

Abstract
Cellular fate depends on the spatio-temporal separation and integration of signaling processes which can be provided by phosphorylation events. In this study we identify the crucial points in signaling crosstalk which can be triggered by discrete phosphorylation events on a single target protein. We integrated the data on individual human phosphosites with the evidence on their corresponding kinases, the functional consequences on phosphorylation on activity of the target protein and corresponding pathways. Our results show that there is a substantial fraction of phosphosites that can play critical roles in crosstalk between alternative or redundant pathways and regulatory outcome of phosphorylation can be linked to a type of phosphorylated residue. These regulatory phosphosites can serve as hubs in the signal flow and their functional roles are directly connected to their specific properties. Namely, phosphosites with similar regulatory functions are phosphorylated by the same kinases and participate in regulation of similar biochemical pathways. Such sites are more likely to cluster in sequence and space unlike sites with antagonistic outcomes of their phosphorylation on a target protein. In addition we found that in silico phosphorylation of sites with similar functional consequences have comparable outcomes on a target protein stability. An important role of phosphorylation sites in biological crosstalk is evident from the analysis of their evolutionary conservation.
Introduction
Recent phosphoproteomic analyses showed that almost half of all proteins in eukaryotic cells are phosphorylated and protein phosphorylation enables cells to dynamically regulate protein activity, subcellular localization, and transmit signals downstream the reaction path1;2. Regulatory mechanisms of phosphorylation are quite diverse. It may be accompanied by changes in local site environment or global conformation, lead to protein activation or inactivation 3. At the same time it can modulate the nature and strength of protein interactions, thereby regulating protein binding and coordinating different pathways 4; 5.
Many proteins contain multiple phosphorylation sites which can control different functions of the target protein and provide an expanded combinatorial repertoire for regulation of functional activity. For example, the binding affinity of tumor suppressor protein p53 to CREB binding protein is modulated by multiple phosphorylation events and its triple phosphorylation results in a ten-fold increase in affinity compared to a single phosphorylation 6. In other cases, phosphorylation at different sites might have an opposite effect on protein activity causing protein activation or inhibition 7; 8. Multiple sites can be (de)phosphorylated by single or different kinases or phosphatases which might serve as a basis of separation or integration of various signals and allow system control by different agonists 9 (Figure 1). Moreover, the mechanism of phosphorylation might define the response kinetics and it is known that sequential phosphorylation may result in steeper response curves while random phosphorylation gives rise to more shallow responses 10; 11.
Figure 1. Scenarios of pathway regulation by single or multiple phosphorylation.
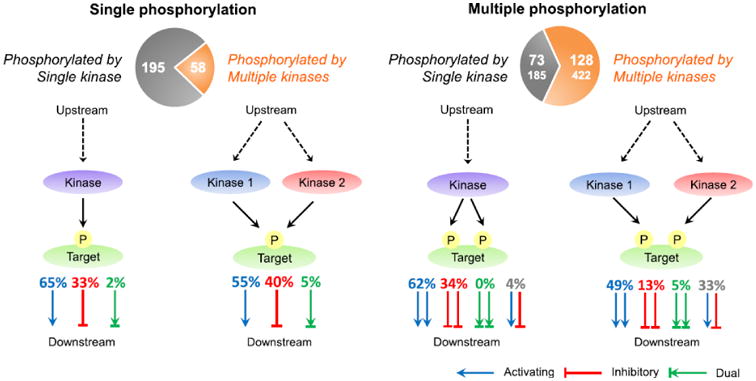
Left and right: regulation by proteins containing single or multiple phosphosites respectively. For each panel two scenarios are illustrated: when site/sites are phosphorylated by one kinase or multiple kinases. Functional effect on a target protein is shown by: activating (blue), inhibitory (red) and dual (green) arrow(s). In the right panel, site pairs with the same function (homo-functional pairs; activating-activating, inhibitory-inhibitory, dual-dual) are shown as a set of two red, blue, or green arrows respectively, while pairs with different function (hetero-functional pairs) are shown as a combination of blue and red arrows. Homo-functional pairs may provide a pathway crosstalk by integrating activating or inhibitory signals. Hetero-functional site pairs may provide a different scenario where phosphorylation of one site leads to activation while phosphorylation of another site on the same protein leads to inhibition. Note that hetero-functional pairs include activating-inhibitory, inhibitory-dual, and dual-activating. Percentage of sites with given functional consequences is listed above the arrows. Pie charts show the number of proteins (sites in parentheses) phosphorylated by single (grey) and multiple (brown) kinases. The numbers were based on the intersection of Function and Kinase sets.
Biological signaling is very complex, involving many states and oftentimes redundant or alternative relationships between the systems components. The signaling complexity in turn may or may not be accompanied by modularity and hierarchical organization 12; 13. It has been argued that such a seemingly unnecessary increase in diversity of regulatory systems might compensate for the variety of inputs and disturbances to provide specific system responses 14. Moreover, cellular fate depends on the spatio-temporal distinction between signaling processes and requires the correct integration and separation of different cellular signals which in turn provides signal amplification and enhances the response sensitivity. At the same time, the signal integration and separation between alternative or redundant pathways may provide better response specificity. There can be multiple points in signaling pathways which mediate such pathway crosstalk when the components and their functional states of one pathway may affect the function of another pathway. In some cases pathway crosstalk may be sustained by single proteins 13 through molecular switches provided by post-translational modifications. Namely different phosphorylation events may lead to inhibition or activation of the target protein and consequently potentially inhibit one pathway and activate another.
There have been numerous studies addressing the topic of topological properties of regulatory networks with the ultimate goal of identifying their hubs and bottlenecks 15; 16. However, a full understanding of how signal propagation is controlled requires an integration of systems and molecular levels of description. In particular, deducing the ubiquitous principles of regulation of protein activity and signal transduction through phosphorylation of individual sites remains an unsolved problem. In this study we try to pinpoint the crucial points in signaling and pathway crosstalk which are triggered by discrete phosphorylation events. We integrate the data on individual phosphosites with the evidence on their corresponding kinases and functional consequences of phosphorylation on the target protein and downstream signaling. Our results show that there are certain patterns in phosphosites' locations and structural/sequence properties which point to their potential role in mediating the communication between different functional states and pathways. Namely, phosphosites having similar regulatory functions, sharing the same kinases and participating in regulation of similar pathways are more likely to cluster in sequence and space. Phosphorylation of sites with similar downstream functional consequences as well as phosphosites regulated by the same kinase have comparable effects on protein stability. The fundamental regulatory role of such phosphorylation sites is also evident from their evolutionary conservation patterns.
Results
Functional effect of phosphorylation can be linked to phosphorylated residue type
Molecular mechanisms of signaling through phosphorylation differ depending on the external signal and internal properties of the regulatory system, biomolecules, their interactions and pathways. In some cases the number, position and combinations of phosphosites determine the functional outcome while in other cases the identities of individual phosphorylated residues are also of extreme importance. First we asked if functional consequences of phosphorylation might be linked with the residue type for those sites which can be phosphorylated in human proteins (Ser, Thr, Tyr). Overall, there were substantially more activating than inhibitory sites for all three types of residues which was consistent with the previous observation that activating signal flows outnumbered the inhibitory signal flows in signal transduction networks17. We found the strongest tendency toward activation for pTyr (the ratio of activating and inhibitory sites was 3.7 for pTyr, 2.6 for pThr and 1.7 for pSer, Figure 2a). Phosphorylation of tyrosine was used considerably more often for activating signals compared to the two other residues taken together (Fisher exact test p-value = 4.3*10-6) whereas pSer was more regularly utilized for inhibition compared to the other two residues (p-value = 1.0*10-6). Although the same tyrosine site can be phosphorylated by different kinases, such cases of promiscuous phosphorylation were found to be less frequent for tyrosine compared to serine and threonine. This supports previous observations about the targeted specificity of tyrosine kinases 18. The results were unchanged even if phosphosites were clustered based on their location in sequence (Table S4).
Figure 2. Regulatory properties of all phosphorylation sites.
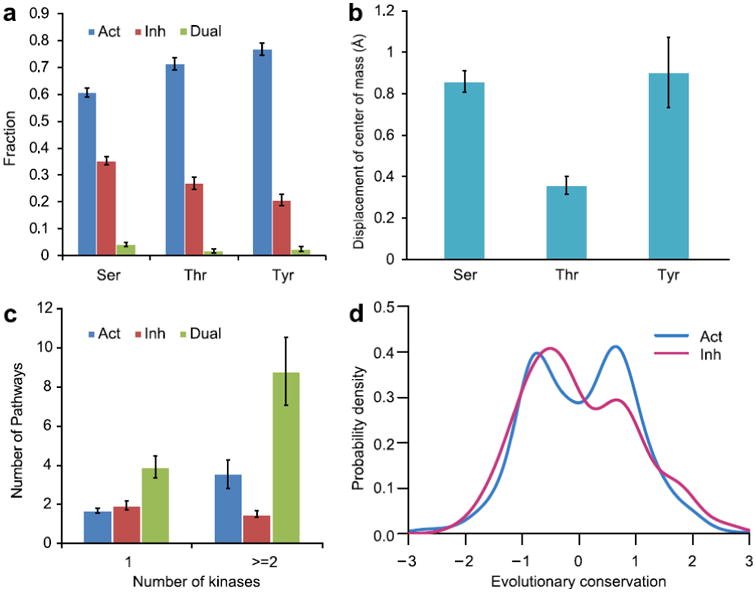
a: fraction of phosphorylation sites with a given functional consequence (Act –activating, 1068 sites; Inh – inhibitory, 489 sites and Dual – dual phosphosites, 52 sites) for different residue types plotted with the standard errors. b: mean value of displacement of side chain center of mass upon phosphorylation; standard error bars are shown. c: relationship between the numbers of kinases and pathways regulated by phosphosites with different functions. d: evolutionary conservation of phosphosites. Positive conservation score corresponds to sites that are more evolutionary conserved compared to other sites in a protein family. Conservation distribution for activating sites can be described by two normal distributions. The distributions were smoothed by the Gaussian kernel density estimation.
We analyzed local structural changes upon phosphorylation by measuring the center of mass displacement of the side chain of target residue before and after phosphorylation (the phosphate group was not considered in the calculation of the center of mass). We found that for large majority of sites phosphorylation produced only small changes in side chain conformation of less than 1 Å. However, there were 19 sites with relatively large displacements of 2-6 Å upon phosphorylation, all of them belonged to Tyrosine sites. Overall phosphorylation of Tyr and Ser in protein structural regions led to the larger structural changes while phosphorylation of Thr produced on average very small changes in local structure (p-value=1.3*10-26) (Figure 2b). Similar to the functional effect, the results on structural changes were not affected by the phosphosite clustering (Table S4).
Distinct phosphorylation sites may mediate crosstalk between signaling pathways
Protein phosphosites may provide points of crosstalk between signaling pathways through a single “junction” protein. The shared “junction” protein may have a single or multiple sites phosphorylated by the same or different kinases from the same or different pathways. In one crosstalk scenario, multiple phosphorylation may lead to separation of signaling events in time or space by inhibiting or activating the target protein and consequently potentially inhibiting one pathway and activating another (site pairs with double arrows of different color, Figure 1). In these cases one might expect a certain functional heterogeneity between phosphorylation sites used for regulation (38(33+5)% and 4(4+0)% phosphosites regulated by multiple and single kinase respectively, Figure 1). In another scenario multiple phosphorylation may support the integration and convergence of different pathways resulting in amplification, reduction or termination of the signal (the majority of sites, red and blue labels/arrows). In such cases the functional homogeneity is expected between the sites.
We analyzed the principles of mediation of pathway crosstalk through phosphorylation. First we found that proteins containing sites phosphorylated by only one kinase participated in a smaller number of pathways (2.4 pathways on average) than proteins phosphorylated by multiple kinases (5.3 pathways, p-value ≪ 0.01). While the number of pathways increased with the diversity of kinases phosphorylating activating and dual sites, for inhibitory sites no such association was observed (Figure 2c). No correlation was found between the phosphoprotein length and number of pathways it controlled.
Different patterns of multiple phosphorylation
Furthermore, we hypothesized that the integration or separation of pathways provided by the shared protein and multiple phosphorylation sites might depend on their intra-molecular distance in space and along the sequence. Overall, the distance between two phosphorylation sites in protein three-dimensional structures correlated very well with the distance along the sequence (Pearson correlation coefficient ρ = 0.63) and almost no long-range spatial contacts between phosphosites separated by large sequence spans were detected. Consistent with the previous observation 19; 20, we found that sites phosphorylated by the same kinase were closer in sequences than those phosphorylated by different kinases (p-value ≪ 0.01, Figure S2). Importantly we observed that two sites which were located closer to each other in sequence usually participated in regulation of the same pathway (p-value=2.7*10-17, Figure S2), although this effect was supported by KEGG and PID but not Reactome pathway databases (Table S3).
Next we analyzed phosphosites with respect to the functional effect (activating, inhibitory or dual) of phosphorylation on the target protein. Sites having the opposite, antagonistic functional effect (hetero-functional site pairs) showed a larger separation along the sequence (p-value =2.0*10-28, Figure 3a) and in space (p-value=6.7*10-5) compared to homo-functional site pairs (see Methods for definitions). After subdividing homo-functional site pairs further into different residue types, we found that activating pairs were less clustered than inhibitory pairs. Moreover, activating pTyr pairs were less clustered than activating Ser/Thr pairs (p-value = 2.3*10-7, Figure 3b) although no such distinction was observed between inhibitory sites of different types of residues. This result helps to understand the previous observation that majority of pSer/pThr sites occur in clusters in sequence 1; 21; 22 while only 19% of pTyr are within 1-4 amino acids from each other 19. Additional inspection of hetero-functional sites showed that they tend to be phosphorylated by different kinases (p-value value ≪ 0.01, Figure 3c). Here “shared” kinases refer to those kinases which can phosphorylate both sites in a pair. Next we analyzed the pathways which could be regulated by phosphorylation of individual sites. If kinase A phosphorylates protein B (A->B) we used a corresponding pathway which contained both these proteins with recorded relationship A->B. We found thatm hetero-functional phosphosite pairs regulated fewer common (the same) pathways (0.6 pathways on average) compared to homo-functional site pairs (2.3 pathways, p-value ≪ 0.01, Figure 3d).
Figure 3. Regulatory properties of multiple phosphorylation sites.
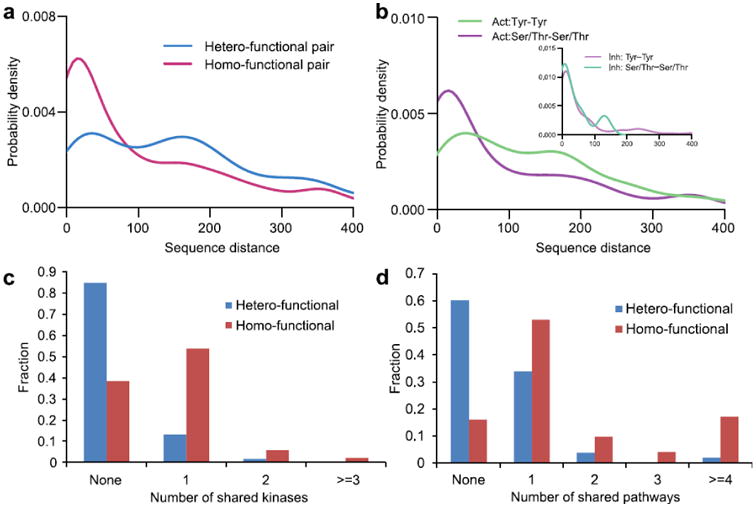
a: probability density function of sequence distance between phosphorylation sites for hetero-functional (blue) and homo-functional (red) site pairs. b: probability density function of sequence distance between phosphorylation sites for activating (main figure) and inhibitory (inset) site pairs. Green: Tyr-Tyr pairs, purple: Ser/Thr-Ser/Thr pairs. c: the histogram of the number of shared (same) kinases which can phosphorylate both sites for hetero- (blue) and homo-functional site (red) pairs. d: the histogram of the number of shared (same) pathways for hetero- (blue) and homo-functional site (red) pairs. The distributions were smoothed by the Gaussian kernel density estimation.
Patterns of evolutionary conservation of phosphosites with different functions
Previous studies showed that protein sites, which could be potentially phosphorylated, were under stronger evolutionary constraints compared to non-phosphorylated surface residues 5; 21. Here we analyzed evolutionary conservation by further distinguishing phosphosites based on their functional outcomes (Figure 2d). Namely, we aligned phosphoprotein sequences from our data set to domain families as described in Methods and calculated evolutionary conservation using the entropy-based measure based on sequence-weighted observed amino acid frequencies for each column in the alignment. Unlike inhibitory sites, activating sites had bimodal conservation distribution which could be well described by two Gaussian distributions according to Lilliefors test (see Suppl Materials). It comprised two distinct fractions of activating sites: one fraction included sites which were less conserved than the rest of the family (negative conservation values, Figure 2d) while the second fraction contained evolutionarily conserved activating sites. (Figure S3B).
Structural and thermodynamic properties of regulatory phosphorylation sites
Phosphorylation can induce conformational changes and affect the stability and binding properties of proteins which in turn can be directly linked to the activity changes 3; 23. Multiple phosphorylation might further amplify this effect. We performed structural modeling as described in Methods and obtained 466 high-quality protein structural models containing 809 phosphosites, among them 161 proteins had several phosphorylation sites on one chain (altogether 582 phosphosite pairs in total). We repeated structural analysis using high quality structural models obtained from the structural templates with more than 90% identity to the query phosphoprotein (537 sites from 304 proteins, “Model-90” set). Next we calculated changes in unfolding free energy (ΔΔG) by attaching the phosphate group to each phosphorylation site in all possible pair combinations on one protein since the order of phosphorylation for these proteins is largely unknown.
We analyzed the effects of multiple phosphorylation events on protein stability in their relation to function using FoldX. Since phosphorylation might stabilize or destabilize protein state, we hypothesize here that sites with similar functional consequences of phosphorylation on a single target protein might have a similar effect on stability of a given protein state. In other words, if phosphorylation outcome is measured in terms of quantitative ΔΔG values, such phosphosites might have the same sign and similar amplitudes of their corresponding ΔΔG values. In support of this hypothesis, phosphosite pairs with the same activating or inhibitory functional consequences (activating-activating, inhibitory-inhibitory) showed more coherent behavior (measured in terms of ΔΔG values) compared to other site pairs. It was evident from measuring the relative differences between their ΔΔG values (Figure S4A, p-value = 0.024). This effect was more pronounced for structural models from Model-90 set (p-value = 0.008) and was mostly attributed to heavily phosphorylated proteins with more than five sites (Table S2). Furthermore, we found a weak but significant linear correlation between ΔΔG values for sites with the same functional outcome (p-value = 0.035) which was further supported by Model-90 set (p-value = 8*10-5). On the contrary, there was no correlation between ΔΔG values for sites with different functions (p-value = 0.5, Figure S5). Interestingly, if two sites shared the same kinase, the effect of their phosphorylation on protein stability was more similar. (Figure S4B, p-value = 0.008 and p-value = 0.02 for Model-90 set).
Multiple phosphorylation of interferon regulatory factors 3 (IRF3) illustrates how phosphorylation sites located in different clusters might have differential effects on IRF3 stability and structure. IRF3 is expressed in cytoplasm as inactive monomer. Viral infection induces the phosphorylation of IRF3 which forms an active oligomer, enters the nucleus and activates the expression of interferon-α/β. Phosphorylation in Ser/Thr sites may activate IRF3 by inducing conformational rearrangements so that the C-terminal segment switches from an autoinhibitory state to an active dimer. According to PhosphositePlus database (“Function” set) seven phosphorylated Ser and Thr residues which are important for the activation are located in two major clusters; sites of each cluster are close to each other in sequence and space. The first cluster includes residues Ser385 and Ser386 which are partially exposed and might trigger or complement subsequent phosphorylation events. According to our analysis of IRF3 autoinhibitory structure (pdb code 1QWT), phosphorylation of Ser385 and Ser386 does not produce large effects on stability and conformation of inactive monomer (ΔΔG = +0.2 - 0.6 kcal/mol and center of mass displacement upon phosphorylation of 0.1 - 0.2 Å) and phosphorylation of these sites might be necessary but not sufficient for IRF3 activation. It was originally proposed and subsequently confirmed by different experimental studies24 that phosphorylation of residues in another cluster (Ser396, Ser398, Ser402, Thr404 and Ser405) releases IRF3 autoinhibition and allows it to interact with the coactivator CBP/p300 to initiate the transcription. Consistent with the authoinhibitory model, we show that phosphorylation of all but one residue destabilizes the autoinhibitory IRF3 structure, the largest destabilizing effect on conformation is observed for phosphorylation of Ser398 and Ser396 residues (ΔΔG = +2.4 and +1.4 kcal/mol respectively; center of mass displacement of 1.3 Å). The critical role of Ser396 was previously confirmed as the minimal phosphoacceptor residue required for the in vivo activation of IRF-3 25. All above mentioned phosphosites belong to homo-functional sites with the coherent outcome of their phosphorylation on IRF3 function and stability.
Discussion
Sequential reactions have certain advantages over single step signaling in providing additional regulatory checkpoints or proofreading steps 26. Moreover, selection of signal through the successive regulatory checkpoints and dichotomic search should be more fast and efficient. Indeed, regulatory reactions are highly non-linear which explains the predominance of indirect consequences of their perturbation and difficulty of functional annotations of many phosphorylation sites 27.
Here we applied the systems approach and analyzed the functional consequences of hundreds of phosphorylation events on target proteins and downstream signaling by crosslinking the data on individual phosphorylation events with the quarrying of biological pathways. We found that multiple phosphorylation and in a few cases single phosphorylation events on the same protein can serve as molecular switches allowing the biological crosstalk between different redundant or alternative pathways. These phosphosites can serve as hubs in the signal flow through the phosphorylation networks and possess special properties. Namely, phosphosites having similar regulatory functions (homo-functional site pairs), sharing the same kinases and participating in regulation of similar pathways are more likely to cluster in sequence and space and this tendency for clustering is more pronounced for pThr/pSer sites. Furthermore, phosphosites with similar downstream functional consequences as well as phosphosites regulated by the same kinase have comparable effects on protein stability. This in turn might point to possible amplification of phosphorylation effects when multiple sites are phosphorylated which can lead to integration of activating or inhibitory signals. Contrariwise, phosphosites with antagonistic regulatory functions tend to be located farther apart in sequence and structure while being phosphorylated by different kinases. Such sites may provide the separation of pathways in time and space.
We observe that there are more activating signaling events associated with Tyr phosphorylation compared to inhibitory flows. While activating role of phosphorylation could be explained in some cases by the evolution of phosphorylation sites from negatively charged amino acids 28, an understanding of its molecular mechanisms could be attained through the “conformational selection” hypothesis 29; 30. Within the framework of this hypothesis, activation by phosphorylation may occur when phosphorylation selects the relevant discrete conformation out of the entire preexisting ensemble, shifting the equilibrium of the conformational ensemble31 and leading to activation of downstream signaling by stabilizing the active or destabilizing the inactive states. Recently it was shown that pathway relations involving activation were characterized by a high percentage of structured regions and low disorder content 32. We hypothesize here that one of the reasons why tyrosine is strongly associated with activation and depleted in inhibitory reactions is that pTyr is usually located in structured regions 33; 34 and its phosphorylation leads to a more specific response. Such fine-tuned specific response is more suitable for activating events which are generally accompanied by precise conformational changes at both local and global levels 35. The association of tyrosine phosphorylation with activating events is consistent with a pivotal role of tyrosine in oncogene signaling, which extensively deregulates tyrosine phosphorylation 36; 17. It was shown recently that phosphorylation in structured regions, especially tyrosine phosphorylation, is related to longer-lasting effects of phosphorylation 34 compared to dynamically regulated phosphorylation in disordered regions for cell cycle pathways. At the same time, reactions involving disorder-order transitions and binding through unstructured regions are usually enriched with pSer and pThr 33; 37 and can be used for activating as well as inhibitory flows.
It has been a controversial topic in the literature regarding the functional neutrality and importance of phosphorylation sites. One might argue that many or even the majority of phosphorylation events do not have phenotypic consequence and might not be vital for the cell survival 27. Different explanations can support these ideas including the necessity to retain a certain level of redundancy in the system to ensure its robustness. However, regulatory phosphorylation sites analyzed here might represent exceptional cases with critical functional properties and severe consequences of their deregulation. Our results show that phosphorylation sites which can potentially provide pathway crosstalk are under selective pressure as they maintain a series of different sequence, structural, and thermodynamic properties consistent with their function. This is evident from their evolutionary conservation: activating phosphosites consist of two fractions of slowly and rapidly evolving sites. The former fraction is under stronger evolutionary constraints compared to the rest of protein family sites.
Overall our study complements and provides the novel insights into the current understanding of design principles of signaling regulation. We argue here that signaling crosstalk does not in principle require a modular network structure and in a considerable number of cases may be achieved at the level of a single protein molecule. Future experimental studies on the individual phosphosites and their functional roles may complete or challenge this view of the phosphorylation networks and their control principles.
Materials and Methods
Linking individual phosphorylation events with their functions and pathways
To compile a comprehensive list of human phosphorylation sites with the functional information on the individual phosphosites in biological pathways, we first extracted data on the locations of phosphorylation sites in human proteins and for each site we found its in vivo responsible kinase and/or its function (“activating”, “inhibitory”) using the PhosphoSitePlus 38 database. Phosphorylation sites were confirmed by at least two independent high-throughput or one low-throughput experimental studies. Next we eliminated phosphorylation sites for which the responsible kinases and the function were unknown. After removing redundancy and excluding proteins with sequence identity of more than 50% (the longest protein was retained in the cluster), we ended up with a set of phosphorylation sites with the known functions of phosphorylation events (“Function” set, 1609 sites on 715 proteins), and a set with in vivo data on the corresponding kinases (“Kinase” set, 2465 sites on 1057 proteins) (see Supplementary Materials and Figure S1 for details). There is an overlap between Function and Kinase sets, which contains 860 sites on 454 proteins.
Next, we derived phosphorylation and protein activation/inhibition events in human-specific pathways from the biological pathway databases: KEGG 39, REACTOME 40 and Pathway Interaction Database (PID) 41. These pathways include biomolecules and their specific experimentally identified relationships such as protein binding, activation, inhibition, and post-translational modifications. In KEGG and PID, target protein-kinase relations may have “activation (active)” or “inhibition (inactive)” attributes assigned to them, therefore indicating how phosphorylation may be associated with activation or inhibition of a target protein. On the other hand, REACTOME uses a reaction-centered ontology to represent cellular processes. Activity consequences of phosphorylation events are represented explicitly through downstream reactions. To infer activity changes from REACTOME, we searched for reactions where a phosphate group was added to a protein. We then looked for reactions that are either catalyzed by the modified state or another state that is produced directly from the modified state – i.e. it is one-step downstream. We annotated a phosphorylation reaction as “activating” if phosphorylation in REACTOME caused activation or prevented inhibition of the target protein or its downstream reactions. Similarly, a phosphorylation was defined as “inhibiting” if it caused inhibition or prevented activation of the target protein or its downstream reactions. The redundancy within the pathways was further removed as described in Supplementary Materials. All data are provided via the ftp site ftp://ftp.ncbi.nih.gov/pub/panch/Phospho/.
To integrate the data on phosphorylation sites, responsible kinases and biochemical pathways, we mapped phosphorylation sites onto pathways via in vivo kinase-substrate relation. Namely, if kinase A phosphorylates protein B (A->B) in vivo in PhosphoSitePlus, we use a corresponding pathway which contains both of these proteins with recorded relationship A->B. Then functional annotation (activating or inhibitory) of phosphorylation events in REACTOME, KEGG and PID pathway databases was used in the pathway analysis in addition to functional annotation derived from PhosphoSitePlus. It should be noted that 81% of phosphosite functions obtained from the pathway databases coincide with the functional outcomes annotated in PhosphoSitePlus. As a result, we obtained 354 phosphorylation sites on 152 non-redundant proteins distributed among 210 non-redundant pathways (“Pathway” set Figure S1). Furthermore, 85 out of 152 proteins with assigned pathways had two or more phosphorylation sites. Since pathway databases may differ in terms of the extent of manual annotation, we repeated all pathway analyses separately for each pathway database, the results are listed in Table S3.
To exclude a possible bias coming from proteins enriched with phosphorylated sites, all analyses were also repeated without heavily phosphorylated proteins. All results reported in the paper were found to be robust with respect to adding or removing heavily phosphorylated proteins (p-values were less than 0.05 even though the dataset was smaller). In addition we repeated all analyses by a more stringent verification of phosphosites by requiring at least two independent low-throughput studies to support a phosphosite location. All results reported in the paper were found to be robust with respect to this additional filter (see tables in Supplementary Materials). We also performed clustering of phosphorylation sites to remove potential redundancy at the phosphosite level for single phosphorylation site analyses (see Table S4 for details).
We further subdivided all phosphosites into three categories depending on their activating or inhibitory effects on the target protein: activating, inhibitory and sites with dual properties (sites that can function as both activating and inhibitory according to PhosphoSitePlus database). For multisite proteins we defined so-called “hetero-functional” phosphosite pairs as pairs between activating and inhibitory, activating and dual, inhibitory and dual sites. All other site pairs were denoted as “homo-functional” site pairs.
Structural modeling and in silico phosphorylation
Structural templates for modeling were chosen from the Protein Data Bank 42 using the BLAST algorithm 43. To eliminate low quality models we selected only those templates with sequence identities of more than 40% to the query protein and with more than 80% of template structures covered by the Blast alignment. These thresholds were chosen according to a previous study which showed that high-quality models can always be built for proteins with sequence identity higher than 40% 44. To build homology models we employed the NEST program from the Jackal package 45 with the option which optimized the configurations of loops and secondary structure regions. The loop prediction program loopy in the Jackal package was used to complement missing coordinate regions of some structural templates. Similarly, the refinement of side-chain conformations was performed by the side-chain program scap. As a result, we obtained 466 structural models containing 809 phosphosites from the union of Function and Kinase sets.
For the next step we phosphorylated the modeled proteins in silico and calculated the stability changes upon single phosphorylation events. Namely we built phosphorylated models by attaching the phosphate to those Ser, Thr and Tyr side chains known to be phosphorylated. To assess the effect of phosphorylation on protein stability we used the FoldX method which performed among the best three methods in predicting the experimental changes in stability produced by amino acid substitutions46. The FoldX program 47 estimates protein stability using an empirical force field; it attaches a phosphate group to Ser/Thr/Tyr, optimizes the side chain conformations of the phosphorylated residue, and calculates the difference in unfolding free energy between the original and phosphorylated complexes (ΔΔG): ΔΔG=GP - ΔGU. Here ΔGP and ΔGU are the unfolding free energies of the phosphorylated and unphosphorylated states, respectively. Positive and negative ΔΔG values correspond to destabilizing and stabilizing effects of phosphorylation.
Evolutionary conservation
We aligned phosphoprotein sequences from our data set to domain families from the Conserved Domain Database 48 and calculated evolutionary conservation using the al2co program 49 with default parameters. Conservation analysis could not be performed for dual phosphosites since the number of dual sites which could be mapped on CDD domains was very small. The conservation score represents the entropy-based measure calculated from sequence-weighted observed amino acid frequencies. The score is normalized by subtracting the mean and dividing by the standard deviation of the score distribution for the whole alignment. Therefore, the conservation score of a given site is negative if the site is less conserved than the average conservation background of protein family, and vice versa. Statistical analyses are performed as described in the Supplementary Materials.
Supplementary Material
Highlights.
Identified and provided the list of phosphosites that serve as switches for pathway crosstalk;
Phosphosites with similar regulatory functions share sequence and structural properties;
Phosphosites with antagonistic functions are located apart and regulated by different kinases;
different patterns of evolutionary conservation for activating and inhibitory sites;
Acknowledgments
We thank Thomas Madej and Yuri Wolf for insightful discussions. This work was supported by the Intramural Research Program of the National Library of Medicine at the U.S. National Institutes of Health. ED was supported by grant 5U41-HG006623-02 of the National Human Genome Research Institute, NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–48. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 3.Johnson LN. The regulation of protein phosphorylation. Biochem Soc Trans. 2009;37:627–41. doi: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- 4.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 5.Nishi H, Hashimoto K, Panchenko AR. Phosphorylation in protein-protein binding: effect on stability and function. Structure. 2011;19:1807–15. doi: 10.1016/j.str.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreon JC, Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc Natl Acad Sci U S A. 2009;106:6591–6. doi: 10.1073/pnas.0811023106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G. Phosphorylation and activation of human cdc25-C by cdc2--cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 1993;12:53–63. doi: 10.1002/j.1460-2075.1993.tb05631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salazar C, Hofer T. Multisite protein phosphorylation--from molecular mechanisms to kinetic models. FEBS J. 2009;276:3177–98. doi: 10.1111/j.1742-4658.2009.07027.x. [DOI] [PubMed] [Google Scholar]
- 9.Cohen P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 10.Patwardhan P, Miller WT. Processive phosphorylation: mechanism and biological importance. Cell Signal. 2007;19:2218–26. doi: 10.1016/j.cellsig.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11:414–26. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 13.Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras AC, Nesvizhskii AI, Tyers M. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–6. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashby WR. Requisite variety and its implications for the control of complex systems. Cybernetica. 1958;1:83–99. [Google Scholar]
- 15.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–2. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 16.Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol. 2007;3:e59. doi: 10.1371/journal.pcbi.0030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Q, Ma Y, Jaramillo M, Bari H, Awan A, Yang S, Zhang S, Liu L, Lu M, O'Connor-McCourt M, Purisima EO, Wang E. A map of human cancer signaling. Mol Syst Biol. 2007;3:152. doi: 10.1038/msb4100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Songyang Z, Carraway KL, 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, et al. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–9. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 19.Schweiger R, Linial M. Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol Direct. 2010;5:6. doi: 10.1186/1745-6150-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yachie N, Saito R, Sugahara J, Tomita M, Ishihama Y. In silico analysis of phosphoproteome data suggests a rich-get-richer process of phosphosite accumulation over evolution. Mol Cell Proteomics. 2009;8:1061–71. doi: 10.1074/mcp.M800466-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boekhorst J, van Breukelen B, Heck A, Jr, Snel B. Comparative phosphoproteomics reveals evolutionary and functional conservation of phosphorylation across eukaryotes. Genome Biol. 2008;9:R144. doi: 10.1186/gb-2008-9-10-r144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–49. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chem Rev. 2001;101:2209–42. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- 24.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10:913–21. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 25.Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, Hiscott J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem. 2003;278:9441–7. doi: 10.1074/jbc.M209851200. [DOI] [PubMed] [Google Scholar]
- 26.Ferrell JE, Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–8. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 27.Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O, Brusniak MY, Roschitzki B, Zhang C, Shokat KM, Schlapbach R, Colman-Lerner A, Nolan GP, Nesvizhskii AI, Peter M, Loewith R, von Mering C, Aebersold R. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal. 2010;3:rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearlman SM, Serber Z, Ferrell JE., Jr A mechanism for the evolution of phosphorylation sites. Cell. 2011;147:934–46. doi: 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–20. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 30.Boehr DD, Wright PE. Biochemistry. How do proteins interact? Science. 2008;320:1429–30. doi: 10.1126/science.1158818. [DOI] [PubMed] [Google Scholar]
- 31.Shen T, Zong C, Hamelberg D, McCammon JA, Wolynes PG. The folding energy landscape and phosphorylation: modeling the conformational switch of the NFAT regulatory domain. FASEB J. 2005;19:1389–95. doi: 10.1096/fj.04-3590hyp. [DOI] [PubMed] [Google Scholar]
- 32.Fong JH, Shoemaker BA, Panchenko AR. Intrinsic protein disorder in human pathways. Mol Biosyst. 2012;8:320–6. doi: 10.1039/c1mb05274h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishi H, Fong JH, Chang C, Teichmann SA, Panchenko AR. Regulation of protein-protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol Biosyst. 2013;9:1620–6. doi: 10.1039/c3mb25514j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyanova S, Cox J, Olsen J, Mann M, Frishman D. Phosphorylation variation during the cell cycle scales with structural propensities of proteins. PLoS Comput Biol. 2013;9:e1002842. doi: 10.1371/journal.pcbi.1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xin F, Radivojac P. Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics. 2012;28:2905–13. doi: 10.1093/bioinformatics/bts541. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Tibiche C, Fu C, Kaneko T, Moran MF, Schiller MR, Li SS, Wang E. The human phosphotyrosine signaling network: evolution and hotspots of hijacking in cancer. Genome Res. 2012;22:1222–30. doi: 10.1101/gr.128819.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 38.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–70. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D'Eustachio P, Stein L. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–7. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the Pathway Interaction Database. Nucleic acids research. 2009;37:D674–9. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y. Protein structure prediction: when is it useful? Curr Opin Struct Biol. 2009;19:145–55. doi: 10.1016/j.sbi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrey D, Xiang Z, Tang CL, Xie L, Gimpelev M, Mitros T, Soto CS, Goldsmith-Fischman S, Kernytsky A, Schlessinger A, Koh IY, Alexov E, Honig B. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins. 2003;53 Suppl 6:430–5. doi: 10.1002/prot.10550. [DOI] [PubMed] [Google Scholar]
- 46.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22:553–60. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 47.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–87. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 48.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41:D348–52. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pei J, Grishin NV. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics. 2001;17:700–12. doi: 10.1093/bioinformatics/17.8.700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.