

Abstract
G-protein-coupled receptor (GPCR) kinases (GRKs) were first identified based on their ability to specifically phosphorylate activated GPCRs. Although many soluble substrates have since been identified, the chief physiological role of GRKs still remains the uncoupling of GPCRs from heterotrimeric G-proteins by promoting β-arrestin binding through the phosphorylation of the receptor. It is expected that GRKs recognize activated GPCRs through a docking site that not only recognizes the active conformation of the transmembrane domain of the receptor but also stabilizes a more catalytically competent state of the kinase domain. Many of the recent gains in understanding GRK-receptor interactions have been gleaned through biochemical and structural analysis of recombinantly expressed GRKs. Described herein are current techniques and procedures being used to express, purify, and assay GRKs in both in vitro and living cells.
1. Introduction
Most activated G-protein-coupled receptors (GPCRs) are subject to homologous desensitization, wherein GPCR kinases (GRKs) phosphorylate serine and threonine residues in the cytoplasmic tails and loops of the receptor, which in turn recruits arrestin and blocks the binding of heterotrimeric G-proteins (Gurevich, Tesmer, Mushegian, & Gurevich, 2012). There are seven GRKs in man (GRK1–7), constituting three subfamilies. The GRK1 subfamily consists of GRK1 (rhodopsin kinase) and 7, which are expressed in the rod and cone cells of the retina. The GRK2 subfamily consists of GRK2 (βARK1) and GRK3 (βARK2), which are ubiquitously expressed. The GRK4 subfamily consists of GRK4–6. GRK5 and GRK6 are ubiquitously expressed, while GRK4 is found primarily in testes and kidneys.
All GRKs have a common catalytic core composed of what is best thought of as a protein kinase domain inserted into the loop of a regulator of G-protein signaling homology domain (Tesmer, 2009). The C-terminal regions of GRKs are their most distinguishing characteristic, but in each GRK, they contain motifs that help target the enzyme to the plasma membrane. The GRK kinase domain belongs to the PKA, PKG, and PKC (AGC) kinase family. As in other AGC kinase domains, it includes a C-terminal extension that contributes residues to the active site and is a locus for posttranslational regulation (Kannan, Haste, Taylor, & Neuwald, 2007). Unlike other AGC kinases, GRKs are not regulated via phosphorylation of the activation loop of the kinase domain. Instead, GRKs have evolved an activation mechanism that relies on docking with the cytoplasmic surface of activated GPCRs (Tesmer, 2011).
The characteristic feature of GRKs is their ability to specifically recognize and phosphorylate activated GPCRs. GRKs phosphorylate activated receptors up to 1000-fold better than the peptide substrates derived from the same receptor, indicating the existence of an allosteric docking site on GRKs (Palczewski & Benovic, 1991). When the C-terminal tail of rhodopsin is proteolytically removed, light-activated rhodopsin (Rho*) enhances the phosphorylation of soluble peptide substrates over 100-fold (Palczewski, Buczylko, Kaplan, Polans, & Crabb, 1991), indicating that GRKs interact with both the C-terminal tail and the transmembrane domain of GPCRs. The recent crystal structure of GRK6 in a closed conformation suggests that the N-terminal helix (αN), which is critical for efficient receptor phosphorylation, and the AGC kinase C-terminal extension coalesce to form a receptor-docking domain that is allosterically coupled to a closed, more active conformation of the kinase domain (Boguth, Singh, Huang, & Tesmer, 2010), consistent with biochemical data from functional analysis of GRK1 (Huang & Tesmer, 2011; Huang, Yoshino-Koh, & Tesmer, 2009).
2. Expression and Purification of GRKs
Structural and functional analysis requires homogenous preparations of GRKs with high-specific activity for structure determination by X-ray crystallography and for unambiguous interpretation of enzymatic data.
2.1. Previous recombinant GRK expression systems
GRK2 and GRK3 were first successfully expressed in Spodoptera frugiperda (Sf9) cells using Autographa californica nuclear polyhedrosis virus as the vector (Kim, Dion, Onorato, & Benovic, 1993). GRK2 and GRK3 were sequentially purified using S-Sepharose, heparin-Sepharose, and Mono S chromatography, generating 5–7 mgl−1 culture of pure GRK. Assuming Michaelis–Menten kinetics, a Vmax of ∼ 3000 nmol min−1 mg−1 and a KM of 14.5 μM for rhodopsin were achieved.
GRK5 and GRK6 were expressed in Sf9 cells using the BacPAK system (Clontech) for the generation of baculoviruses (Benovic & Gomez, 1993; Kunapuli & Benovic, 1993). GRK5 was purified to homogeneity using S-Sepharose and Mono S chromatography, exhibiting a Vmax of ∼ 1000 nmol min−1 mg−1 and a KM of 14.5 μM for rhodopsin (Kunapuli, Onorato, Hosey, & Benovic, 1994; Pronin, Loudon, & Benovic, 2002). GRK6 was purified to homogeneity using SP-Sepharose and heparin-Sepharose columns, exhibiting a Vmax of ∼51 nmol min−1 mg−1 and a KM of ∼10 μM for rhodopsin (Loudon & Benovic, 1994). Note that GRK2–5 were all purified in the presence of 0.02% (v/v) Triton X-100, which is known to stabilize GRK2 (Benovic, 1991) but is also thought to be detrimental to crystallization. Thus, a purification protocol in the absence of detergent was used to purify GRK2 for the purpose of structural analysis (Lodowski, Pitcher, Capel, Lefkowitz, & Tesmer, 2003).
Recombinant GRK1 and GRK4 were generated using the pVL1393 transfer vector and BaculoGold transfection (Pharmingen) (Cha, Bruel, Inglese, & Khorana, 1997; Ohguro et al., 1996; Premont et al., 1996). GRK1, which contains a farnesylation site, was purified by various combinations of heparin-Sepharose and anion and cation exchangers in the presence of detergent from insect cells lysed in either Tween 80 or dodecylmaltoside (Cha et al., 1997; Ohguro et al., 1996). A relatively low-specific activity of 94 nmol min−1 mg−1 was reported using 20 μM rhodopsin. GRK1 undergoes both heterogeneous autophosphorylation and prenylation (Cha etal., 1997). More recently, a higher-specific activity, C-terminally truncated GRK1 was expressed and purified from High Five cells, yielding an enzyme with a Vmax of 2300 nmol min−1 mg−1 and a KM of 2.1 μM (Singh, Wang, Maeda, Palczewski, & Tesmer, 2008). GRK4α, which contains C-terminal palmitoylation sites, was extracted from homogenates with 1% Lubrol PX and partially purified by Mono Q anion exchange chromatography (Premont et al., 1996).
GRK7, which contains a geranylgeranylation site, was expressed in the presence of supplemental mevalonolactone in High Five insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen) but was not purified (Chen et al., 2001).
GRK1 and GRK7 have also been N-terminally FLAG tagged, expressed in HEK293 cells and used in Michaelis–Menten kinetic studies after extraction in 0.5% dodecylmaltoside and purification by anti-FLAG affinity chromatography (Horner, Osawa, Schaller, & Weiss, 2005). Vmax values for FLAG-GRK1 and FLAG-GRK7 were ∼ 1130 and ∼920 nmol min−1 mg−1, respectively, and KM values were ∼3.5 and ∼2 μM, respectively.
Whereas all GRKs can be expressed in insect cells, at least some active GRK2 reportedly can be expressed in E. coli BL21(DE3) cells using the Studier system (Studier & Moffatt, 1986). The reported Vmax and KM values for rhodopsin were ∼ 450 nmol min−1 mg−1 and ∼ 8 μM, respectively (Gan et al., 2000).
2.2. Baculovirus-mediated expression of C-terminal hexahistidine-tagged GRKs
In our labs, Sf9 cells are used to generate baculoviruses, whereas High Five cells, which in our hands and have a faster doubling time, are used for baculovirus infection and GRK purification. Our preferred baculovirus expression system for the past decade has been Invitrogen's Bac-to-Bac. The use of hexahistidine-tagged variants now greatly facilitates GRK purification to levels suitable for most purposes even after one chromatography step, provided sufficient levels of expression. Because the N-termini of GRKs are known to be critical to their function, the pFastBac Dual vector (Invitrogen) was modified to instead express the open reading frames that encode a C-terminal hexahistidine tag. Two complementary primers encoding the hexahistidine tag and a TAA stop codon and generating 5′ Sal I and 3′ SpeI overhangs were annealed and ligated into SalI/SpeI-cut pFBDual vector. This modification leaves most of the polylinker downstream of the polyhedrin promoter intact (Fig. 19.1). cDNAs from full-length or truncated GRKs can therefore be amplified with primers containing BamHI and SalI restriction sites and ligated into the pFBDual-H6 vector. The following GRKs have been expressed using this system: bovine GRK1 (1–535-H6) (Singh et al., 2008), bovine GRK2 (1–689-H6) (Huang et al., 2009), and human GRK6 (1–531-H6) (Boguth et al., 2010). In the case of GRK1 and GRK6, C-terminal lipid modification sites were truncated in order to improve expression and solubility and facilitate crystallization. Consistent with loss of C-terminal farnesylation, bovine GRK1 (1-535-H6) had a sixfold higher KM for rhodopsin and a twofold lower Vmax compared to wild type (Singh et al., 2008).
Figure 19.1.
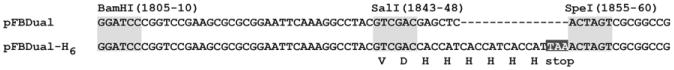
Modification of Invitrogen's pFastBac Dual vector to allow expression of carboxyl terminal hexahistidine-tagged proteins (see text).
We refer readers to the 2010 Invitrogen Bac-to-Bac manual for most details of how we produce baculovirus for GRK expression, although we use the less expensive bacmid purification described in the 2002 manual. Below, we only describe the steps that are specific to analysis of GRKs and use GRK2-H6 as an example GRK.
2.2.1 Checking GRK2-H6 expression in transfected cells
We harvest the VS0 baculovirus (or the P1 viral stock) after 3 days, and the expression of GRK-H6 is surveyed in the soluble fraction of cell lystates.
Solutions, reagents, and equipment
Microprocessor-based probe sonicator equipped with microprobe (e.g., Fisher Sonic Dismembrator F550 Ultrasonic Homogenizer).
Cell Wash Buffer (20 mM HEPES, pH 7.5, 150 mM NaCl).
Nickel Column Base Buffer (20 mM HEPES, pH 8, 300 mM NaCl).
Nickel Column Lysis Buffer (NCLB): base buffer with protease inhibitors and reducing agent added before use (20 mM HEPES, pH 8, 300 mM NaCl, 1 mM DTT, 1 mM PMSF, 10 μg ml−1 leupeptin, 200 μg ml−1 benzamidine).
Dithiothreitol (DTT, Sigma, 1 M aliquots stored at −20 °C).
Benzamidine (Sigma; 20 mg ml−1 stock in water stored at −20 °C).
Leupeptin (Sigma or ThermoFisher; 10 mg ml−1 stock in dimethyl sulfoxide).
Phenylmethylsulfonyl fluoride (PMSF, Sigma; prepared fresh at 100 mM in 100% isopropanol).
2-ml conical sonication tube (e.g., Axygen MCT-200-C).
Trypan Blue (select carcinogen).
Protocol
NCLB with protease inhibitors and reducing agent (NCLB/PI/DTT; enough for 400 μl/sample) is prepared and stored on ice. Virus- containing media are removed from cells, clarified at 300 × g for 5 min and stored in the presence of 2% FBS. Cells are washed with 2 ml cold Cell Wash Buffer on the 6-well plate, resuspended with 1 ml wash buffer, delivered to 1.5-ml microfuge tubes, and spun 500 × g for 5 min at 4 °C (cell pellets may be frozen at −20 °C at this stage). Cells are resuspended in 400 μl NCLB/PI/DTT and transferred to 2-ml conical sonication tubes, where they are sonicated (with probe placed 2–3 mm from the top of the lysate) in an ice-water bath for three rounds of 30 cycles consisting of 1-s pulses. Lysis is checked under a microscope with trypan blue, and viscosity is assessed by pipetting. The lysate is transferred to a microfuge tube and spun 14,000 × g at 4 °C, and the supernatant fraction is pipetted to a fresh tube. The protein concentration is determined (usually 2–3 mg ml−1), and the presence of the target protein is determined by analyzing 20 μg of lysate by SDS-PAGE and Coomassie staining (or immunoblotting). Clarified lysates are stored at −20 °C. If no convincing signal is observed, the pellet fraction from the 14,000 × g spin can be solubilized in SDS sample buffer and analyzed to determine whether the target protein has aggregated. Sufficient viral titers can also be verified (Kitts & Green, 1999).
2.2.2 Optimizing expression kinetics with the VS1 virus
We have generally infected 20 ml of High Five cells with 2–3 concentrations (0.5–1.5 ml) of VS1 virus (amplified VS0) and found that optimal GRK2-H6 expression in various mutants and baculovirus preparations occurs between 44 and 56 h. Three 125-ml baffled Erlenmeyer flasks closed with BugStoppers (Whatman) containing 20 ml of High Five cells at 1 × 106 cells/ml are infected with 0.5, 1, or 1.5 ml of VS1 viral supernatant, and the infection is allowed to proceed for 44, 48, 52, and 56 h. At each time point, 1 ml of culture is removed, cells are pelleted at 300 × g for 5 min, the supernatant is poured off, and the cells are washed with 1 ml Cell Wash Buffer. After removal of buffer, the cell pellets are stored at −20 °C. Heterologous expression equal to the major High Five proteins in the soluble fraction is indicative of strong expression. One-quarter of that expression may lead to ∼ 4 mg purified GRK2 protein per liter of culture. If expression is weak, viral titer should be determined and a broader range of virus concentrations (0.1–10 MOI) for infection should be used. From the results above, a VS1 volume and infection time are determined. If expression is strong, we have found that 200 ml infection is sufficient. For GRKs with weaker expression, we typically carry out 2 × 250 ml infections.
2.3. GRK2-H6 purification
2.3.1 Day 1: harvest, Ni-NTA agarose chromatography
Reagents, solutions, and equipment
Rotor–stator homogenizer ideally equipped with external speed control.
Nickel Column Base Buffer (20 mM HEPES, pH 8, 300 mM NaCl).
Peristaltic Pump.
Fraction Collector and rack for 13 × 100 test tubes.
Low-speed refrigerated centrifuge and an ultracentrifuge and accompanying rotors (e.g., Fiberlite F21S and Beckman 70Ti) to accommodate ∼30 ml of lysate.
5 ml Ni-NTA agarose (Qiagen) column (e.g., 1 × 10 cm Bio-Rad glass Econo column (737-1012) fitted with a flow adapter (738-0015) and equilibrated in nickel column base buffer.
DTT (Sigma, 1 M aliquots stored at −20 °C).
Benzamidine (Sigma; 20 mg ml−1 stock in water stored at −20 °C).
Leupeptin (Sigma or ThermoFisher; 10 mg ml−1 stock in dimethyl sulfoxide).
PMSF (Sigma; prepared fresh at 100 mM in isopropanol).
NCLB base buffer with protease inhibitors and reducing agent added before use (20 mM HEPES, pH 8, 300 mM NaCl, 1 mM DTT, 1 mM PMSF, 10 μg ml−1 leupeptin, and 200 μg ml−1 benzamidine. Fresh protease inhibitors are added immediately before lysis and before the Ni2+ -NTA agarose column.)
Imidazole (Sigma, prepared fresh in NCLB at 150 mM; this stock used to make 20 and 40 mM column buffers).
Trypan Blue (a select carcinogen).
Cell pellets from 0.2 to 0.5 l cultures are resuspended in 15 ml of NCLB and lysed in an ice-water bath using a mounted rotor–stator homogenizer (usually three bursts of 30 s at ∼ 20,000 rpm). Lysis is verified under a microscope using Trypan Blue staining. The lysate is clarified first by centrifugation at 17,000 × g for 20 min and then by centrifugation at 100,000 × g for 45 min. The 100k supernatant fraction is collected and then the protein concentration is determined by Bradford assay and adjusted to 5 mg ml−1 with NCLB. Imidazole is added to the lysate to 20 mM from a 150 mM stock in NCLB. If the GRK is sensitive to proteolytic degradation, 1 mM EDTA can be added to the NCLB to inhibit metalloproteases but should be diluted to ∼100 μM before loading on to Ni-NTA columns.
The column is equilibrated with three column volumes of NCLB supplemented with imidazole to 20 mM at a flow rate of 0.5 ml min−1 for this and subsequent steps. In the fraction collector, 13 × 100 test tubes are used to hold 1.5-ml microfuge collection tubes. The clarified lysate is loaded on the column, the column is washed with three column volumes of NCLB plus 20 mM imidazole, washed with three more column volumes of NCLB plus 40 mM imidazole, and then the hexahistidine-tagged protein is elutedin 1 ml fractions with 40 ml of NCLB plus 150 mM imidazole. Fifteen microliters of selected column fractions are combined with 5 μl 4 × SDS sample buffer (8% (w/v) SDS, 250 mM Tris, pH 6.8, 20 mM EDTA, 40% glycerol, trace bromophenol blue, and 10 mM DTT), and 10 μl of this mixture is analyzed by 8% SDS polyacrylamide gels and stained with Coomassie Blue (Fig. 19.2A). Fractions are pooled and stored overnight at 4 °C.
Figure 19.2.

Purification of GRK2-H6 from baculovirus-infected High Five cells. (A) Clarified lysates from a 0.5-l culture were chromatographed on a 5-ml Ni-NTA column. HSS, high-speed supernatant; imid, imidazole. (B) GRK2-H6-containing fractions (6-14) were pooled and purified over a 1-ml Econo-Pac High Capacity High S column. Column fractions were analyzed by 8% SDS-PAGE and Coomassie staining.
2.3.2 Day 2: cation exchange chromatography
A cation exchange step has proven to be greatly useful not only for achieving higher purity but also as a diagnostic tool. Some GRKs, such as GRK1, express with different N-terminal truncations, and a strong cation exchanger can help to separate these pools (Singh et al., 2008). The cation exchanger can also discriminate between different phosphorylation states. For example, MAP kinase-mediated phosphorylation of GRK2 at Ser670 was first revealed by cation exchange chromatography (Pitcher et al., 1999). For this reason, the GRK2-S670A mutant is often used for GRK2 preparations.
Reagents, solutions, and equipment
1 ml High S and High Q columns (Econo-Pac High Capacity, Bio-Rad).
Gradient maker (e.g., Hoeffer SG 30).
No salt buffer (20 mM HEPES, pH 7.5, 5 mM EDTA, 1 mM DTT, 0.02% (v/v) Triton X-100, with protease inhibitors; stored at 4 °C without DTT and protease inhibitors).
Low salt buffer (20 mM HEPES, pH 7.5, 50 mM NaCl, 5 mM EDTA, 1 mM DTT, 0.02% (v/v) Triton X-100, with protease inhibitors; stored at 4 °C without DTT and protease inhibitors).
High salt buffer (20 mM HEPES, pH 7.5, 600 mM NaCl, 5 mM EDTA, 0.02% (v/v) Triton X-100, with protease inhibitors; stored at 4 °C without DTT and protease inhibitors).
For GRK2, the High Q column simply acts as a prefilter for the removal of contaminants, including other protein kinases (Benovic, Mayor, Staniszewski, Lefkowitz, & Caron, 1987; Gan et al., 2004; Sterne-Marr et al., 2009). This property applies to GRK5 and GRK6, but not to GRK1 or GRK4α, which bind to both cation and anion exchangers (Ohguro et al., 1996; Premont et al., 1996; Sallese et al., 1997). The nickel column pool is diluted sixfold in 20 mM HEPES, pH 7.5, 5 mM EDTA, and 0.02% TX-100, with protease inhibitors to lower the NaCl concentration to 50 mM. The High Q and High S columns are hooked up in tandem and equilibrated in low salt buffer at a flow rate of 1 ml min−1. The nickel column pool is loaded on the tandem columns, and then after a one-column volume wash, the High Q column is removed. The High S column is washed with 5–10 column volumes of low salt buffer and then eluted with a 20 ml linear gradient of 50–600 mM NaCl. Column fractions are visualized (Fig. 19.2B) as described above, and high GRK2 fractions (e.g., 7–11) and lower GRK fractions (e.g., 6, 12–15) are each pooled and combined with one-third volume of glycerol (25% glycerol stocks), distributed in aliquots, and stored at −20 or −80 °C. We generally obtain 10–20 mg pure GRK2-H6 per liter of insect cell culture.
2.3.3 Polishing by size exclusion chromatography
Although at this point hexahistidine-tagged GRKs are often >90% pure, sometimes a buffer exchange step is required or additional purification is needed. Size exclusion chromatography achieves both purposes. Typically, two tandem Superdex S200 columns (GE Healthcare) mounted on a high-pressure liquid chromatography system such as a Biologic Duo-Flow (Bio-Rad) are used to separate monodisperse GRKs from aggregates and other minor contaminants. The standard running buffer for the column is 20 mM HEPES, pH 8.0, 100–200 mM NaCl, and 1–2 mM DTT.
2.3.4 Crystallization of GRKs
Currently, there are 23 crystal structures deposited in the Protein Data Bank representing bovine GRK1, human and bovine GRK2 (alone and in complex with heterotrimeric G-proteins), and human GRK6. Ligand-free structures diffract to lower resolution and their electron density maps suggest a high degree of structural heterogeneity (Lodowski et al., 2005; Singh et al., 2008). Crystallization is typically performed at 4 °C using the hanging drop method, and, with only one exception, all GRKs have been crystallized using polyethylene glycol (typically 3350 molecular weight) as the precipitating agent at low ionic strength (typically 100 mM NaCl). The exception is the recent structure of GRK6 in complex with the inhibitor sangivamycin, which used ammonium sulfate as the precipitating agent (Boguth et al., 2010). Most GRK crystals were obtained with well solutions adjusted to acidic pH (4.3–6.5).
3. GRK Functional Assays
Three assays are typically used to characterize GRK function in vitro: the rhodopsin (Rho) phosphorylation assay, the peptide C phosphorylation assay, and the activation assay. Endogenous Rho isolated from bovine rod outer segments (ROSs) is the most readily available and convenient GPCR substrate for these assays. All phosphorylation sites are found in the carboxyl tail of the receptor. However, the C-terminal Rho peptide, known as peptide C, is a very poor substrate for GRKs. Thus, the peptide C phosphorylation assay is thought to measure the intrinsic ability of the kinase to assume an activated conformation in the absence of activated receptor. In contrast, the activation assays measures the ability of truncated Rho* that lacks the phosphoacceptor sites in the C-terminal tail (G329 -Rho*) to enhance peptide C phosphorylation. Residues that when mutated show defects in the activation assay (but not in the peptide C phosphorylation assay, are thought to be involved in direct interactions with the receptor substrate. Residues that show defects in both are thought to impair formation of the receptor-docking site) as this is believed to be allosterically coupled to closure of the kinase domain (Boguth et al., 2010; Huang, Orban, Jastrzebska, Palczewski, & Tesmer, 2011).
3.1. Rho* phosphorylation assay
ROSs are isolated from dark-adapted bovine retinas (W. L. Lawson Co., Omaha, NE). One hundred retinas are processed at a time under dim red light by published procedures (Kuhn & Wilden, 1982; Papermaster, 1982), typically yielding 10–20 mg of Rho. For the standard assay, final assay conditions are ∼40 nM GRK2, 8 μM Rho, 200 μM ATP, 0.5–1 dpm/fmol γ- 32P-ATP, 20 mM Tris, pH 7.5,2 mM EDTA, and 7.5 mM MgCl2. Stocks of Rho and GRK2 are prepared at 32 mM and 80 nM, respectively, in GRK assay buffer. A “4 × ATP Hot Mix” is prepared with 800 μM ATP and 1–2 dpm/pmol 32P-ATP (MP Biomedical) in GRK assay buffer. The 4 × ATP Hot Mix is diluted 100-fold and 5 μl are counted in a scintillation counter to determine the specific radioactivity. Five microliters of GRK2 and 2.5 μl 4 × ATP Hot Mix are combined in 0.5-ml PCR tubes. Samples are taken to a dark room where Rho is resuspended by vortex mixing, and 2.5 ml are delivered to the reaction tube under dim red light. Samples are transferred to a 30 °C incubator for 30 s, and then a desk lamp with a 100 W bulb positioned over the incubator is illuminated. After a 3 min incubation, samples are transferred to an ice-water bath and reactions are quenched with 14 μl 4 × SDS/DTT sample buffer (8% (w/v) SDS, 250 mM Tris, pH 6.8, 20 mM EDTA, 40% glycerol, trace bromophenol blue, and 50 mM DTT). Samples are mixed by tapping periodically during a 30 min incubation at 65 °C and briefly spun at 500 × g before separation on 10% SDS-PAGE. Afterward, the bottom of the gel with the running dye (and free ATP) is removed, the gel is stained with Coomassie dye, destained, the Rho bands are excised (from the hydrated gel), and the radioactivity is determined in a scintillation counter. For steady state kinetics studies, 2 × concentrations of Rho in GRK assay buffer are prepared, and the reaction contains 5 μl Rho, 2.5 μl 80 nM GRK2, and 2.5 μl 4 × ATP Hot Mix.
3.2. Peptide C phosphorylation assay
Peptide C contains residues 330-DDEASTTVSKTETSQVA-346 of bovine Rho. Three arginines are often appended to the peptide sequence shown above to facilitate binding to negatively charged filters (such as P81 paper) in a filter-based kinase assay, but we have found that the triargininyl peptide C can be separated from free 32P-ATP on 18% SDS poly-acrylamide gels where the radioactivity can be more confined. Peptide C phosphorylation reactions (∼500 nM GRK2, 1 mM peptide C, 100 μM ATP, 1.5dpm/fmol 32P-ATP, 20 mM Tris, pH 7.5, 2 mM EDTA, and 7.5 mM MgCl2) are carried out for 50 min at 30 °C before quenching the reaction, loading 10 μl on an 18% gel, and quantifying phosphorylation as described above.
3.3. 329G-Rho preparation and activation assay
Endoprotease Asp-N cleavage of Rho to prepare G329 -Rho has been previously described (Palczewski et al., 1991). After inactivation of the protease by the addition of EDTA and DTT to 2 mM, 12% SDS-PAGE/Coomassie staining is used to verify reaction completion. The ROS are stripped with 5 M urea and washed three times with 50 mM Tris, pH 7.5. Following the second wash, an aliquot is removed for protein concentration determination, and the final G329 -Rho is resuspended at 12 μM in GRK assay buffer.
The activation assay is carried out in 12 μl reactions with 100 nM GRK, 2 μM 329G-Rho, 100 μM peptide C, and 100 μM ATP (1.5 dpm/fmol 32P-ATP). Sixfold concentrated stocks of GRK, 329G-Rho, and peptide C are prepared in GRK assay buffer. Two microliters each of GRK2 and peptide C are combined with 6 μl 200 μM ATP (3 dpm/fmol 32P-ATP) in 0.5-ml PCR tubes. Two microliters of 12 μM 329G-Rho is added under dim red light. Two identical sets of reactions are prepared to allow for reaction in the light and dark for 30 min at 30 °C with periodic tap mixing. Reactions are quenched and analyzed as described for the peptide C assay.
3.4. Cell-based β2-adrenergic receptor (β2AR) phosphorylation assay
Cell-based detection of GPCR phosphorylation has emerged as a sensitive and direct assessment of GRK activity in intact cells. GRK2 phosphorylates Ser355, 356, and 364 of the β2AR in living cells (Seibold et al., 2000). Based upon the success of detecting in vivo phosphorylation of chemokine receptor type 5 using monoclonal phosphosite antibodies (Pollok-Kopp, Schwarze, Baradari, & Oppermann, 2003), a similar approach was used to measure GRK2 phosphorylation of the β2AR (Tran et al., 2004). This methodology generates a robust agonist-induced and dose-dependent phosphorylation signal in HEK293 cells stably expressing hexahistidine-tagged β2AR.
Unfortunately, using the above protocol we were unable to detect any dependence of phosphorylation on transiently transfected GRK2, as was also reported by others for exogenously supplied GRK2 (Shenoy et al., 2006). We have, therefore, modified the procedures of Tran et al. (2004) to allow detection of exogenous GRK2 promoted phosphorylation of the transiently transfected β2AR in COS-7 cells. This procedure does not require enrichment of β2AR by affinity chromatography or immunoprecipitation, and therefore untagged receptors can be used.
3.5. Days 1-3: DNA transfection
Reagents
FuGENE® HD transfection (Promega).
Transfection Quality DNA (Qiagen midi preps).
DNAs
pcDNA3.1-HA-hβ2AR (HA-tagged human β2AR from Drs. Marc Caron and Larry Barak).
pcDNA3-bGRK2 (bovine GRK2 from Dr. Jeffrey Benovic).
pcDNA3.1-Gαs (human Gαs long, UMR cDNA Resource Center).
pcDNA3.1-Gβ1 (human Gβ1, UMR cDNA Resource Center).
pcDNA3.1-Gγ2 (human Gγ2, UMR cDNA Resource Center).
Approximately 3 × 106 COS-7 cells/well are plated in 6-well plates (∼25% confluency) and incubated overnight in a humidified atmosphere at 37 °C/ 5% CO2. In general, DNA:FuGENE® HD complexes are prepared with 2 μg of total DNA and 8 μl of FuGENE® HD. Transfection mixes are prepared by the addition of DNA to 100 μl serum-free and antibiotic-free DMEM in microfuge tubes followed by tap mixing. FuGENE® HD is added, the transfection mix is vortexed for 1 s, and then incubated for 15 min. Transfection mixes are added drop-wise to each well of the 6-well plate, and the cells are incubated for 48 h in a humidified atmosphere at 37 °C/5% CO2.
3.6. Day 4: agonist (isoproterenol)/inverse agonist (alprenolol) treatment, cell harvest
Reagents and Equipment
Cell scrapers for 6-well tissue culture plate.
Microprocessor-based probe sonicator equipped with microprobe (e.g., Fisher Sonic Dismembrator F550 Ultrasonic Homogenizer).
Cell Wash Buffer (20 mM HEPES, pH 7.5, 150 mM NaCl), 4 °C.
Receptor Solubilization Buffer (RSB, 200 μl/well; 20 mM HEPES, pH 7.4, 150 mM NaCl, 10 mg ml−1 n-dodecyl-β-d-maltoside (DDM), 10 mM DTT (reducing agent), 1 mM PMSF, 10 μg ml−1 leupeptin, 200 μg ml−1 benzamidine (protease inhibitors, PI), 20 mM tetrasodium pyrophosphate, 10 mM sodium fluoride, and ±100 nM okadaic acid (phosphatase inhibitors, PhI)).
(−)Isoproterenol (ISO, Sigma, prepared fresh as described below).
Alprenolol (ALP, Sigma, 1 mM stock in ethanol, stored at −20 °C).
DDM (Anatrace Inc., Maumee, OH; prepared fresh at 10 mg ml−1 in RSB).
DTT (Sigma, 1 M aliquots stored at −20 °C).
Benzamidine (Sigma; 20 mg ml−1 stock in water stored at −20 °C).
Leupeptin (Sigma or ThermoFisiier; 10 mg ml−1 stock in dimethyl sulfoxide).
PMSF (Sigma; prepared fresh at 100 mM in 100% isopropanol).
Sodium pyrophosphate (Sigma; 0.5 M stocks stored at −20 °C and thawed at 65 °C).
NaF (Sigma; 0.5 M stocks stored at −20 °C).
Okadaic acid (Axxora, suspected carcinogen, 100 μM stocks in ethanol stored at −20 °C).
Peptide N-glycosidase F (PNGase; New England Biolabs, Beverly, MA, 500 U μl−1).
On the day of cell harvest, the cell lysis/RSB is first prepared and stored on ice. The 30-min serum starvation of cells is initiated and then ISO and ALP are prepared. Because there are many additives to the RSB, we make a 2 × stock of the base buffer (40 mM HEPES, pH 7.4, 300 mM NaCl), add the DTT, protease inhibitors (PMSF, benzamidine, leupeptin) and phosphatase inhibitors (sodium fluoride, sodium pyrophosphate, okadaic acid), and bring the volume up with cold deionized water. DDM is added as a solid and dissolved on a nutator at 4 °C.
The media are removed from the transfected cells and replaced with 1 ml serum-free media. Approximately, 1 mg of ISO is dissolved in 1 mM ascorbic acid (an antioxidant to protect the vicinal hydroxyls on the catecholamine) to generate a 20 mM isoproterenol stock. This is further diluted to 1 mM in 1 mM ascorbic acid and stored on ice. The 20 mM ALP is diluted to 1 mM in ethanol.
Cells are treated for 5 min by staggering the addition of 10 μM ISO or ALP to each well every 15 s to allow equal treatment. Following the 5-min incubation, the media are aspirated and the cells are washed two times with cold cell wash buffer. Buffer is removed, and each well is scraped in the presence of 200 μl RSB on ice. The lysate is transferred to a 1.5-ml microfuge tube and sonicated using a 90-cycle regimen of 1 s on/2 s off. The receptor is further solubilized by mixing 30 min on a nutator at 4 °C and then clarified by centrifugation at 14,000 × g at 4°C. The soluble fraction, 3–5 mg ml−1 protein, may be stored at −20 °C.
3.7. Days 4 and 5: Peptide N-glycosidase F (PNGase) treatment and immunoblotting
Reagents and Equipment
Electrophoresis and immunoblotting equipment.
Nutator rocker/shaker at 4 °C.
4 × SDS receptor sample buffer/DTT (8% SDS, 250 mM Tris, pH 6.8, 20 mM EDTA, 40% glycerol, trace bromophenol blue, and 40 mM DTT).
Precision Plus WesternC Standards for Chemiluminescent detection (Bio-Rad).
Rocking platform at 4 °C
β2AR GRK phosphosite antibodies (Santa Cruz Biotechnology sc-16719-R).
β2AR carboxyl tail affinity purified polyclonal antibodies (Santa Cruz Biotechnology, sc-569).
GRK2 polyclonal antibody (Santa Cruz Biotechnology, sc-562).
Goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody.
StrepTactin HRP Conjugate (Bio-Rad).
Tris buffered saline/Tween (TBS/Tween; 50 mM Tris, pH 7.5, 150 mM NaCl, 0.1 % (v/v) Tween-20).
Blocking Buffer (TBS/Tween with 5% nonfat dried milk).
Immunoblot stripping buffer (25 mM glycine, pH 2, 1% SDS).
West Pico, Chemiluminescent Reagent (or West Femto if necessary; Thermo Scientific).
The stock PNGase is diluted fivefold to 100 U μl−1 in RSB (or 20 mM HEPES, pH 7.5,150 mM NaCl if PNGase treatment occurs on a day other than the day of harvest). Thirty microliters of the soluble fraction are treated with 1 μl 100 U μl−1 PNGase for 2 h at 37°C. Ten microliters of SDS sample buffer containing DTT at a final concentration of 40 mM is added, and the samples are incubated at 65 °C for 30 min with tap mixing every 5 min. Twenty microliters of the PNGase-treated samples are separated on 10% acrylamide SDS-PAGE alongside Precision Plus WesternC (Bio-Rad) molecular weight standards that can be visualized by chemiluminescent detection. Standard immunoblotting procedures are used employing 1:666 dilution of the primary antibody overnight with rocking at 4°C. A 1:2000 dilution of the goat anti-mouse HRP secondary antibody is used in conjunction with a 1:20,000 dilution of the StrepTactin HRP Conjugate. We develop our blots with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and image using the Bio-Rad ChemiDoc XRS System. The molecular weight standards are cut off and stored in TBS/Tween at 4 °C, whereas the remainder of the blot is stripped at room temperature for 5 min in stripping buffer. The blot is washed, reblocked, and probed with the β2AR C-tail antibody diluted 1:1000, 1 h at room temperature, and then the secondary antibody and StrepTactin HRP as described above. The blot is developed with chemiluminescent reagent in the presence of the molecular weight standards, imaged, stripped, and reblocked. Finally, the blot is probed with the GRK2 antibody (1:1000, 1 h incubation at room temperature) followed by goat anti-mouse HRP secondary antibody (1:2000) and StrepTactin HRP (1:10,000) incubation as described above.
Densitometry is used to quantify in vivo phosphorylation of the β2AR by transfected GRK2. The pSer and C-tail signals are normalized to the lane that contains transfected β2AR alone treated with the inverse agonist, ALP. The ratio of pSer/C-tail is obtained and yields a measure of agonist-inducible receptor phosphorylation. To assess the effect of transfected G-proteins or mutant GRK, the pSer/C-tail ratio is normalized to the level of GRK in each GRK-transfected sample.
We found that the absence of okadaic acid in the RSB did not negatively impact the detection of pSer β2AR (Fig. 19.3A), but that PNGase treatment is absolutely essential to allow detection of phosphorylated β2AR at its monomer molecular weight (Fig. 19.3B). Exogenous GRK2 stimulated the ISO-dependent phosphorylation ∼ 10-fold (Fig. 19.4). Cotransfection of GRK2 with Gβγ enhanced the GRK2-dependent pSer signal, but the ALP-treated samples also displayed elevated signal. Therefore, the strongest GRK2-dependent signal is observed with Gβγ, but the strongest agonist-dependent signal is observed with GRK2 alone.
Figure 19.3.
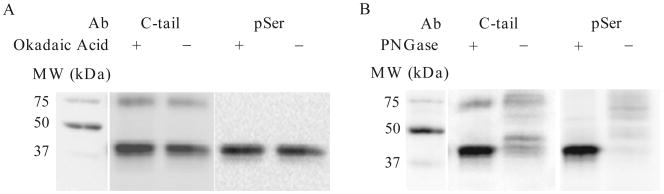
Test of the role of okadaic acid and PNGase F on agonist-induced β2AR phosphorylation in intact cells using GRK phosphosite antibodies. COS-7 cells were grown in 6-well plates and transfected with cDNA encoding β2AR, GRK2, Gβ1, and Gγ2 at a ratio of 0.7:0.7: 0.2: 0.2 μg. After 48 h, cells were treated with 10 μM isoproterenol for 5 min. (A) Cell lysates were prepared in the presence of reducing agent, protease inhibitors, phosphatase inhibitors sodium pyrophosphate, and sodium fluoride, but in the absence or presence of okadaic acid, and treated with PNGase F (PNGase). (B) Cell lysates were prepared in the presence of reducing agent, protease inhibitors, phosphatase inhibitors including okadaic acid and then treated in the absence or presence of PNGase F. Samples were separated by 10% SDS-PAGE and transferred to nitrocellulose. Agonist-induced phosphorylation of the β2AR was detected with a phosphosite antibody that recognizes phosphorylated Ser355 and Ser356 (pSer), and total β2AR was detected with an antibody that recognizes the carboxyl tail of the receptor independent of its phosphorylation status (C-tail).
Figure 19.4.

Role of exogenous GRK2 and heterotrimeric G-proteins on agonist-induced β2AR phosphorylation in intact cells. COS-7 cells were grown in 6-well plates and transfected with 0.8 μg β2AR cDNA in the absence or presence of cDNAs encoding GRK2 (0.3 μg), Gαs (0.3 μg), Gβ1 (0.3 μg), and Gγ2 (0.3 μg) as indicated. Cells were treated for 5 min with 10 μM alprenolol (ALP) or isoproterenol (ISO), and the detection of phosphorylated β2AR was carried out as described in the legend to Fig. 19.3 and the text. The pSer and C-tail signals were normalized to that of the ALP-treated β2AR lane and phosphorylation of the β2AR was determined by taking the pSer/C-tail ratio. The levels of GRK2, when transfected, were with 20% of each other. The results of this experiment are representative of four similar experiments.
Acknowledgments
This work was supported by National Science Foundation grants MCB0315888 and MCB0744739 (R. S. M.) and National Institute of Health grants HL071818, HL086865, and GM081655 (J. T.). We thank Dick Clark and Faiza Baameur (University of Texas Health Science Center, Houston) for advice and phosphorylated β2AR controls, Valerie Tesmer (University of Michigan) for advice on insect cell culture, and Amber Cutter, Katelynn Mannix, Alex Leahey, Tim Clarke, and Devin McDonald (Siena College) for their contributions to these projects.
References
- Benovic JL, Gomez J. Molecular cloning and expression of GRK6. A new member of the G protein-coupled receptor kinase family. The Journal of Biological Chemistry. 1993;268:19521–19527. [PubMed] [Google Scholar]
- Benovic JL. Purification and characterization of beta-adrenergic receptor kinase. Methods in Enzymology. 1991;200:351–362. doi: 10.1016/0076-6879(91)00152-m. [DOI] [PubMed] [Google Scholar]
- Benovic JL, Mayor F, Jr, Staniszewski C, Lefkowitz RJ, Caron MG. Purification and characterization of the β-adrenergic receptor kinase. The Journal of Biological Chemistry. 1987;262:9026–9032. [PubMed] [Google Scholar]
- Boguth CA, Singh P, Huang CC, Tesmer JJ. Molecular basis for activation of G protein-coupled receptor kinases. The EMBO Journal. 2010;29:3249–3259. doi: 10.1038/emboj.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha K, Bruel C, Inglese J, Khorana HG. Rhodopsin kinase: expression in baculovirus-infected insect cells, and characterization of post-translational modifications. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10577–10582. doi: 10.1073/pnas.94.20.10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Zhang K, Church-Kopish J, Huang W, Zhang H, Chen YJ, et al. Characterization of human GRK7 as a potential cone opsin kinase. Molecular Vision. 2001;7:305–313. [PubMed] [Google Scholar]
- Gan X, Ma Z, Deng N, Wang J, Ding J, Li L. Involvement of the C-terminal proline-rich motif of G protein-coupled receptor kinases in recognition of activated rhodopsin. The Journal of Biological Chemistry. 2004;279:49741–49746. doi: 10.1074/jbc.M407570200. [DOI] [PubMed] [Google Scholar]
- Gan XQ, Wang JY, Yang QH, Li Z, Liu F, Pei G, et al. Interaction between the conserved region in the C-terminal domain of GRK2 and rhodopsin is necessary for GRK2 to catalyze receptor phosphorylation. The Journal of Biological Chemistry. 2000;275:8469–8474. doi: 10.1074/jbc.275.12.8469. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacology & Therapeutics. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner TJ, Osawa S, Schaller MD, Weiss ER. Phosphorylation of GRK1 and GRK7 by cAMP-dependent protein kinase attenuates their enzymatic activities. The Journal of Biological Chemistry. 2005;280:28241–28250. doi: 10.1074/jbc.M505117200. [DOI] [PubMed] [Google Scholar]
- Huang CC, Orban T, Jastrzebska B, Palczewski K, Tesmer JJ. Activation of G protein-coupled receptor kinase 1 involves interactions between its N-terminal region and its kinase domain. Biochemistry. 2011;50:1940–1949. doi: 10.1021/bi101606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Tesmer JJ. Recognition in the face of diversity: interactions of heterotrimeric G proteins and G protein-coupled receptor (GPCR) kinases with activated GPCRs. The Journal of Biological Chemistry. 2011;286:7715–7721. doi: 10.1074/jbc.R109.051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yoshino-Koh K, Tesmer JJ. A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. The Journal of Biological Chemistry. 2009;284:17206–17215. doi: 10.1074/jbc.M809544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Haste N, Taylor SS, Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1272–1277. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CM, Dion SB, Onorato JJ, Benovic JL. Expression and characterization of two β-adrenergic receptor kinase isoforms using the baculovirus expression system. Receptor. 1993;3:39–55. [PubMed] [Google Scholar]
- Kitts PA, Green G. An immunological assay for determination of baculovirus titers in 48 hours. Analytical Biochemistry. 1999;268:173–178. doi: 10.1006/abio.1998.3042. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Wilden U. Assay of phosphorylation of rhodopsin in vitro and in vivo. Methods in Enzymology. 1982;81:489–496. doi: 10.1016/s0076-6879(82)81066-5. [DOI] [PubMed] [Google Scholar]
- Kunapuli P, Benovic JL. Cloning and expression of GRK5: A member of the G protein-coupled receptor kinase family. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5588–5592. doi: 10.1073/pnas.90.12.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunapuli P, Onorato JJ, Hosey MM, Benovic JL. Expression, purification, and characterization of the G protein-coupled receptor kinase GRK5. The Journal of Biological Chemistry. 1994;269:1099–1105. [PubMed] [Google Scholar]
- Lodowski DT, Barnhill JF, Pyskadlo RM, Ghirlando R, Sterne-Marr R, Tesmer JJ. The role of Gβγ and domain interfaces in the activation of G protein-coupled receptor kinase 2. Biochemistry. 2005;44:6958–6970. doi: 10.1021/bi050119q. [DOI] [PubMed] [Google Scholar]
- Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. Keeping G proteins at bay: A complex between G protein-coupled receptor kinase 2 and Gβγ. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- Loudon RP, Benovic JL. Expression, purification, and characterization of the G protein-coupled receptor kinase GRK6. The Journal of Biological Chemistry. 1994;269:22691–22697. [PubMed] [Google Scholar]
- Ohguro H, Rudnicka-Nawrot M, Buczylko J, Zhao X, Taylor JA, Walsh KA, et al. Structural and enzymatic aspects of rhodopsin phosphorylation. The Journal of Biological Chemistry. 1996;271:5215–5224. doi: 10.1074/jbc.271.9.5215. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Benovic JL. G-protein-coupled receptor kinases. Trends in Biochemical Sciences. 1991;16:387–391. doi: 10.1016/0968-0004(91)90157-q. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Buczylko J, Kaplan MW, Polans AS, Crabb JW. Mechanism of rhodopsin kinase activation. The Journal of Biological Chemistry. 1991;266:12949–12955. [PubMed] [Google Scholar]
- Papermaster DS. Preparation of antibodies to rhodopsin and the large protein of rod outer segments. Methods in Enzymology. 1982;81:240–246. doi: 10.1016/s0076-6879(82)81037-9. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Tesmer JJ, Freeman JL, Capel WD, Stone WC, Lefkowitz RJ. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signal-regulated kinases. The Journal of Biological Chemistry. 1999;274:34531–34534. doi: 10.1074/jbc.274.49.34531. [DOI] [PubMed] [Google Scholar]
- Pollok-Kopp B, Schwarze K, Baradari VK, Oppermann M. Analysis of ligand-stimulated CC chemokine receptor 5 (CCR5) phosphorylation in intact cells using phosphosite-specific antibodies. The Journal of Biological Chemistry. 2003;278:2190–2198. doi: 10.1074/jbc.M209844200. [DOI] [PubMed] [Google Scholar]
- Premont RT, Macrae AD, Stoffel RH, Chung N, Pitcher JA, Ambrose C, et al. Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. The Journal of Biological Chemistry. 1996;271:6403–6410. doi: 10.1074/jbc.271.11.6403. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Loudon RP, Benovic JL. Characterization of G protein-coupled receptor kinases. Methods in Enzymology. 2002;343:547–559. doi: 10.1016/s0076-6879(02)43157-6. [DOI] [PubMed] [Google Scholar]
- Sallese M, Mariggio S, Collodel G, Moretti E, Piomboni P, Baccetti B, et al. G protein-coupled receptor kinase GRK4. Molecular analysis of the four isoforms and ultrastructural localization in spermatozoa and germinal cells. The Journal of Biological Chemistry. 1997;272:10188–10195. doi: 10.1074/jbc.272.15.10188. [DOI] [PubMed] [Google Scholar]
- Seibold A, Williams B, Huang ZF, Friedman J, Moore RH, Knoll BJ, et al. Localization of the sites mediating desensitization of the β2-adrenergic receptor by the GRK pathway. Molecular Pharmacology. 2000;58:1162–1173. doi: 10.1124/mol.58.5.1162. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. The Journal of Biological Chemistry. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJ. Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. The Journal of Biological Chemistry. 2008;283:14053–14062. doi: 10.1074/jbc.M708974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Marr R, Leahey PA, Bresee JE, Dickson HM, Ho W, Ragusa MJ, et al. GRK2 activation by receptors: Role of the kinase large lobe and carboxyl-terminal tail. Biochemistry. 2009;48:4285–4293. doi: 10.1021/bi900151g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of Molecular Biology. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ. Structure and function of regulator of G protein signaling homology domains. Progress in Molecular Biology and Translational Science. 2009;86:75–113. doi: 10.1016/S1877-1173(09)86004-3. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ. Activation of G protein-coupled receptor (GPCR) kinases by GPCRs. In: Giraldo J, Pin JP, editors. G protein-coupled receptors: From structure to function RSC Drug Discovery Series 8. London: The Royal Society of Chemistry, London; 2011. pp. 297–315. [Google Scholar]
- Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the β2-adrenergic receptor using phosphoserine-specific antibodies. Molecular Pharmacology. 2004;65:196–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]