

Abstract
Idiopathic pulmonary fibrosis (IPF) is one of the most common and severe interstitial lung diseases. Epithelial-to-mesenchymal transition (EMT) is a process whereby epithelial cells undergo transition to a mesenchymal phenotype. This process has been shown to contribute to IPF. MicroRNAs (miRNAs) are small non-coding RNAs of 18 to 24 nucleotides in length which regulate gene expression. Several studies have implicated miRNAs in EMT; however, specific miRNAs that regulate EMT in IPF have not yet been identified. In this study, we identified 6 up-regulated and 3 down-regulated miRNAs in a human lung epithelial cell EMT model using miRNA microarray and real-time PCR. Overexpression of one of these up-regulated miRNAs, miR-424, increased the expression of α-smooth muscle actin, an indicator of myofibroblast differentiation, but had no effects on the epithelial or mesenchymal cell markers. miR-424 enhanced the activity of the TGF-β signaling pathway, as demonstrated by a luciferase reporter assay. Further experiments showed that miR-424 decreased the protein expression of Smurf2, a negative regulator of TGF-β signaling, indicating that miR-424 exerts a forward regulatory loop in the TGF-β signaling pathway. Our results suggest that miR-424 regulates the myofibroblast differentiation during EMT by potentiating the TGF-β signaling pathway, likely through Smurf2.
Keywords: Epithelial-to-mesenchymal transition, idiopathic pulmonary fibrosis, miR-424, TGF-β, Smurf2, myofibroblast differentiation
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common type of interstitial lung disease. IPF is characterized by deterioration of respiratory functions due to fibrosis of the lung interstitium. The pathologic features of IPF include heterogenic injury and fibrosis, foci of fibroblast/myofibroblast accumulation beneath flattened alveolar epithelial cells, and inflammation [1, 2]. The prevalence and incidence of IPF is estimated to be 42.7 per 100,000 and 16.3 per 100,000, respectively [3]. Although the etiology of the disease is still unknown, some genetic abnormalities of alveolar epithelial cells, including mutations of surfactant protein A2, ELMOD2 and MUC5B, have been described in some IPF patients [4–6]. Potential environmental risk factors include: cigarette smoking, exposure to wood and metal dust (pine, brass and steel), and viral infections (Epstein-Barr virus and herpes simplex virus) [7–9]. Recent studies have demonstrated that injury to alveolar epithelial cells is the primary driving force of fibrosis [5, 10, 11]. In response to epithelial cell injury, resident fibroblasts proliferate and migrate to various sites in the lung. Other sources of fibroblasts and myofibroblasts in fibrotic foci include circulating fibrocytes from bone marrow, and alveolar epithelial cells themselves via epithelial-to-mesenchymal transition (EMT) [12–18]. Fibroblasts aggregate at fibrotic foci and differentiate into myofibroblasts, which are more active than fibroblasts in the extracellular matrix (ECM) production [19–21].
During EMT, epithelial cells lose phenotypic features, such as apical-basal polarity and specialized cell-cell contacts, and acquire mesenchymal cell properties such as increased cell mobility [22]. EMT is essential for the early stages of development of most tissues. It enables the development of the mesoderm from the epithelium during gastrulation, which is necessary for the formation of organs such as the lung and heart. While EMT is an integral process during development, it also contributes to fibrosis in fully developed organs and to invasion and metastasis of carcinomas. In cancer, tumor cells become more invasive after undergoing EMT, which facilitates the metastasis and progression of the tumor [23]. EMT also occurs after tissue injury and/or stress. Many studies provide convincing evidence to support the hypothesis that a significant portion of the myofibroblasts in tissue fibrosis come from epithelial cells which have undergone EMT [2, 12, 22, 24–27]. Indeed, cells from fibrotic foci in the lungs of IPF patients have features of both epithelial and mesenchymal cells [28]. In vitro and in vivo studies suggest that alveolar epithelial cells can acquire mesenchymal characteristics in response to injury [2, 15, 17, 18, 28].
Many signaling pathways are known to trigger EMT, including the TGF-β, Wnt and Notch pathways. These pathways do not operate independently; rather, crosstalk between them ensures EMT completion. The signaling pathway that conveys TGF-β inputs from membrane receptors to target genes is well studied. TGF-β binds to trans-membrane serine-threonine kinase receptors on the cell surface, leading to the formation of a bi-dimeric receptor complex composed of receptor type I (TβRI) and type II (TβRII). TβRII phosphorylates and activates TβRI, which then phosphorylates cytoplasmic Smad2/3 transcription factors, allowing them to translocate into the nucleus. Smad4 acts as a partner with receptor-activated Smads to facilitate this process. In contrast, Smad6 and Smad7 inhibit activated receptor-regulated Smads [29, 30]. Three families of the transcription factors (Snail, ZEB and bHLH) are all involved in the TGF-β transcription program. Their expression is regulated either directly through a Smad-dependent pathway, or indirectly through other transcription factors. These transcription factors repress epithelial genes (e.g., E-cadherin and zonula occludens-1) and activate mesenchymal genes (e.g., N-cadherin and α-smooth muscle actin) [31]. Functionally, the TGF-β signaling pathway plays a predominant role in tissue fibrosis by influencing processes such as EMT, fibroblast recruitment, fibroblast contractility, myofibroblast differentiation, and ECM deposition [32–35].
MicroRNAs (miRNAs) are a group of small non-coding RNAs that are 18–24 nt in length and regulate gene expression at the post-transcriptional level. miRNAs are first transcribed from miRNA genes by RNA polymerase II, then cleaved by Drosha/Dicer to become mature miRNAs. The mature single-stranded miRNA is incorporated into the RNA-induced silencing complex (RISC), which binds to target mRNAs to inhibit protein translation or to cleave the mRNAs. miRNAs regulate both physiological and pathological processes [36–40].
A number of miRNAs have been reported to be involved in fibrotic diseases [41]. The miR-200 family and miR-205 target ZEB1 and SIP1, two important transcriptional factors in the TGF-β pathway, and inhibit EMT in renal epithelial cells [42, 43]. Recently, miR-424 has been shown to induce EMT in cancer cells [44]. Several other miRNAs are also implicated in fibrotic lung diseases. TGF-β1 induces miR-21 expression, and miR-21 in turn promotes activation of fibroblasts by inhibiting Smad7 [45]. miR-155 targets the keratinocyte growth factor in lung fibroblasts to induce fibrosis [46]. Knock-down of miR-29 increases several pro-fibrotic genes in lung fetal fibroblasts [47]. A decrease in Let-7d is observed in the lungs of IPF patients, and studies in rat lung epithelial cells suggest that Let-7d contributes to EMT through its target, HMGA2 [43]. However, no direct evidence has been presented to show that miRNAs regulate EMT in human lung epithelial cells. In this study, we performed a miRNA microarray analysis on a human lung epithelial cell model of EMT. Six of the up-regulated and the down-regulated miRNAs were identified. Of these, only up-regulated miR-424 induced the expression of α-smooth muscle actin (α-SMA) during EMT. Moreover, miR-424 expression was increased by TGF-β, and miR-424 decreased Smurf2 protein expression. Together, our studies support a role for miR-424 in myofibroblast differentiation during EMT.
Materials and Methods
Culture of human A549 cells
Human lung epithelial cells (A549) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 1% penicillin/streptomycin and 10% heat-inactivated fetal bovine serum (FBS). Cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
Cell model of EMT
A549 cells (1.6 × 105 per well) were cultured in DMEM, supplemented with 10% charcoal-stripped FBS in 6-well plates. After reaching 60% confluence, the cells were treated with 5 ng/ml of recombinant human TGF-β1 (R&D Systems, Minneapolis, MN) or with vehicle alone for 4, 8 and 12 days. The concentration of TGF-β1 used in our study (5 ng/ml) was based on the report from literature [16] and was physiologically relevant [48]. To avoid confluence, the cells were split every 4 days using trypsin. One third of the cells were reseeded in the original plates, while the other two thirds were collected for mRNA and protein analyses.
Real-time PCR
Total RNA was isolated from cultured A549 cells using TRI-Reagent following the manufacturer’s instructions. RNA was treated with TURBO DNase (Ambion, Austin, TX) to remove genomic DNA contamination. One μg of RNA was reverse-transcribed into cDNA using M-MLV reverse transcriptase, random primers, and oligo dT. Real-time PCR was carried out on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) using SYBR Green I detection. The primers were designed using Primer Express software (Applied Biosystems) and are listed in Table 1. Data was normalized to 18S rRNA.
Table 1.
Real-time PCR primers for human mRNAs
| CDH1 Forward | 5′ TGCCCAGAAAATGAAAAAGG |
| CDH1 Reward | 5′ GTGTATGTGGCAATGCGTTC |
| CDH2 Forward | 5′ CTGCACAGATGTGGACAGGATT |
| CDH2 Reward | 5′ TTCTTTATCCCGGCGTTTCAT |
| α-SMA Forward | 5′ GAGAAGAGTTACGAGTTGCCTGA |
| α-SMA Reward | 5′ TGTTAGCATAGAGGTCCTTCCTG |
| ZEB1 Forward | 5′ AGCAGTGAAAGAGAAGGGAATGC |
| ZEB1 Reward | 5′ GGTCCTCTTCAGGTGCCTCAG |
| SIP1 Forward | 5′ CCACCACCTACAAGCTCACTCC |
| SIP1 Reward | 5′ AATGGCGATGGCGAGGAGAC |
| E2A Forward | 5′ TGTGCCAACTGCACCTCAA |
| E2A Reward | 5′ CCGTTTCAAACAGGCTGCTT |
| Id2 Forward | 5′ CGTGAGGTCCGTTAGGAAAA |
| Id2 Reward | 5′ AGGCTGACAATAGTGGGATG |
| Id3 Forward | 5′ ACTCACTCCCCAGCATGAAG |
| Id3 Reward | 5′ AAGCTCCTTTTGTCGTTGGA |
| Snail1 Forward | 5′ AGGATCTCCAGGCTCGAAAG |
| Snall1 Reward | 5′ GTAGCAGCCAGGGCCTAGAG |
| Snail2 Forward | 5′ CTGCGGCAAGGCGTTTTCCAGA |
| Snail2 Reward | 5′ CAGATGAGCCCTCAGATTTGAC |
| Twist Forward | 5′ CGGACAAGCTGAGCAAGATT |
| Twist Reward | 5′ CCTTCTCTGGAAACAATGAC |
| CTGF Forward | 5′ CAGCATGGACGTTCGTCTG |
| CTGF Reward | 5′ AACCACGGTTTGGTCCTTGG |
| FN Forward | 5′ AGCCTCGAAGAGCAAGAGG |
| FN Reward | 5′ CAAAACTTCAGCCCCAACTT |
| Smurf1 Forward | 5′ AGATCCGTCTGACAGTGTTATGT |
| Smurf1 Reward | 5′ AGATCCGTCTGACAGTGTTATGT |
| Smurf2 Forward | 5′ GGCAATGCCATTCTACAGATACT |
| Smurf2 Reward | 5′ CCACTTTGGATCAAGCGTATTCT |
| Smad7 Forward | 5′ ATAGCTAGCGCTTTACCGTGCAGATCAGCTT |
| Smad7 Reward | 5′ ATAGTCGACTTAATGGAACATAAACTCCTTT |
| 18S rRNA Forward | 5′ CGTTGATTAAGTCCCTGCCCTT |
| 18S rRNA Reward | 5′ TCAAGTTCGACCGTCTTCTCAG |
Real-time PCR for miRNAs was carried out as previously described [49, 50]. Briefly, two μg of total RNA, without DNase treatment, was polyadenylated using a Poly(A) polymerase tailing kit (Epicentre, Madison, WI). One μg of the poly(A) tailed RNA was then reverse-transcribed into cDNA using M-MLV reverse transcriptase, oligo dT and miRNA RT primers, including a universal reverse primer and a specific forward primer for each target miRNA listed in Table 2. Real-time PCR was performed using miRNA PCR primers (Table 2). Data were normalized to RNU6B small RNA.
Table 2.
Primers for miRNA Reverse transcription and real-time PCR
| Reverse transcription Primers | |
| has-miR-Universe | GTCGTGTCCAGTCGTGTGTT |
| hsa-miR-1183 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12GCCCA |
| hsa-miR-23b-5p | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12AAATCA |
| hsa-miR-1224-3p | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12CTGAGG |
| hsa-miR-155 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12ACCCCT |
| hsa-miR-298 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12GGGAG |
| hsa-miR-933 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12GGGAGA |
| hsa-miR-31 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12AGCTAT |
| hsa-miR-424 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12CAAA |
| hsa-miR-216b | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12CACAT |
| hsa-miR-219-2-3p | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12ACAGCT |
| hsa-miR-190 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12ACCTAA |
| hsa-miR-1273 | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12AAGAAA |
| hsa-miR-582-5p | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12AGTAAC |
| hsa-miR-487a | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12AACTGG |
| hsa-miR-136-3p | GCGAGCACAGAATTAATACGACTCACTATAGG(T)12AGACTC |
| Real-time PCR primers | |
| hsa-miR-1183 | ACTGACCACTGTAGGTGATGGT |
| hsa-miR-23b-5p | CAATTATGGGTTCCTGGCATGC |
| hsa-miR-1224-3p | ATTAATCCCCACCTCCTCTCTC |
| hsa-miR-155 | GCCGGCTTAATGCTAATCGTGA |
| hsa-miR-298 | TACATAGCAGAAGCAGGGAGG |
| hsa-miR-933 | ATTATATGTGCGCAGGGAGACC |
| hsa-miR-31 | AGTCGTAGGCAAGATGCTGGC |
| hsa-miR-424 | GCTCGACAGCAGCAATTCATGT |
| hsa-miR-216b | CGAGCTAAATCTCTGCAGGCAA |
| hsa-miR-219-2-3p | CTGCATAGAATTGTGGCTGGAC |
| hsa-miR-190 | CCGCGCTGATATGTTTGATATA |
| hsa-miR-1273 | TCTAGTGGGCGACAAAGCAAGA |
| hsa-miR-582-5p | CGACGGTTACAGTTGTTCAACC |
| hsa-miR-487a | CGTGCGAATCATACAGGGACAT |
| hsa-miR-136-3p | GCTCGGCATCATCGTCTCAAAT |
miRNA microarray
The miRNA microarray slides were printed in-house, as previously described [51], using oligos from the miRCURY LNA miRNA array v.11-has, mmu and rne (Exiqon, Woburn, MA), which contains 1,700 capture probes consisting of human, mouse and rat mature miRNAs. Of the 1,700 probes, there were 1,282 human miRNAs (854 from miRbase 11.0 and 428 from proprietary miRPlus miRNAs identified by Exiqon using cloning and sequencing) and 80 human viral miRNAs. Total RNA (300 ng) from control and 8 d TGF-β1-treated A549 cells were extracted and labeled with Hy3 and Hy5 using the microRNA Array Power labeling kit (Exiqon). The two labeled samples were co-hybridized to a miRNA microarray slide at 56°C for 16 h. There were three biological replications. A dye flip was performed to eliminate the dye bias. The slides were then scanned with a ScanArray Express scanner (PerkinElmer Life and Analytical Sciences, Boston, MA), and the images were analyzed using GenePix 5.0 software (Axon Instruments, Union City, CA). Data were analyzed using our in-house RealSpot software [52]. miRNAs with an average quality index (QI) <1 were excluded. Data of the remaining miRNAs were analyzed using Student’s t-test (p < 0.05).
miRNA over-expression
These studies utilized lentiviral miRNA expression vectors containing three elements, CMV-driven EGFP, followed by the mature miRNA sequence plus flanking sequences, which were then followed by the SV40 PolyA terminal sequence. The mature miRNA sequence and its 5′ and 3′ flanking sequences of approximately 250 nt were PCR-amplified from human genomic DNA and cloned into the pLVX (Lenti-X) vector (Clontech, Mountain View, CA). The lentivirus was generated in HEK 293T cells using Lenti-X HTX packaging mix (Clontech). To determine the titer of viruses, different dilutions of viral stocks were used to infect 293T cells, followed by counting GFP-positive infected cells. A549 cells were infected with miRNAs or control lentivirus at MOI=50 for 24 h. After infection, virus-containing medium was removed and the cells were cultured for an additional 24 h in fresh medium.
TGF-β reporter luciferase assay
Four million HEK 293T cells were seeded on a 96-well plate. Fifty ng of a TGF-β reporter plasmid (SABiosciences, Valencia, CA), which contained a Smad transcriptional response element and a firefly luciferase reporter gene, and 100 ng of miR-424 lentiviral expression plasmid were co-transfected into HEK 293T cells using Lipofectamine 1000. A lentiviral plasmid containing a scrambled sequence was used as a control. After 24 h of incubation, the cells were treated with TGF-β1 (5 ng/ml) or vehicle for 24 h and assayed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI).
3′-UTR pmirGLO vector construction
3′-UTR segments of three predicted targets, Smad7, Smurf1 and Smurf2 of miR-424, containing restriction sites NheI and SalI on the 5′ and 3′ ends, were amplified using PCR. Three PCR products and the pmirGLO empty vector (Promega, Madison, WI) were double-digested using NheI and SalI restriction endonucleases (New England Biolabs, Ipswich, MA). The 3′-UTR segments were then inserted into the pmirGLO vector using T4 DNA ligase (New England Biolabs, Ipswich, MA). The inserts were confirmed by sequencing.
Western blotting
The cells were harvested and lysed in a lysis buffer (PIERCE, Rockford, IL). Twenty μg of protein were separated on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. The membrane was incubated overnight at 4°C with mouse anti-Smad7 antibodies (1:200, SIGMA, St. Louis, MO), mouse anti-Smurf1 antibodies (1:500, SIGMA), or rabbit anti-Smurf2 antibodies (1:500, Novus Biologicals, Littleton, CO) after being blocked with 10% non-fat milk. Then the membrane was washed and incubated with goat anti-mouse or goat anti-rabbit secondary antibodies (1:2000) for 1 h. The blots were then developed with enhanced chemiluminescence (ECL) reagents (PIERCE, Rockford, IL).
Statistical analysis
Results were analyzed by the Student’s t-test for two independent groups, one-way ANOVA followed by Tukey’s posthoc test or two-way ANOVA with Bonferroni posttests for multiple comparisons.
Results
TGF-β1 reduces epithelial cell marker expression and increases mesenchymal cell marker expression
In order to investigate the role of miRNAs in EMT, we used a TGF-β1-induced EMT cell model [16]. We treated human lung epithelial A549 cells with 5 ng/ml of TGF-β1 for 4, 8, or 12 days, and determined epithelial and mesenchymal cell mRNA marker expression. E-cadherin (CDH1), an epithelial marker, was down-regulated in a time-dependent manner. It dramatically decreased on day 4 and was barely detectable on day 12 (Fig. 1A). N-cadherin (CDH2), a mesenchymal marker, was increased on day 4, peaked on day 8 and returned to the day 4 level by day 12 (Fig. 1B). α-smooth muscle actin (α-SMA), a myofibroblast marker, was marginally increased on day 8 (Fig. 1C), consistent with a previous report [16]. These results indicate that TGF-β causes EMT, but is not sufficient for myofibroblast differentiation.
Fig. 1. TGF-β1 induces EMT in human alveolar epithelial cells.

Human A549 cells were treated with TGF-β1 (5 ng/ml) for 4, 8 and 12 days (D4, D8 and D12). The mRNA expressions of the cell markers were determined by real-time PCR. (A) Epithelial cell marker, E-cadherin (CDH1). (B) Mesenchymal cell marker, N-cadherin (CDH2). (C) Myofibroblast marker, α-smooth muscle actin (α-SMA). Results were normalized to 18S rRNA and expressed as a ratio of the TGF-β1 treated group to the control group at each time point. Results are means ± s.e. from the three independent experiments, which were performed in triplicate. *P<0.05 v.s. without TGF-β1; **P<0.01 v.s. without TGF-β1; ***P<0.001 v.s. without the TGF-β1. Student’s t-test.
We further examined genes that encode key transcription factors in the TGF-β signaling pathway. We found that mRNA for ZEB1, a repressor of epithelial genes, peaked on Day 8 (Fig. 2A). However, SIP1, another repressor of epithelial genes, was not significantly affected at any time point by TGF-β (Fig. 2B). Among other transcription factors in the TGF-β signaling pathway which were tested, Snail2 mRNA showed the greatest increase by TGFβ, while Id2 (an inhibitor of the E-cadherin repressor, E2A) showed the greatest decrease (Fig. 2C).
Fig. 2. Effects of TGF-β on transcription factors.

A549 cells were treated with TGF-β1 (5 ng/ml) for 4, 8 and 12 days (D4, D8 and D12). The mRNA levels of ZEB1 (A) and SIP1 (B) were measured by real-time PCR and expressed as a ratio of the TGF-β1 treated group to the control group at each time point. The mRNA levels of E2A, Id2, ID3, Snail1, Snail2, and Twist on day 8 after TGF-β1 were also determined by real-time PCR and expressed as delta delta CT (C). Data were normalized to 18S rRNA. Results are means ± s.e. from the three independent experiments, which were performed in triplicate. *P<0.05 v.s. without TGF-β1; **P<0.05 v.s. without TGF-β1. Student’s t-test.
Six miRNAs are up-regulated and three miRNAs are down-regulated during EMT
Since the mesenchymal cell marker, CDH2, and the transcription factor, ZEB1, peaked on day 8 of TGF-β1 treatment, we chose this time point for miRNA microarray analysis as described in our previous studies [51–53], with a goal of identifying miRNAs involved in the process of EMT. We used in-house printed miRNA microarray slides containing 1,700 miRCURY LNA probes for human, rat and mouse miRNAs (EXIQON). After quality control and statistical analysis, six miRNAs were found to be up-regulated, while nine miRNAs were down-regulated. Since miRNA microarray is semi-quantitative [36], we used real-time PCR to verify the miRNA microarray data. We verified 9 miRNAs, including 6 up-regulated (miR-31, miR-190, miR-424, miR-136-3p, miR-487a and miR-582-5p) and 3 down-regulated (miR-1224-3p, miR-23b-5p and miR-933) miRNAs (Fig. 3).
Fig. 3. miRNAs changed during EMT.
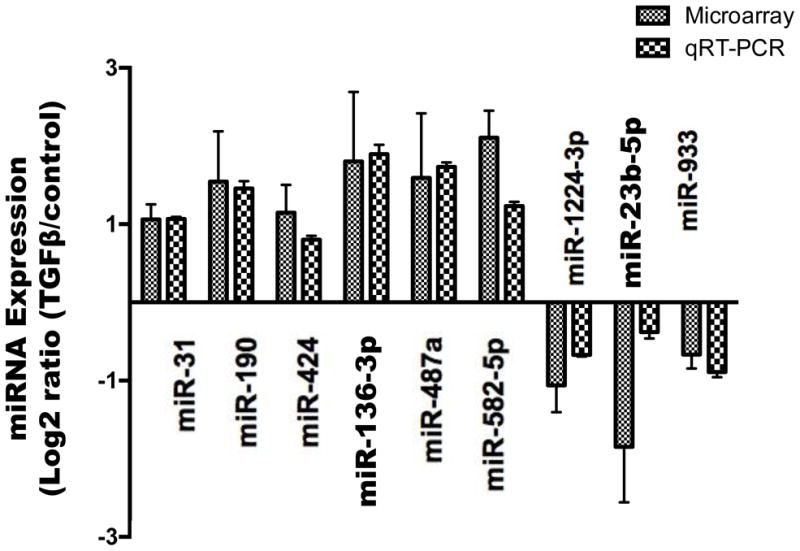
A549 cells were treated with 5 ng/ml of TGF-β1 for 8 days. miRNA microarray and real-time PCR were performed to identify the miRNAs changed during EMT. All real-time PCR data were normalized to RNU6B small RNA. The results were expressed as log2 ratio (TGF-β1-treated vs control). Data shown are means ± s.e. All microarray and real-time PCR results were statistically significant (P<0.05). n=3 cell preparations. Student’s t-test.
Over-expression of miR-424 promotes myofibroblast differentiation
We designed two experiments to test whether the identified 9 miRNAs affected EMT. If TGF-β1-mediated EMT is due to the down-regulation of inhibitory miRNAs, the over-expression of these down-regulated miRNAs should inhibit TGF-β1-induced EMT. On the other hand, if the up-regulated miRNAs during EMT cause EMT, the over-expression of these miRNAs should mimic the effect of TGF-β1 treatment. We over-expressed the 3 down-regulated miRNAs in A549 cells using lentiviral miRNA expression vectors, treated the cells with TGF-β1, and determined the epithelial and mesenschymal cell marker expression. Real-time PCR analysis revealed that over-expression of those miRNAs had no effect on the expression of the epithelial marker CDH1 (Fig. 4A) compared to the virus control. However, expression of the mesenchymal marker CDH2 was increased by 2 of the 3 miRNAs tested (miR-1224-5p and miR-23b), which was an effect opposite to that expected if these miRNAs have a role in EMT (Fig. 4B). Furthermore, miR-23b and miR-933 also slightly increased α-SMA expression (Fig. 4C). These results suggest that the 3 down-regulated miRNAs tested are likely not involved in EMT.
Fig. 4. Effect of over-expressing miRNAs on the expression of epithelial cell marker and mesenchymal cell markers.

A549 cells were infected with a miRNA lenti-virus at a MOI of 50 for 2 days and then treated with TGF-β1 (5 ng/ml) (A-C) or nothing (D-F) for 3 days. mRNA levels of CDH1, CDH2 and α-SMA were measured by real-time PCR and normalized to 18S rRNA. Results are means ± s.e. (two cell preparations, measured in duplicate), except miR-424 which had 5 cell preparations. *p<0.05 v.s. VC. BC: Blank control, VC: Virus control.
Next, we overexpressed the 6 up-regulated miRNAs in A549 cells without TGF-β1 treatment to see whether they could induce EMT. miR-136, miR-190, miR-31 and miR-424 had no effect on either the CDH1 or CDH2 expression (Fig. 4D, E). In contrast, miR-487a and miR-582 increased both epithelial (CDH1) and mesenchymal (CDH2) marker expression. This is not consistent with EMT, in which epithelial cell markers are decreased and mesenchymal cell markers are increased. However, miR-424 significantly increased α-SMA expression (Fig. 4F).
We further examined the effects of miR-424 on the myofibroblast differentiation during TGF-β1-induced EMT. The miR-424 level was increased by 25 fold when A549 cells were treated with a miR-424 lentivirus (Fig. 5A). TGF-β1 further increased the miR-424 level in the miR-424 lentivirus-treated cells. While TGF-β1 decreased CDH1 and increased CDH2 mRNA expression, miR-424 had no effect on CDH1 or CDH2 mRNA expression in either the presence or the absence of TGF-β1 (Fig. 5B, C). However, over-expression of miR-424 increased α-SMA mRNA expression. The combination of miR-424 over-expression with TGF-β1 treatment further increased α-SMA expression compared to TGF-β1 or miR-424 over-expression alone (Fig. 5D). The same effects were observed for connective tissue growth factor (CTGF), another myofibroblast marker (Fig. 5E). Although miR-424 overexpression alone had no effect on the fibronectin (FN) mRNA level, the combination of TGF-β1 and miR-424 over-expression increased FN expression (Fig. 5F). The results suggested that miR-424 may enhance myofibroblast differentiation, but not EMT.
Fig. 5. Over-expression of miR-424 promotes myofibroblast differentiation during EMT.

A549 cells were infected with miR-424 lentivirus at a MOI of 50 for 24 h, followed by TGF-β1 treatment (5 ng/ml) for 72 h. miR-424 and mRNA expressions were determined using real-time PCR. (A) miR-424, (B) CDH1, (C) CDH2, (D) α-SMA, (E) CTGF, and (F) FN. miR-424 expression was normalized to RNU6B, while mRNA expression was normalized to 18S rRNA. BC: Blank control, VC: Virus control, CTGF: Connective tissue growth factor, FN: Fibronectin. Results are means ± s.e. (n=3). *P<0.01 v.s. VC. #P<0.05 v.s. non-TGFβ1 treated control. Two-way ANOVA with Bonferroni post-test.
miR-424 enhances activity of the TGF-β signaling pathway
Since miRNAs form many positive and negative regulatory loops, and miR-424 is regulated by TGF-β signaling, we examined whether miR-424 affects the activity of the TGF-β pathway using a TGF-β reporter luciferase assay. TGF-β1 increased the reporter activity in the vector control group. miR-424 alone enhanced the reporter activity to almost the same level as that in the TGF-β1-treated vector control group (Fig. 6). The combination of miR-424 over-expression and TGF-β1 stimulation further increased the reporter activity. This result indicates that miR-424 potentiates the TGF-β signaling pathway.
Fig. 6. miR-424 increases TGF-β signaling activity.

HEK 293T cells were co-transfected with 50 ng of the TGF-β signaling reporter plasmid and 100 ng of miR-424 expression plasmid by using Lipofectamine. After 24 h of incubation, the cells were treated with TGF-β1 (5 ng/ml) for 24 h and assayed for dual-luciferase activities. The results shown are mean ± s.e. (n=3). *P<0.001 v.s. vector control (VC). #P<0.001 v.s. non-TGF-β1 treated control.
Smurf2 is a target of miR-424
Since miRNAs negatively regulate their target genes, we would expect that miR-424 targets negative regulators in the TGF-β signaling pathway to enhance TGF-β signaling activity. Using Web- based target predication software (TargetScan, miRand and DIANA-microT), we identified three negative regulators of the TGF-β/Smad signaling (Smad7, Smurf1 and Smurf2) as potential targets of miR-424 (Table 3). To verify the prediction, we first used a 3′-UTR reporter assay in HEK293 cells. As shown in Fig. 7A, miR-424 inhibited the 3′-UTR reporter activities of Smad7, Smurf1 and Smurf2. Since miRNA-mediated effects may be cell content-dependent, we further studied whether miR-424 could reduce the endogenous expression of these proteins in A549 cells. We increased the miR-424 expression level in A549 cells using a miR-424 lentivirus at a MOI of 50. Western blotting revealed that the Smurf2, but not Smurf1 or Smad7 protein levels, was significantly decreased in the miR-424-over-expressed cells in comparison with virus control or blank control groups (Fig. 7B, C). The mRNA expression levels of the three predicted targets were not affected by miR-424 (Fig. 7D), suggesting that miR-424 directly inhibits protein translation of Smurf2.
Table 3.
Predicated binding sites of miR-424
| Smad7 | Smurf1 | Smurf2 | |
|---|---|---|---|
| miRRanda | 42–62 | 2904–2925 | 210–232 |
| TargetScan 6.2 | 55–62 | 2918–2925 (conserved) 451–457 (poorly conserved) |
225–231 |
| DIANA microT 3.0 | 33–61 | 2896–2924 931–959 1368–1396 |
N/A |
Fig. 7. Effects of miR-424 on the expression of Smurf2.

(A) 3′-UTR reporter assay. 3′-UTR reporter plasmids of Smad7, Smurf1 or Smurf2 were co-transfected with a miR-424 expression vector, or its control vector (Con), into HEK293 cells. The dual luciferase activities were measured and expressed a ratio of firefly to Renilla luciferase activity. The results were normalized to the empty vector control without 3′-UTR (EVC) from the two independent experiments, each with 3 replications. *P<0.005 v.s. control. (B-D) A549 cells were infected with a miR-424 lentivirus at a MOI of 50 for 48 h. Protein and mRNA levels of Smad7, Smurf1 and Smurf2 were determined using Western blotting and real-time PCR. (B) Western blot showed the protein expressions of Smurf1, Smurf2 and Smad7. GAPDH was used as a loading control. (C) The quantitation of protein levels from Western blotting using Image J. Data was normalized to GAPDH. (D) mRNA levels of Smurf1, Smurf2 and Smad7 were determined by real-time PCR and normalized to 18S rRNA. BC: Blank control, VC: Virus control. The results shown are means ± s.e. (n=3). *P<0.05 v.s. VC. Two-way ANOVA with bonferroni post-test.
Discussion
The purpose of this study was to identify specific miRNAs that have roles in EMT during IPF. First, we identified 6 up-regulated and 3 down-regulated miRNAs in the TGF-β-induced EMT model of human lung epithelial cells through miRNA microarray analysis. One of these up-regulated miRNAs, miR-424, enhanced the expression of α-SMA without affecting epithelial or mesenchymal cell markers. miR-424 also increased the activity of the TGF-β signaling pathway and decreased the protein expression of Smurf2, a negative regulator of TGF-β signaling. Our results suggest that miR-424 regulates the myofibroblast differentiation during EMT by potentiating the TGF-β signaling pathway, likely through Smurf2.
miRNAs have been reported to regulate EMT in a variety of cell lines. Previous studies have shown that the miR-200 family and miR-205 are gradually down-regulated during TGF-β1-mediated EMT. The manipulation of their levels affects EMT in renal epithelial cells by targeting ZEB1 and SIP1 [42]. The miR-200 family is also down-regulated in IPF patients, and regulates EMT in rat alveolar epithelial cells (AECs) [54]. A number of other miRNAs have also been reported to be involved in lung fibrotic diseases, including miR-21, miR-155, miR-29 and let-7d [41, 43, 45–47]. However, direct miRNA microarray analysis of EMT in lung epithelial cells has not been reported yet. In this study, we identified 9 miRNAs which were changed during EMT of the lung epithelial cells. Among these 9 miRNAs, only miR-31 has been shown to have a role in EMT by inhibiting the activation of lung fibroblasts through directly targeting integrin α(5) and RhoA [55].
The changes in miRNAs during EMT do not necessarily mean that the altered miRNAs have functional roles in EMT. Indeed, using the gain-of-function approach, we found that only miR-424 markedly increased α-SMA expression. α-SMA is considered to be a marker for myofibroblasts and an indicator for myofibroblast differentiation [45, 56, 57]. In lung fibrotic diseases, the persistence of myofibroblasts is an important indicator of the end stage of the disease [20, 21, 58]. Myofibroblasts have stronger contractile activity and are more responsive to inflammatory factors than fibroblasts [59]. Thus, the increase in myofibroblasts in the diseased lung may reduce lung compliance, enhance extracellular matrix production, disrupt the basement membrane and cause inflammation and epithelial injury [60, 61]. It is still not clear what causes myofibroblast differentiation. Several factors have been studied including mechanical stretch, specialized matrix proteins, inflammatory factors, and the TGF-β signaling pathway [58, 62, 63].
A number of studies have characterized regulators of α-SMA transcription. Smad3, a key transcription factor in the Smad-dependent TGF-β signaling pathway, induces α-SMA expression through its binding with two CAGA motifs, called Smad3-binding elements (SBEs), in the promoter of the α-SMA gene [64]. Smad3-independent signaling via p38 and MEK/ERK also contributes to α-SMA regulation [65]. Sp1/Sp3 binding to the MCAT enhancer is also considered to be necessary for α-SMA expression [66]. In addition to the TGF-β signaling pathway, other mechanisms have also been reported to regulate the expression of α-SMA [58]. The Notch/CSL pathway regulates α-SMA expression through its promoter activity [67]. CCAAT/enhancer binding protein β (C/EBPβ) also induces α-SMA expression in lung fibroblasts [68]. The transcription repressor, Gut-enriched Krüppel-like factor, negatively regulates α-SMA by preventing the binding of Smad3 to SBEs [69].
miR-424 has not previously been reported to be involved in any of the pathways mentioned above. Our studies found that miR-424 enhanced the activity of Smad-dependent TGF-β signaling, as revealed by a TGF-β signaling reporter assay. This result suggests that the up-regulation of α-SMA produced by miR-424 likely occurs through potentiating TGF-β signaling. The reporter plasmid we used has a transcription response element (TRE) which can be activated by the Smad2/3/4 complexes, and thus the activity of the reporter represents the activity of the Smad-dependent TGF-β signaling pathway. The Smads are well-studied intracellular effectors of the TGF-β signaling pathway. There are three groups of Smads: receptor-activated Smads (R-Smads, Smad2/3), a common Smad (Smad4), and inhibitory Smads (Smad7). Upon ligand binding, R-Smads are phosphorylated by phosphorylated type II receptors. The phosphorylation of R-Smads on their C-terminal SXS motif results in changes of their conformation, causing the exposure of interacting interfaces and two R-Smads, either homomeric or heteromeric, and one common Smad form a trimeric complex, which then dissociates from the receptors and is translocated into the nucleus, where it acts as a transcription factor regulating numerous transcriptional activities [30]. The inhibitory Smad, Smad7, inhibits the TGF-β signaling pathway by competitively interfering with R-Smad recruitment and phosphorylation by TGF-β receptors [30, 70]. The TGF-β signaling pathway forms an inhibitory feedback loop through the induction of Smad7 expression [71].
Smurf2, another known TGF-β signaling negative regulator, belongs to the family of Smad ubiquitin regulatory factors (Smurfs), which are HECT-domain-containing E3 ligases. They are748 amino acids long and have several distinctive structural features as compared to other E3 ligases, including a phospholipid/calcium-binding C2 domain and WW domains [72]. Smurf2 has been reported to directly interact with Smad2 and Smad3 through the linker region containing a PY motif, which is recognized by the WW domain, causing proteasome-dependent degradation of Smad2 and Smad3 [73–75]. In addition, Smurf2 can also form a complex with Smad7, which then is exported from the nucleus to the cytoplasm and targets the activated TGF-β type I receptor to proteasomes for degradation through ubiquitination [75, 76].
By using web-based target prediction databases, we found that three inhibitory regulators of the TGF-β signaling pathway, Smad7, Smurf1 and Smurf2, were potential targets of miR-424. Western blotting showed that the protein level of Smurf2, but not Smurf1 or Smad7, was significantly decreased after over-expression of miR-424. Furthermore, the mRNA level of Smurf2 was unchanged by miR-424, indicating that the protein change of Smurf2 was not due to mRNA cleavage but because of translation, which is the major mechanism of miRNAs in animals. Although Smurf1 and Smurf2 share most of the same sequence, Smurf2 acts as a specific negative regulator for the Smad signaling pathway [72]. This explains why miR-424 can enhance TGF-β signaling by only decreasing the protein level of Smurf2, but not Smurf1.
miR-424 promoted myofibroblast differentiation, but did not promote EMT in response to TGF-β treatment, even though the TGF-β signaling pathway regulates both processes. The mechanisms remain to be identified. We speculate that the disparate effects of miR-424 on these two processes are due to the specificity of Smad2 and Smad3 in downstream gene expression. Knockout of Smad2, but not Smad3, prevents EMT [77], indicating that Smad2 is involved in EMT. On the other hand, as mentioned above, Smad3 is believed to be the main regulator of TGF-β mediated α-SMA expression [58, 64, 69]. Although Smurf2 has been shown to cause the degradation of TβRI, the Smad2 and Smad3 in vitro [73–76] and in vivo study of Smurf2 knockout mice reveals that Smurf2 does not cause the degradation of Smad2, Smad3 or the TGF-β type I receptor. However, it does induce the multiple mono-ubiquitination of the Smad3 MH2 domain to inhibit the formation of Smad3 complexes [72]. This suggests that Smurf2 acts by specifically inhibiting Smad3. Thus, the reduction of Smurf2 by miR-424 enhances the activity of Smad3, leading to enhanced α-SMA expression after EMT.
In summary, we demonstrated a two-step process of EMT: first, human lung epithelial cells lose epithelial characteristics and gain mesenchymal markers to become fibroblast-like cells, which can be induced by TGF-β treatment alone; and second, the fibroblast-like cells differentiate into myofibroblasts, in which miR-424 potentiates TGF-β signaling.
Highlights.
Identified 6 up-regulated and 3 down-regulated miRNAs in lung epithelial cell EMT.
miR-424 increased the expression of α-smooth muscle actin.
miR-424 enhanced the activity of the TGF-β signaling pathway.
miR-424 decreased the protein expression of Smurf2.
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute under Award Number R01 HL116876 and R03 HL-95383 (to LL), and the National Institute of General Medical Sciences under Award number P20GM103648 (Molecular Biology Core) and grants from Oklahoma Center for Adult Stem Cell Research (to LL, MH and PL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. XX was supported by a pre-doctoral fellowship from the American Heart Association (12PRE12060188).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gross TJ, Hunninghake GW. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 2.Chapman HA. Annu Rev Physiol. 2011;73:413–435. doi: 10.1146/annurev-physiol-012110-142225. [DOI] [PubMed] [Google Scholar]
- 3.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Am J Respir Crit Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 4.Hodgson U, Pulkkinen V, Dixon M, Peyrard-Janvid M, Rehn M, Lahermo P, Ollikainen V, Salmenkivi K, Kinnula V, Kere J, Tukiainen P, Laitinen T. Am J Hum Genet. 2006;79:149–154. doi: 10.1086/504639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khalil N, Parekh TV, O’Connor R, Antman N, Kepron W, Yehaulaeshet T, Xu YD, Gold LI. Thorax. 2001;56:907–915. doi: 10.1136/thorax.56.12.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, Evans CM, Garantziotis S, Adler KB, Dickey BF, du Bois RM, Yang IV, Herron A, Kervitsky D, Talbert JL, Markin C, Park J, Crews AL, Slifer SH, Auerbach S, Roy MG, Lin J, Hennessy CE, Schwarz MI, Schwartz DA. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubbard R, Lewis S, Richards K, Johnston I, Britton J. Lancet. 1996;347:284–289. doi: 10.1016/s0140-6736(96)90465-1. [DOI] [PubMed] [Google Scholar]
- 8.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Am J Respir Crit Care Med. 1997;155:242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 9.Kelly BG, Lok SS, Hasleton PS, Egan JJ, Stewart JP. Am J Respir Crit Care Med. 2002;166:510–513. doi: 10.1164/rccm.2103058. [DOI] [PubMed] [Google Scholar]
- 10.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 11.Xu YD, Hua J, Mui A, O’Connor R, Grotendorst G, Khalil N. Am J Physiol Lung Cell Mol Physiol. 2003;285:L527–L539. doi: 10.1152/ajplung.00298.2002. [DOI] [PubMed] [Google Scholar]
- 12.Kalluri R, Neilson EG. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thannickal VJ, Toews GB, White ES, Lynch JP, III, Martinez FJ. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 15.Yao HW, Xie QM, Chen JQ, Deng YM, Tang HF. Life Sci. 2004;76:29–37. doi: 10.1016/j.lfs.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wynn TA. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hardie WD, Glasser SW, Hagood JS. Am J Pathol. 2009;175:3–16. doi: 10.2353/ajpath.2009.081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araya J, Nishimura SL. Annu Rev Pathol. 2010;5:77–98. doi: 10.1146/annurev.pathol.4.110807.092217. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y, Massague J. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiery JP. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 26.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 27.Thiery JP, Acloque H, Huang RY, Nieto MA. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Marmai C, Sutherland RE, Kim KK, Dolganov GM, Fang X, Kim SS, Jiang S, Golden JA, Hoopes CW, Matthay MA, Chapman HA, Wolters PJ. Am J Physiol Lung Cell Mol Physiol. 2011;301:L71–L78. doi: 10.1152/ajplung.00212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massague J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 30.Feng XH, Derynck R. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 31.Peinado H, Olmeda D, Cano A. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 32.Warner DR, Greene RM, Pisano MM. FEBS Lett. 2005;579:3539–3546. doi: 10.1016/j.febslet.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Thiery JP, Sleeman JP. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 34.Willis BC, Borok Z. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 35.Katoh Y, Katoh M. Int J Mol Med. 2008;22:271–275. [PubMed] [Google Scholar]
- 36.Sun W, Julie Li YS, Huang HD, Shyy JY, Chien S. Annu Rev Biomed Eng. 2010;12:1–27. doi: 10.1146/annurev-bioeng-070909-105314. [DOI] [PubMed] [Google Scholar]
- 37.Berezikov E, Cuppen E, Plasterk RH. Nat Genet. 2006;38(Suppl):S2–S7. doi: 10.1038/ng1794. [DOI] [PubMed] [Google Scholar]
- 38.Majoros WH, Ohler U. BMC Genomics. 2007;8:152. doi: 10.1186/1471-2164-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erson AE, Petty EM. Clin Genet. 2008;74:296–306. doi: 10.1111/j.1399-0004.2008.01076.x. [DOI] [PubMed] [Google Scholar]
- 40.Stefani G, Slack FJ. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 41.Pandit KV, Milosevic J, Kaminski N. Transl Res. 2011;157:191–199. doi: 10.1016/j.trsl.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 42.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 43.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, Richards T, Selman M, Watkins SC, Pardo A, Ben-Yehudah A, Bouros D, Eickelberg O, Ray P, Benos PV, Kaminski N. Am J Respir Crit Care Med. 2010;182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banyard J, Chung I, Wilson AM, Vetter G, Le BA, Bielenberg DR, Zetter BR. Sci Rep. 2013;3:3151. doi: 10.1038/srep03151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pottier N, Maurin T, Chevalier B, Puissegur MP, Lebrigand K, Robbe-Sermesant K, Bertero T, Lino Cardenas CL, Courcot E, Rios G, Fourre S, Lo-Guidice JM, Marcet B, Cardinaud B, Barbry P, Mari B. PLoS ONE. 2009;4:e6718. doi: 10.1371/journal.pone.0006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, Thannickal VJ, Cardoso WV, Lu J. Am J Respir Cell Mol Biol. 2010;45:287–294. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakefield LM, Letterio JJ, Chen T, Danielpour D, Allison RS, Pai LH, Denicoff AM, Noone MH, Cowan KH, O’Shaughnessy JA. Clin Cancer Res. 1995;1:129–136. [PubMed] [Google Scholar]
- 49.Shi R, Chiang VL. Biotechniques. 2005;39:519–525. doi: 10.2144/000112010. [DOI] [PubMed] [Google Scholar]
- 50.Weng T, Mishra A, Guo Y, Wang Y, Su L, Huang C, Zhao C, Xiao X, Liu L. Biochem Biophys Res Commun. 2012;422:586–589. doi: 10.1016/j.bbrc.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Weng T, Gou D, Chen Z, Chintagari NR, Liu L. BMC Genomics. 2007;8:29. doi: 10.1186/1471-2164-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z, Liu L. Physiol Genomics. 2005;21:284–291. doi: 10.1152/physiolgenomics.00236.2004. [DOI] [PubMed] [Google Scholar]
- 53.Chen Z, Chen JW, Weng T, Jin N, Liu L. BMC Genomics. 2006;7:47. doi: 10.1186/1471-2164-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang S, Banerjee S, De FA, Sanders YY, Ding Q, Matalon S, Thannickal VJ, Abraham E, Liu G. Am J Pathol. 2012;180:484–493. doi: 10.1016/j.ajpath.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang S, Xie N, Cui H, Banerjee S, Abraham E, Thannickal VJ, Liu G. FASEB J. 2012;26:3790–3799. doi: 10.1096/fj.11-202366. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Duffy HS. J Cardiovasc Pharmacol. 2011;57:373–375. doi: 10.1097/FJC.0b013e3182155a38. [DOI] [PubMed] [Google Scholar]
- 57.Kis K, Liu X, Hagood JS. Expert Rev Mol Med. 2011;13:e27. doi: 10.1017/S1462399411001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Mol Biol Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hinz B. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 61.Selman M, King TE, Pardo A. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 62.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 63.Darby IA, Hewitson TD. Int Rev Cytol. 2007;257:143–179. doi: 10.1016/S0074-7696(07)57004-X. [DOI] [PubMed] [Google Scholar]
- 64.Hu B, Wu Z, Phan SH. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 65.Ramirez AM, Shen Z, Ritzenthaler JD, Roman J. Am J Transplant. 2006;6:2080–2088. doi: 10.1111/j.1600-6143.2006.01430.x. [DOI] [PubMed] [Google Scholar]
- 66.Cogan JG, Subramanian SV, Polikandriotis JA, Kelm RJ, Jr, Strauch AR. J Biol Chem. 2002;277:36433–36442. doi: 10.1074/jbc.M203232200. [DOI] [PubMed] [Google Scholar]
- 67.Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Circ Res. 2006;98:1468–1470. doi: 10.1161/01.RES.0000229683.81357.26. [DOI] [PubMed] [Google Scholar]
- 68.Hu B, Ullenbruch MR, Jin H, Gharaee-Kermani M, Phan SH. J Pathol. 2007;211:455–462. doi: 10.1002/path.2119. [DOI] [PubMed] [Google Scholar]
- 69.Hu B, Wu Z, Liu T, Ullenbruch MR, Jin H, Phan SH. Am J Respir Cell Mol Biol. 2007;36:78–84. doi: 10.1165/rcmb.2006-0043OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten DP. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 71.Miyazono K. J Cell Sci. 2000;113(Pt 7):1101–1109. doi: 10.1242/jcs.113.7.1101. [DOI] [PubMed] [Google Scholar]
- 72.Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C, Deng CX, Cheng SY, Zhang YE. EMBO J. 2011;30:4777–4789. doi: 10.1038/emboj.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin X, Liang M, Feng XH. J Biol Chem. 2000;275:36818–36822. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Proc Natl Acad Sci U S A. 2001;98:974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Massague J, Seoane J, Wotton D. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 76.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 77.Nawshad A, LaGamba D, Polad A, Hay ED. Cells Tissues Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]