

Abstract
BACKGROUND
Insulin resistance (IR) increases Alzheimer’s disease (AD) risk. IR is related to greater amyloid burden post-mortem and increased deposition within areas affected by early AD. No studies have examined if IR is associated with an in vivo index of amyloid in the human brain in late middle-aged participants at risk for AD.
METHODS
Asymptomatic, late middle-aged adults (N=186) from the Wisconsin Registry for Alzheimer’s Prevention (WRAP) underwent [C-11] Pittsburgh Compound B (PiB) Positron Emission Tomography. The cross-sectional design tested the interaction between insulin resistance and glycemic status on PiB DVR in three regions of interest (frontal, parietal, and temporal).
RESULTS
In participants with normoglycemia but not hyperglycemia, higher insulin resistance corresponded to higher PiB uptake in frontal and temporal areas, reflecting increased amyloid deposition.
CONCLUSIONS
This is the first human study to demonstrate that insulin resistance may contribute to amyloid deposition in brain regions affected by AD.
Keywords: Insulin resistance, amyloid, PiB, Alzheimer’s disease, cognitively normal, prefrontal
1. Background
The etiopathogenesis of Alzheimer’s disease (AD) is partly characterized by extracellular β-amyloid (Aβ) aggregation and medial temporal lobe atrophy (1). Insulin resistance (IR) is associated with brain amyloidosis in rodents and humans (2–6). IR is characterized by loss of tissue responsivity to insulin and progressive compensatory peripheral hyperglycemia. Some studies suggest that higher IR is present in AD (1), increases AD risk (7), and is associated with post-mortem Aβ plaques (8). IR may increase Aβ oligomerization and potentiate brain atrophy via neuroinflammation or other downstream effects (9). Intranasal insulin therapy has been found to conversely increase plasma Aβ40/42 ratios and improve cognition (10).
No study has examined the direct association between IR and an in vivo marker of amyloid load in AD-sensitive brain areas in late middle-aged participants. Regions of interest (ROIs) include inferior and medial temporal lobe, ventral prefrontal cortex, and posteriomedial cortex (6). Insulin resistance and type 2 diabetes involve multiple mechanistic pathways, and the effect of IR on neural health may differ depending on whether or not hyperglycemia is present (7, 11, 12).
In this study, we examined if IR was associated with amyloid binding in three AD-sensitive ROIs based on glycemic status, as found in our previous study on IR and glucose uptake (13). We hypothesized that higher IR, indexed by the Homeostatic Model Assessment of Insulin Resistance (HOMA-IR)(14), would predict greater amyloid burden using [C-11]Pittsburgh Compound B (PiB)(15) positron emission tomography (PET). On an exploratory basis, we also investigated sub-regions of these areas to provide greater spatial specificity.
2. Methods
2.1. Participants
One hundred and eighty-six late middle-aged adults from the Wisconsin Registry for Alzheimer’s Prevention (WRAP) underwent PiB-PET scanning. Demographics are shown in Table 1. Details about selection criteria, recruitment sources, and other aspects are directly discussed elsewhere (16). Briefly, this on-going study examines genetic and biological factors that contribute to the development of dementia-related cognitive decline and neural dysfunction. Participants were classified as either having a positive (FH+) or negative family history (FH−) of AD. FH+ was defined as having one or both parents with autopsy-confirmed or probable AD as defined by research criteria (17), based on review of medical records and autopsy reports when available. FH− was defined as no formal diagnosis of AD or other significant cognitive decline in either parent, based on information provided by telephone interviews with participants. The inclusion criteria for this study consisted of: normal cognitive function determined by neuropsychological evaluation and consensus meeting similar to NINCD-SADRDA recommendations (17), no contraindication for PET or magnetic resonance imaging (MRI) and a subsequently normal MRI scan, no current diagnosis of major psychiatric disease or other major medical conditions (e.g., myocardial infarction, recent history of cancer), and no history of head trauma. The University of Wisconsin Institutional Review Board approved all procedures related to this study. Each participant gave full informed consent prior to study participation.
Table 1.
Participant Demographics
| N | 186 |
| Age in years (mean ± SD) | 60.37 ± 5.63 |
| Gender | |
| Female | 129 (69.4%) |
| Male | 57 (30.6%) |
| Education (mean ± SD) | 16.61 ± 2.94 |
| Family History of AD | |
| Negative | 53 (28.5%) |
| Positive | 133 (71.5%) |
| APOE4 Status | |
| Non-APOE4 | 114 (61.3%) |
| APOE4 | 72 (38.7%) |
| Diabetes Status | |
| Normoglycemia (<100 mg/dL) | 135 (72.6%) |
| At Risk/Pre-Diabetes (100–125 mg/dL) | 43 (23.1%) |
| Type 2 Diabetes (>125 mg/dL) | 8 (4.3%) |
| DBP | 74.11 ± 8.98 |
| SBP | 124.70 ± 15.19 |
| BMI (mean ± SD) | 28.20 ± 5.22 |
| Glucose (mg/dL) | 95.01 ± 10.28 |
| Insulin (μU/mL) | 9.53 ± 7.60 |
| HOMA-IR (mean ± SD) | 2.33 ± 2.19 |
| Total Cholesterol (mg/dL) | 202.39 ± 34.79 |
| MMSE (mean ± SD) | 29.3 ± 0.96 |
| Speed and Flexibility (mean ± SD) | 0.13 ± 0.86 |
| Working Memory (mean ± SD) | 0.20 ± 1.11 |
| Verbal Learning (mean ± SD) | 0.16 ± 0.95 |
| Immediate Memory (mean ± SD) | 0.15 ± 1.07 |
The four cognitive factors at the bottom of Table 1 are Z-scores. Number of subjects is listed unless otherwise specified in parentheses. AD = Alzheimer’s disease; APOE4 = Apolipoprotein ε4 allele; BMI = Body Mass Index; DBP = Diastolic Blood Pressure; HOMA-IR = Homeostatic Model Assessment of Insulin Resistance; MMSE = Mini-Mental State Examination; SBP = Systolic Blood Pressure.
2.2. Neuropsychological testing
To confirm that participants in this sample were cognitively normal, the MMSE and neuropsychological factor scores from the full battery (16) were used (see Table 1). Four cognitive domain factors were derived as described previously (18): Immediate Memory, Verbal Learning & Memory, Working Memory, and Speed and Flexibility. The individual tests which loaded onto the factors were as follows: 1) Rey Auditory Verbal Learning Test (19), Trials 1 and 2 loaded onto Immediate Memory; 2) Rey Auditory Verbal Learning Test (19) Trials 3–5 and Delayed Recall Trial loaded onto Verbal Learning & Memory; 3) Wechsler Adult Intelligence Scale – 3rd edition (20), Digit Span, Arithmetic, and Letter-Numbering Sequencing subtests loaded onto Working Memory; and 4) the interference trial from the Stroop Test (21), and Trail Making Test A and B (22) loaded onto Speed & Flexibility. For the Mini-Mental State Examination (MMSE), a cutoff score of 26 was used based on recommended thresholds from the Alzheimer’s Disease Neuroimaging Initiative (23). It is emphasized here that consensus committee meetings in line with NINCD-SADRDA recommendations (17) were used to confirm if participants were cognitively normal, rather than just the MMSE and other neuropsychological tests.
2.3. APOE genotype
Apolipoprotein (APOE) genotyping has been described previously (24). Participants were categorized as “Non-APOE4” (no ε4 alleles) or “APOE4” (at least one ε4 allele).
2.4. HOMA-IR, BMI, glycemic status, and other factors
Participants were fasted for 12 hours prior to blood collection. Basal glucose and insulin were measured in serum. As described previously (11), insulin was quantified by standard ELISA (R&D Systems, Minneapolis, MN). HOMA-IR was calculated by taking the product of basal glucose (mg/dL) and basal insulin (μU/mL) and dividing by 405 (14). HOMA-IR was treated as a continuous variable for analyses. Body mass index (BMI) was calculated by dividing the participant’s weight in kilograms by the square of their height in meters. Hyperglycemia was defined as either 1) having type 2 diabetes or being at risk for type 2 diabetes (i.e., pre-diabetes) based on American Diabetes Association criteria (i.e., fasting blood glucose levels of 100 mg/dL or greater); or 2) taking medication to control type 2 diabetes. Finally, as described previously (16), diastolic and systolic blood pressure (DBP, SBP) and total cholesterol were acquired.
2.5. MRI: anatomical
A 3-dimensional fast spoiled gradient-echo (3D EFGRE) T1-weighted image was acquired using a 3T General Electric (Waukesha, WI) scanner. Sequence parameters were: TR=8.1 ms; TE=3.2 ms; TI=450 ms; flip angle = 12°; 256×256 matrix; FOV=260 mm; slice thickness= 1.0 mm. Images were inspected for artifact or abnormalities that would exclude them from use in PET analyses. No scans were excluded.
2.6. Regions of Interest: freesurfer segmentation and parcellation
Our analysis focused on broad regions of interest (ROIs) to limit the number of statistical comparisons made. In order to derive the ROIs, T1-images were pre-processed using FreeSurfer 5.1 software (http://surfer.nmr.mgh.harvard.edu/). This software package corrects images for intensity bias, removes the skull, and then uses a probabilistic atlas to segment gray and white matter and classify different sub-cortical structures, as well as parcellate and label cortical regions (25). The 2009 Destrieux atlas was used, which delineates between sulci and gyri prior to regional classification and offers a more precise method for interpolating surface topology relative to other Freesurfer atlases. As depicted in Figure 1, we created three broad ROIs that encompassed ventral frontal, inferior and medial temporal, or posterior parietal regions. Figure S1 depicts the 17 Freesurfer ROIs (“sub-ROIs”) used to construct the three broad ROIs.
Figure 1. Neural Regions of Interest.
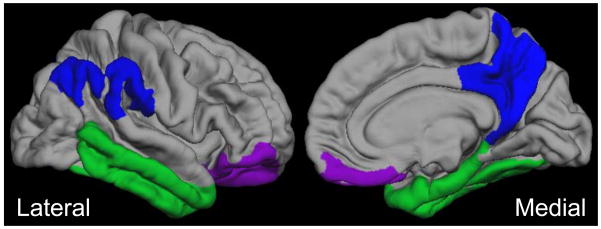
Amyloid binding was broadly assessed in three bilateral regions of interest (ROI): 1) a ventral frontal area in frontal lobe (“frontal ROI”); 2) an inferior and medial temporal area in temporal lobe (“temporal ROI”); and 3) a posterior medial parietal area in parietal lobe (“parietal ROI”).
2.7. PET: [C-11]PiB synthesis, scanning, and distribution volume ratio maps
Details regarding [C-11]PiB radiochemical synthesis and scanning have been described previously (18). Briefly, [C-11]PiB was synthesized using a captive solvent method (26). Typical yields of final [C-11]PiB product were in excess of 2 GBq, with specific activities of 150 – 600 GBq/umol. [C-11]PiB PET data were acquired in 3D mode. A 6-minute transmission scan was acquired for attenuation correction. A 70 minute dynamic [C-11]PiB PET acquisition was then initiated with the injection of a 550 MBq of [C-11]PiB bolus, injected over 30 s. 17 dynamic acquisition frames were collected. The PET data were reconstructed using a filtered back-projection algorithm (DIFT) with sinogram trimming to a voxel size of 2.57mm × 2.57mm × 2.43mm and matrix dimension of 128 × 128 × 63 and corrected for random events, attenuation of annihilation radiation, deadtime, scanner normalization and scatter radiation. The reconstructed time series PET data were realigned using SPM8 (www.fil.ion.ucl.ac.uk/spm) to correct for subject motion during the course of the study. A denoising algorithm, HighlY constrained backPRojection (HYPR) technique with backprojections to local regions of interest (HYPR-LR), was then applied to the voxel-based time series (27). The PET time-series was coregistered into the T1-weighted MRI scan using mutual information.
The [C-11]PET data were then transformed into voxel-based parametric images representing [C-11]PiB binding, using the cerebellar cortex as a reference region of negligible binding (28). Voxel-based parametric images using Logan graphical analysis were created as described by Lopresti and colleagues (28). For the Logan graphical method (29), linear regression was applied to the transformed data using the 35- to 70-min (7 points) interval and a mean efflux constant of 0.149 min−1. The resulting distribution volume ratio (DVR) images were inspected for quality. The three broad gray matter ROIs were used as masks to extract the bilateral mean value from the left and right hemispheres.
2.8. Statistics
For each ROI, a linear mixed model approach was used. The dependent measure was mean PiB signal extracted from one of the 3 ROIs, reflecting amyloid deposition. The fixed effect independent variable of interest was HOMA-IR by Glycemic Status. Fixed effect covariates included age at time of the scan, sex, FH status, APOE4 status, BMI, and type 2 diabetes medication, as well as the main effects of Glycemic Status and HOMA-IR. Additional models were run that also incorporated education, diastolic and systolic blood pressure, or total cholesterol as covariates of secondary interest. The random effect of subject was modeled in all analyses. While we included the main effects of glycemic status and HOMA-IR to test for their interaction, we only anticipated an interaction between HOMA-IR and Glycemic Status. Indeed, previous work has underscored the importance of examining HOMA-IR among participants with normoglycemia or hyperglycemia (13). Holm-Bonferroni correction (30) was used to control type 1 error for the three ROI comparisons. This is closed test procedure maintains a family-wise alpha of .05 by requiring unadjusted p-values of .017, .025, and .05 among the 3 null hypotheses tested. If the HOMA-IR by Glycemic Status interaction was significant, normoglycemia and hyperglycemia groups were separately tested with the same mixed model structure and covariates as above. HOMA-IR was the independent variable of interest in these post-hoc analyses.
A logarithmic transformation was used to normalize HOMA-IR, which originally had positive skew (3.436). Non-parametric Mann-Whitney U tests were used to compare demographic characteristics between participants classified as PiB positive (PiB+) or PiB negative (PiB−), as described below. Finally, to confirm that participants with high values did not bias HOMA-IR and PiB associations, a separate set of non-parametric analyses were conducted. Specifically, ROI values were first covariate-adjusted using the same covariates as above, followed by Spearman correlations for each glycemic group. All analyses were conducted using SPSS 21.0 (Chicago, IL).
3. Results
3.1. Demographics and PiB deposition characteristics
Sample characteristics are listed in Table 1. We have previously classified and reported on WRAP participants that appear to be PiB+, PiB intermediate, and PiB−, as well as corresponding neural and cognitive features (18). Based on a mean DVR of 1.1 or higher over any of the three broad ROIs in this pre-clinical, middle-aged cohort, 18 participants were classified as PiB+ compared to 168 PiB− participants. Based on the Mann-Whitney U statistic, PiB+ corresponded to significantly higher mean BMI [32.4 vs. 27.7, U(184)=932, Z=2.670, p=.008], glucose [114.1 vs. 93.0, U(184)=196, Z=6.067, p<.001], insulin [19.6 vs. 8.5, U(184)=779.5, Z=3.390, p=.001], and HOMA-IR [4.04 vs. 1.65, U(184)=645.5, Z=3.992, p<.001]. PiB+ participants were older [63.6 vs. 60.0 years, U(184)=961.5, Z=2.54, p=.011] but not more likely to have been FH+ versus FH− [U(184)=1431, Z=0.477, p=.633], APOE4+ versus APOE4− [U(184)=1236, Z=1.507, p=.132], or have a different sex ratio [U(184)=1467, Z=0.260, p=.795]. This pattern of results remained identical when using t-tests (data not shown).
3.2. IR, hyperglycemia, and amyloid deposition in ROIs
Figure 2 depicts raw PiB uptake images for illustration purposes. Additional models incorporating education, DBP and SBP, or total cholesterol showed nearly identical results (data not shown). For the frontal ROI, a significant HOMA-IR * Glycemic Status interaction was found [F(1,186)=10.580, p<.001], while the main effects were non-significant for HOMA-IR [F(1,186)=0.131, p=.718] and Glycemic Status [F(1,186)=3.031, p=.084]. The post-hoc mixed model for each glycemic group indicated that the normoglycemia group [F(1,135)=5.429, p=.021], but not hyperglycemia group [F(1,51)=2.532, p=.118], showed a significant, modest relationship between higher HOMA-IR and PiB uptake of R2=.071 (Figure 3). A similar HOMA-IR * Glycemic Status interaction was found for the temporal ROI [F(1,186)=6.109, p=.014]. Post-hoc testing indicated that the normoglycemia group had a small but significant association [F(1,135)=4.751, p=.031] between higher HOMA-IR and higher PiB uptake (R2=.036), whereas the association was non-significant for the hyperglycemia group [F(1,51)=1.389, p=.244]. While the HOMA-IR * Glycemic Status interaction was significant for the parietal ROI [F(1,186)=5.056, p=.026], neither the normoglycemia [F(1,135)=2.697, p=.103] or hyperglycemia [F(1,51)=2.599, p=.113] groups showed a significant HOMA-IR main effect during post-hoc testing. In a set of follow-up analyses, coavariate-adjusted Spearman correlations between HOMA-IR and PiB in the 3 ROIs were conducted for the normoglycemia and hyperglycemia groups. We confirmed that the normoglycemia group showed modest associations between higher HOMA-IR and higher PiB in the frontal [r=.195, p=.012] and temporal [r=.150, p=.020] ROIs, but not the parietal ROI [r=.090, p=.149]. The hyperglycemia group showed no significant associations.
Figure 2. Pittsburgh Compound B Images and Insulin Resistance.
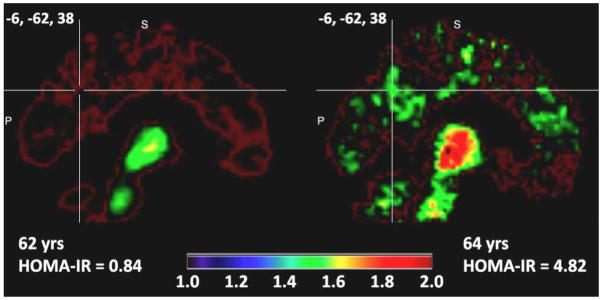
Amyloid uptake images in age- and sex-matched representative participants, who varied by the Homeostatic Model Assessment of Insulin Resistance (HOMA-IR). Two representative sagittal PiB images are shown for a participant with low IR (HOMA-IR < 2) or high IR (HOMA-IR ≥ 2). The color bar depicts the Pittsburgh Compound B (PiB) Distribution Volume Ratio, a quantitative index of PiB uptake.
Figure 3. Insulin Resistance and Frontal Amyloid Deposition.

The association between higher Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) and higher a Pittsburgh Compound B (PiB) frontal Distribution Volume Ratio (DVR) in participants with normal glucose values. DVR values were adjusted by the following covariates: age, sex, apolipoprotein ε4 status, family History of Alzheimer’s disease, taking type 2 diabetes medication, body mass index, and the main effects of HOMA-IR and glycemic status.
3.3. IR, hyperglycemia, and amyloid deposition in sub-ROIs
On an exploratory basis without type 1 error correction, we also investigated bilateral sub-ROIs depicted in Figure S1 that composed the 3 broad ROIs. Specifically, among 17 sub-ROIs, we tested the relationship between higher HOMA-IR and higher PiB in the normoglycemia and hyperglycemia group. The hyperglycemia group showed no associations (data not shown). For the normoglycemia group, Table S1 indicates that the frontal ROI result may be explained by gyrus rectus. For the temporal ROI, significant sub-regions included inferior temporal gyrus and sulcus, as well as medial temporal lobe, where HOMA-IR explained 14.7% of the variance in inferior temporal gyrus. For the parietal ROI, no sub-ROI was statistically significant. We emphasize that these exploratory analyses should be viewed with caution, given the lack of error correction.
4. Discussion
The aim of this study was to determine the association of IR with glycemic status on PiB uptake in cognitively healthy, late-middle aged adults. This is the first study to illustrate an association between higher IR and an in vivo index of amyloid burden. This association was found only in participants without elevated glucose values, which we briefly discuss below. While this relationship was modest for the broad ROIs, it is compelling to consider that these associations were found in an asymptomatic cohort only beginning to show amyloid deposition, given that there is wide variation in the presence and degree of amyloid aggregates present even in aged adults without AD (31). A similar amyloid pattern is seen in sporadic late onset AD (9). IR has been linked with amyloidosis in both humans and animals (3, 4, 9), although others have found no relationship between IR and amyloid scans or histopathology (32). Given that neural amyloidosis is one of the first salient features in preclinical AD (23), it may be of clinical relevance to test whether interventions that reduce IR may also reduce amyloid deposition and other aspects of disease pathogenesis in similar cohorts. However, one cannot infer causality or directionality about IR and amyloid burden from this study.
It should be cautioned that one should not extrapolate results from middle-aged participants without clinical impairment to MCI and AD or cognitively normal, aged participants. Indeed, it is noteworthy to mention that these results differ from Thambisetty and colleagues (32), who recently found that longitudinal insulin resistance derived from oral glucose tolerance tests did not predict PiB uptake or amyloid at autopsy in aged subjects. The PiB imaging component of their study had a comparatively smaller sample size than ours and did not covary typical AD risk factors. Furthermore, multiple insulin kits were used to analyze values over the course of the study, which might cause significant variation. Given that baseline HOMA-IR may only predict AD conversion out to three years (7), longitudinal HOMA-IR may also be less important than HOMA-IR collected near scan time. Finally, Talbot and colleagues (8) found post-mortem that central IR progressively increased from cognitively normal to AD participants, and was related to higher amyloid plaque load.
It has often been speculated that IR induces atrophy in part through amyloid neurotoxicity (33) and decreased glucose uptake (34), the latter of which can induce protein glycation and subsequent reactive oxygen species production. Tau aggregation and hyperphosphorylation may be one mechanism to consider, although tau pathology is hypothesized to occur later in the disease process (23). IR also dysregulates vasodilation and damages microvascular architecture (35, 36), which can cause microangiopathy. These events can precede type 2 diabetes onset (37, 38). If IR influences amlyoid aggregation, it may only begin to affect brain volume later in life or during early AD, although again it is not appropriate to infer from the study that IR will predict amyloid deposition in AD.
Our primary analysis did not show a statistical effect among participants with hyperglycemia. These results may be due to the limited range of hyperglycemia in our sample, particularly the relatively few participants with type 2 diabetes. Hyperglycemia may also impact amyloid burden independent of IR. Given that type 2 diabetes increases AD risk (7), larger studies of amyloid binding in patients with frank diabetes, both treated and untreated, will be needed to further examine these associations.
Several limitations should be discussed. On average, the DVRs for [C11]PiB were low. However, similarly low DVR and Mean Cortical Binding Potential have been found in studies focused on a slightly older middle-aged cohort (39) or that in part examined middle-aged participants (40). By extension, it was not feasible to examine PiB deposition in control regions unlikely to be affected by IR, because there is not a high enough DVR signal to be considered reliable in this healthy, middle-aged cohort. While additional areas of interest could have been chosen for our analysis, we chose a small number of broad ROIs in which early amyloidosis or AD pathology is found, primarily to reduce and control type 1 error. The exploratory analysis of sub-ROIs within these broad ROIs especially warrants caution, as regional analyses were done without type 1 error correction. The associations between HOMA-IR and PiB DVR in broad ROIs were modest. However, Reiman and colleagues (39) found in several ROIs similar to ours that heterozygous or homozygous ε4 carriers had significant but similarly modest increases in PiB DVR, although they also found larger increases in other ROIs. It should again be noted that causal inferences cannot be drawn based on our study. Finally, because not all cognitively normal participants who show amyloid deposition will develop AD, we reiterate that it is inappropriate to infer that higher HOMA-IR is a risk factor for amyloid deposition in MCI and AD patients. In conclusion, cognitively normal middle-aged participants with high HOMA-IR had modestly higher amyloid binding in several regions impacted by early AD. IR may be an important biomarker for early amyloid deposition in middle-aged participants at higher risk for AD.
Supplementary Material
Systematic Review.
We searched for articles in PubMed using the following keywords: insulin resistance; amyloid; PiB; Pittsburgh compound B; Aβ1–42; positron emission tomography; type 2 diabetes; metabolic syndrome; and Alzheimer’s disease. We examined in vivo and ex vivo work in rodents and humans, to determine to what degree insulin resistance predicts amyloidosis and may serve as a biomarker for amyloid accumulation in tissue.
Interpretation.
This is the first human study to demonstrate that insulin resistance is associated with amyloid deposition in brain regions affected by AD. These associations were found in late middle-aged participants only beginning to show amyloidosis, and the pattern reflects early AD progression.
Future Directions.
To confirm and expand on our work, it will be important to further examine IR and amyloid deposition in cognitively healthy and impaired older adults. Intervention studies are also needed to test the protective effect of IR reduction on AD prevention.
Acknowledgments
GRECC MS #2013-10. The authors gratefully acknowledge Nancy Davenport-Sis, Amy Hawley, Sandra Harding, Caitlin Cleary, Chuck Illingworth, and the support of researchers and staff at the Waisman Center, University of Wisconsin-Madison, for their assistance in recruitment, data collection, and data analysis. Above all, we wish to thank our dedicated volunteers for their participation in this research. This project was supported by the National Institute on Aging (R01 AG027161 [MAS], ADRC P50 AG033514 [SA], R01 AG021155 [SCJ], the University of Wisconsin Institute for Clinical and Translational Research, funded through a National Center for Research Resources/National Institutes of Health Clinical and Translational Science Award, 1UL1RR025011, a program of the National Center for Research Resources, United States National Institutes of Health), and by the facilities and resources at the Geriatric Research, Education, and Clinical Center (GRECC) of the William S. Middleton Memorial Veterans Hospital, Madison, WI. Barbara Bendlin had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Financial Disclosures: The authors report no disclosures such as conflicts of interest directly pertaining this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/jad-2005-7106. [DOI] [PubMed] [Google Scholar]
- 2.Arab L, Sadeghi R, Walker DG, Lue LF, Sabbagh MN. Consequences of Aberrant Insulin Regulation in the Brain: Can Treating Diabetes be Effective for Alzheimer’s Disease. Current neuropharmacology. 2011;9:693–705. doi: 10.2174/157015911798376334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4162–7. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. Faseb J. 2004;18:902–4. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 5.Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24:11120–6. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craft S, Cholerton B, Baker LD. Insulin and Alzheimer’s disease: untangling the web. J Alzheimers Dis. 2013;33(Suppl 1):S263–75. doi: 10.3233/JAD-2012-129042. [DOI] [PubMed] [Google Scholar]
- 7.Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam Study. Neurology. 2010;75:1982–7. doi: 10.1212/WNL.0b013e3181ffe4f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. The Journal of clinical investigation. 2012;122:1316–38. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72:49–66. doi: 10.2165/11597760-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–8. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- 11.Willette AA, Xu G, Johnson SC, Birdsill AC, Jonaitis EM, Sager MA, et al. Insulin Resistance, Brain Atrophy, and Cognitive Performance in Late Middle-Aged Adults. Diabetes care. 2012 doi: 10.2337/dc12-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benedict C, Brooks SJ, Kullberg J, Burgos J, Kempton MJ, Nordenskjold R, et al. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes care. 2012;35:488–94. doi: 10.2337/dc11-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Archives of neurology. 2011;68:51–7. doi: 10.1001/archneurol.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 15.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 16.Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer’s disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer’s Prevention. J Geriatr Psychiatry Neurol. 2005;18:245–9. doi: 10.1177/0891988705281882. [DOI] [PubMed] [Google Scholar]
- 17.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson SC, Christian BT, Okonkwo OC, Oh JM, Harding S, Xu G, et al. Amyloid burden and neural function in people at risk for Alzheimer’s Disease. Neurobiol Aging. 2013 doi: 10.1016/j.neurobiolaging.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spreen O, Strauss E. A compendium of neuropsychological tests : administration, norms, and commentary A compendium of neuropsychological tests : administration, norms, and commentary. 2. Place Oxford University Press; 1998. [Google Scholar]
- 20.Wechsler D. WAIS-III: Wechsler Adult Intelligence Scale WAIS-III: Wechsler Adult Intelligence Scale Place. Psychological Corporation; 1997. [Google Scholar]
- 21.Trenerry MR. Psychological Assessment Resources. 1989. Stroop Neuropsychological Screening Test Manual Stroop Neuropsychological Screening Test Manual Place. [Google Scholar]
- 22.Reitan RM, Wolfson D. Neuropsychology Press. 1993. The Halstead-Reitan Neuropsychological Test Battery: Theory and Clinical Interpretation The Halstead-Reitan Neuropsychological Test Battery: Theory and Clinical Interpretation Place. [Google Scholar]
- 23.Weiner MW, Aisen PS, Jack CR, Jr, Jagust WJ, Trojanowski JQ, Shaw L, et al. The Alzheimer’s disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:202–11. e7. doi: 10.1016/j.jalz.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson SC, La Rue A, Hermann BP, Xu G, Koscik RL, Jonaitis EM, et al. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE epsilon3/epsilon3 genotype. Alzheimers Dement. 2011;7:456–65. doi: 10.1016/j.jalz.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- 26.Wilson AA, Garcia A, Jin L, Houle S. Radiotracer synthesis from [(11)C]-iodomethane: a remarkably simple captive solvent method. Nucl Med Biol. 2000;27:529–32. doi: 10.1016/s0969-8051(00)00132-3. [DOI] [PubMed] [Google Scholar]
- 27.Christian BT, Vandehey NT, Floberg JM, Mistretta CA. Dynamic PET denoising with HYPR processing. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2010;51:1147–54. doi: 10.2967/jnumed.109.073999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2005;46:1959–72. [PubMed] [Google Scholar]
- 29.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–40. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6:65–70. [Google Scholar]
- 31.Rabinovici GD, Jagust WJ. Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol. 2009;21:117–28. doi: 10.3233/BEN-2009-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thambisetty M, Metter EJ, Yang A, Dolan H, Marano C, Zonderman AB, et al. Glucose Intolerance, Insulin Resistance, and Pathological Features of Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA neurology. 2013 doi: 10.1001/jamaneurol.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schioth HB, Craft S, Brooks SJ, Frey WH, 2nd, Benedict C. Brain insulin signaling and Alzheimer’s disease: current evidence and future directions. Mol Neurobiol. 2012;46:4–10. doi: 10.1007/s12035-011-8229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–78. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 35.Hoyer S. The aging brain. Changes in the neuronal insulin/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini-review. J Neural Transm. 2002;109:991–1002. doi: 10.1007/s007020200082. [DOI] [PubMed] [Google Scholar]
- 36.Murray IV, Proza JF, Sohrabji F, Lawler JM. Vascular and metabolic dysfunction in Alzheimer’s disease: a review. Exp Biol Med (Maywood) 2011;236:772–82. doi: 10.1258/ebm.2011.010355. [DOI] [PubMed] [Google Scholar]
- 37.Tooke JE, Goh KL. Endotheliopathy precedes type 2 diabetes. Diabetes care. 1998;21:2047–9. doi: 10.2337/diacare.21.12.2047. [DOI] [PubMed] [Google Scholar]
- 38.Convit A. Links between cognitive impairment in insulin resistance: an explanatory model. Neurobiol Aging. 2005;26(Suppl 1):31–5. doi: 10.1016/j.neurobiolaging.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 39.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6820–5. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.