209–17. doi: 10.1016/j.ab.2008.03.035. Epub 2008/04/15. [DOI] [PubMed] [Google Scholar]
- 52.Kawakami T, Ishizawa T, Fujino T, Reid PC, Suga H, Murakami H. In Vitro Selection of Multiple Libraries Created by Genetic Code Reprogramming To Discover Macrocyclic Peptides That Antagonize VEGFR2 Activity in Living Cells. ACS chemical biology. 2013 doi: 10.1021/cb300697h. Epub 2013/03/23. [DOI] [PubMed] [Google Scholar]
- 53.Ylera F, Harth S, Waldherr D, Frisch C, Knappik A. Off-rate screening for selection of high-affinity anti-drug antibodies. Analytical biochemistry. 2013;441(2):208–13. doi: 10.1016/j.ab.2013.07.025. Epub 2013/08/03. [DOI] [PubMed] [Google Scholar]
- 54.Yu H, Dong J, Gu Y, Liu H, Xin A, Shi H, et al. The novel human beta-defensin 114 regulates lipopolysaccharide (LPS)-mediated inflammation and protects sperm from motility loss. The Journal of biological chemistry. 2013;288(17):12270–82. doi: 10.1074/jbc.M112.411884. Epub 2013/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan Y, Qu K, Ouyang Z, Chang HY. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nature protocols. 2013;8(5):849–69. doi: 10.1038/nprot.2013.045. Epub 2013/04/06. [DOI] [PubMed] [Google Scholar]
- 56.Navarro JR, Werts MH. Resonant light scattering spectroscopy of gold, silver and gold-silver alloy nanoparticles and optical detection in microfluidic channels. The Analyst. 2013;138(2):583–92. doi: 10.1039/c2an36135c. Epub 2012/11/23. [DOI] [PubMed] [Google Scholar]