

Abstract
Neurodegeneration after traumatic brain injury (TBI) is facilitated by innate and adaptive immunity and can be harnessed to effect brain repair. In mice subjected to controlled cortical impact (CCI) we show that treatment with granulocyte macrophage colony stimulating factor (GM-CSF) affects regulatory T cell numbers coincident with decreased lesion volumes and increased cortical tissue sparing. This paralleled increases in neurofilament and diminished reactive microglial staining. Transcriptomic analysis showed that GM-CSF induces robust immune neuroprotective responses seven days following CCI. Together, these results support the therapeutic potential of GM-CSF for TBI.
Keywords: Traumatic brain injury, immunity, microglia, neuroinflammation, T cell
1. Introduction
Traumatic brain injury (TBI) leads to chronic neurodegeneration driven, in part, by innate and adaptive immune responses. Both influence neuronal injury (Jin et al., 2012). Brain shearing forces affect mechanical trauma causing release of purinergic signaling molecules (Davalos et al., 2005), excitatory neurotransmitters (Faden et al., 1989, Palmer et al., 1993), and other damage-associated factors (Laird et al., 2014). These all affect resident glial (astrocytes and microglia) function leading to the production of pro-inflammatory mediators. These include reactive oxygen species, cytokines and pro-apoptotic proteins; all known to perpetuate neural injury (Burda and Sofroniew, 2014). Notably, the traumatic event can damage the neurovasculature and attract immunocytes, such as neutrophils, lymphocytes and blood-borne macrophages, to sites of brain injury (Carlos et al., 1997, Schwarzmaier et al., 2010, Soares et al., 1995). Once in the parenchyma, the cells can become immune competent, affecting glial responses and accelerating ongoing neural damage. Such changes can affect microglial responses, where a pro-inflammatory, classically activated state (M1) and an anti-inflammatory, alternatively activated state (M2) evolve in a temporally ordered fashion over prolonged time periods (Loane et al., 2014). Indeed, long-term immune response changes were observed in human brains up to 17 years after brain injury (Ramlackhansingh et al., 2011).
The role of the adaptive immunity in the pathobiology of TBI is heralded by a steady increase in the number of lymphocytes that enter the brain, but not in draining lymph nodes or spleen (Clausen et al., 2009, Jin et al., 2012). Such cellular shifts occur as early as one day following injury and can continue for up to 28 days after the traumatic event. Whether the cells serve to accelerate damage or perform homeostatic functions are not understood. Previous work by our group and others, demonstrated that regulatory T cells (Treg) can attenuate microglial pro-inflammatory activities leading to robust neuroprotective responses in models of stroke, HIV-1 encephalitis, amyotrophic lateral sclerosis, myasthenia gravis and Parkinson’s disease (PD) (Gendelman and Appel, 2011, Kosloski et al., 2013, Li et al., 2013, Liesz et al., 2009, Reynolds et al., 2007, Reynolds et al., 2010, Reynolds et al., 2009, Sheng et al., 2008).
Unlike neurodegenerative and neuroinflammatory disorders, immune activity changes with time following TBI (Jin et al., 2012, Kox et al., 2008, Loane et al., 2014). The dynamics of immune mediated injuries versus neuroprotection are also controlled by environmental events and immune regulatory activities (Burda and Sofroniew, 2014). The transformation of innate microglial and astrocyte activities together with the emergence of an altered adaptive immune response can certainly aggravate neural damage (Jin et al., 2012). Such events are operative immediately following injuries but, interestingly, evolve over time leading to compensatory neuroprotective outcomes (Ziebell et al., 2014). On the cellular level, astrocytes contribute to these complex immunoregulatory events by affecting blood-brain barrier integrity and microglial secretory responses that include the production of neuroregulatory and neuroprotective factors to speed neuronal repair (Segev-Amzaleg et al., 2013). The degree and timing of cell-based cross-talk is, nonetheless limited for TBI, leaving open the notion that pharmacological measures that strengthen neurotrophic immune responses could significantly affect disease outcomes. To these ends, we investigated the use of granulocyte macrophage colony-stimulating factor (GM-CSF), an approved adjunctive therapy for chemotherapy-induced granulocytopenia (Buchsel et al., 2002). GM-CSF, a hematopoietic cell growth and differentiation factor, is produced by macrophages, T cells, mast cells, endothelial cells, and fibroblasts. GM-CSF, in turn, stimulates the mobilization of hematopoietic progenitor cells, including Treg (Schabitz et al., 2008, Sheng et al., 2011, Zou et al., 2011), which affect neuroprotective responses as we recently demonstrated in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of Parkinson’s disease (Kosloski et al., 2013). We now demonstrate that GM-CSF can have broad neuroprotective responses following a controlled cortical impact (CCI) injury. The CCI model was used to investigate GM-CSF neuroprotective activities as it shows high fidelity for many aspects of human head injury (Xiong et al., 2013). When injured, mice develop a lesion in the injured cortex that is identified as early as six hours following trauma and evolves over time to frank neurodegeneration (Hall et al., 2008). Lesion size remains relatively unchanged at 48 hours with peak area of degenerative neurons observed at this time point as determined by silver staining. By seven days, lesion volume has decreased slightly from a peak at 24 hours post-injury. Herein, neuroprotection was readily seen in mice following CCI and treatment with GM-CSF. This, in part, was linked to the induction of Treg populations that serve to transform innate immune responses (Liesz et al., 2013). Supporting these findings were a combination of histology, immunohistochemistry, flow cytometry, and molecular transcriptomic tests to evaluate broad immune regulatory responses of GM-CSF that serve to facilitate neural repair following TBI.
2. Material and methods
2.1. Animals
Male C57BL/6 mice (20 – 24 g; Charles River Laboratories, Wilmington, MA) were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited animal facility at the University of Nebraska Medical Center. The animal housing facility was on a 12-hour light/dark cycle held at constant temperature (21 – 25°C) and humidity (45 – 50%). Animals were allowed free access to food and water. All experimental procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center.
2.2. Controlled cortical impact injury and treatment paradigm
Mice were randomized and subjected to CCI injury using a Precision Systems and Instrumentation (Fairfax, VA) TBI-0310 Impactor (Boulet et al., 2013, Kelso et al., 2009, Kelso et al., 2006). Briefly, anesthesia was induced in a Plexiglass chamber with 5% inhaled isoflurane on an O2 carrier and was maintained at 2–2.5% via a nose cone during the procedure. The head was shaved and the animal was secured in a stereotactic frame (David Kopf, Tujunga, CA). After disinfecting the skin, the skull was exposed by a midline scalp incision. A left parietal craniotomy was performed midway between bregma and lambda and lateral to the midline using a high-speed dental drill so as to expose the somatosensory cortex. Care was taken not to disturb the underlying dura. The stereotaxic frame was positioned so that the exposed brain was placed directly under an electronically controlled, air-driven piston that delivered the impact. This compressed the cortical surface by 0.5 mm (3.0 mm tip diameter, 3.5 m/s velocity, 200 ms dwell time). After impact, Surgicel (Johnson & Johnson, Dallas, TX) was applied to the dura, the skullcap was replaced and affixed with dental adhesive and the incision closed with wound clips. Mice were then placed back into the home cage to recover from anesthesia. By six hours after injury, recovered animals were administered either GM-CSF (50 μg/kg; Peprotech, Rocky Hill, NJ) or an equal volume of vehicle (sterile phosphate buffered saline with 0.1% bovine serum albumin) by intraperitoneal (ip) injection as previously reported (Kosloski et al., 2013). Injections were administered daily until the time of sacrifice at either seven or fourteen days post-injury, except for the PCR array when animals were sacrificed at two, seven, or fourteen days post-injury. For sham treatment, animals’ were treated as CCI-injured animals, but received no CCI injury.
2.3. Flow cytometric analysis of peripheral lymphocyte populations
To determine T cell phenotypes, we harvested and prepared single cell suspensions from cervical lymph nodes and spleens of sham mice (mice underwent an identical surgical procedure except that an impact was not performed; n = 10), and animals treated with GM-CSF (n = 5 treated for 7 and 14 days), or vehicle (n = 5 for 7 and 14 days). Monoclonal antibodies (eBiosciences, San Diego, CA, USA) for CD4 [fluorescein isothiocyanate (FITC)], CD25 [phycoerythrin (PE)] and FoxP3 [allophycocyanin (APC)] were used to distinguish T cell subsets (Hori et al., 2003). Briefly, lymphoid organs were minced and suspended in Hanks’ Balanced Saline Solution (HBSS). Erythrocytes were lysed from the samples using an Ammonium-Chloride-Potassium (ACK) lysis solution (0.155 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). The erythrocyte-free samples were then double labeled with 1 μg antibody (PE-anti-CD25 and FITC-anti-CD4)/1 million cells for 30 minutes. One set of lymphocytes were permeabilized using a FoxP3 T regulatory cell staining kit (BioLegend, San Diego, CA) and incubated with APC-anti-FoxP3 for 30 minutes at 4°C. Stained cell suspensions were analyzed using a FACSCalibur flow cytometer (Becton-Dickinson ImmunoCytometry Systems, San Jose, CA). Data analysis was performed with FACSDiva software.
2.4. Tissue preparation and measurements of cortical tissue sparing
Animals were terminally-anesthetized with a lethal overdose of sodium pentobarbital (150 mg/kg body weight) by intraperitoneal injection. Anesthetized mice were transcardially perfused with 50 mL of chilled (4°C) 0.1M PBS, pH 7.4, followed by 75 mL of chilled 4% paraformaldehyde in 0.1 M PBS. The brains were rapidly removed and cryoprotected in 30% sucrose in PBS for an additional 24 hours. Brains were sectioned by the use of Hacker-Bright cryostat into 20 μm and 12 μm thick coronal slices. Every 6th 20 μm section was mounted onto a glass slide and stained with 0.1% cresyl violet. The slides were blinded and cresyl violet-stained cortical areas were measured with Image J software (NIH, Bethesda, MD). The area of ipsilateral cortical tissue was compared with the area of cortical tissue contralateral to the injury; results were expressed in percentage cortical tissue spared (Kelso et al., 2009, Kelso et al., 2006). The values from each group were then averaged and compared using Student’s t-test.
2.5. Immunohistochemistry, image acquisition and analyses
Four equally-spaced cryosections (12 μm) taken throughout the lesion area were sequentially treated with blocking solution (10% normal goat serum in Tris-buffered saline/Tween 20) for 1 h, incubated with polyclonal antibodies against anti-ionized calcium binding adaptor molecule 1 (Iba1, 1:1000; Wako, Richmond, VA), glial fibrillary acidic protein (GFAP, 1:1000; Dako, Carpenteria, CA), microtubule associated protein-2 (MAP-2, 1:500; Millipore, Billerica, MA), or with monoclonal antibodies against neurofilament (1:500; Dako) and were reacted with secondary anti-rat or anti-rabbit conjugated to fluorescent probes (Alexa Fluor 488 or Alexa Fluor 568). To ensure uniform staining for each cell type, tissues were reacted en masse with the same antibody dilution for the same period of time. Slides were cover slipped with ProLong Gold anti-fade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Thermo Fisher Scientific Corporation, Carlsbad, CA), allowed to dry for 24 h at room temperature then stored at −20°C for future analysis. Slides were evaluated by fluorescence microscopy (Leica DMRXA2, North Central Instruments, Plymouth, MN) and digital image analysis, whereby images of identical area were obtained around the cortical lesion penumbra and mirrored on the contralateral cortex were acquired under a 40x objective at wavelengths encompassing the emission spectra of the probes. Fluorescence was quantified by multispectral imaging/image analysis using a Nuance EX system (Cambridge Research Instruments, Woburn, MA) and ImagePro Plus image analysis software (Media Cybernetics, Rockville, MD) (Dash et al., 2011).
2.6. Real-time PCR analysis
To further investigate changes in inflammatory genes following induction of traumatic injury and subsequent treatment with GM-CSF, an Inflammatory Cytokines and Receptors PCR array (QIAGEN, Valencia, CA) was employed along with the First Strand cDNA synthesis kit and the SYBR green double-stranded DNA detection kit. The Inflammatory Cytokines and Receptors PCR array consists of 84 genes involved with the inflammatory response including chemokines (Ccl1, Ccl11, Ccl12, Ccl17, Ccl19, Ccl2, Ccl20, Ccl22, Ccl24, Ccl3, Ccl4, Ccl5, Ccl6, Ccl7, Ccl8, Ccl9, Cxcl1, Cxcl10, Cxcl11, Cxcl12, Cxcl13, Cxcl15, Cxcl5) and chemokine receptors (Ccr1, Ccr10, Ccr2, Ccr3, Ccr4, Ccr5, Ccr6, Ccr8, Cxcr2, Cxcr3, Cxcr5), interleukins (Il11, Il13, Il15, Il16, Il17a, Il17b, Il17f, Il1a, Il1b, Ilrn, Il21, Il27, Il3, Il33, Il4, Il5, Il7) and interleukin receptors (Il10ra, Il10rb, Ilr1, Il2rb, Il5ra, Il6ra, Il6st), and other cytokines (Aimp1, Bmp2, CD40lg, Csf1, Csf2, Csf3, Fasl, lfng, Lta, Ltb, Mif, Nampt, Osm, Pf4, Spp1, Tnf, Tnfsf10, Tnfsf11, Tnfsf13, Tnfsf13b, Tnfsf4, Vegfa) along with other cytokine receptors (Tnfrsf11b). Total RNA was extracted with a 3 mm biopsy punch from the cortical area adjacent to the lesion site from naïve (animals that did not undergo surgical manipulation; n = 4), vehicle- (n = 3 – 4 per time point) and GM-CSF-treated (n = 3 – 4 per time point) animals using a RNeasy isolation kit (Qiagen, Valencia, CA, USA) and cDNA synthesis was performed on 0.1 μg of total RNA using RT2 first strand kit (Qiagen). This PCR-based array is performed similar to real-time PCR, except that the primers are preloaded into the PCR plate wells. The PCR was performed on Eppendrof Mastercycler® ep realplex 2 and the data were analyzed using the RT2 Profiler PCR Array Data Analysis (SABiosciences, available online at http://www.sabiosciences.com).
2.7. Statistical analyses
All values are expressed as the mean ± standard error of the mean (SEM). Differences between the Vehicle and GM-CSF groups for the tissue sparing analysis, MAP2 immunohistochemistry and NF68 immunohistochemistry were analyzed by Student’s t-test. For the Iba1 and GFAP immunohistochemistry experiments where large intra-animal differences were observed as well as inter-animal differences, a two-way ANOVA was performed followed by a Bonferroni post-hoc test. Flow cytometric experiments were analyzed with a one-way ANOVA. All analyses were performed using GraphPad Prism 6 (La Jolla, CA).
3. Results
3.1. Neuroprotection and neuronal integrity
Herein, we investigated the effects of GM-CSF on macroscopic changes in the cortical regions by seven days post-injury. The work parallels prior pre-clinical studies that have shown that Il-10 expressing T-cells peak at seven days following experimental stroke (Liesz et al., 2013) and human pharmacodynamics studies have shown that GM-CSF induces a T cell response over prolonged periods of administration (Liljefors et al., 2008). Seven days post-injury, CCI produced cortical tissue loss at and around the impact site in all animals (Fig 1a). Notably, GM-CSF administration significantly attenuated TBI-induced cortical tissue loss, increased the amount of cortical tissue remaining from approximately 66% of the contralateral cortex of vehicle-treated animals to approximately 85% of the contralateral cortex of GM-CSF-treated animals (p < 0.001, Fig 1b).
Fig. 1. GM-CSF administration is neuroprotective seven days following CCI.

a) Representative photomicrographs of coronal brain sections stained with cresyl violet show a large lesion in the ipsilateral hemisphere that is reduced by GM-CSF administration. b) Lesion areas were quantified using Image J and demonstrated approximately 19% (p < 0.001) more cortical tissue (relative to the contralateral hemisphere) for GM-CSF treated animals (n = 7) compared to vehicle-treated animals (n = 6). c) Representative photomicrographs of NF68 immunoreactivity in the lesion area of vehicle and GM-CSF treated animals. d) GM-CSF treatment significantly increased NF68 staining by approximately 100% in the lesion area compared to vehicle treatment (p < 0.01). e) Representative photomicrographs of MAP2 immunoreactivity in the lesion area of vehicle and GM-CSF treated animals. f) There was no significant difference in MAP2 staining between vehicle and GM-CSF treated animals in the region of cortical damage.
To further characterize the neuroprotective effects of GM-CSF, we performed immunohistochemistry to determine expression of the axonal cytoskeleton protein neurofilament 68 kDa (NF68) and microtubule-associated protein 2 (MAP2), a cytoskeletal protein found in the dendrite and perikarya. We observed increased NF68 immunoreactivity in the lesion area of GM-CSF-treated animals compared to vehicle-treated animals seven days following injury (Fig 1c). Neurofilament (NF68) immunoreactivity from sections of vehicle-treated animals was approximately half of GM-CSF-treated animals (p < 0.01; Fig 1d). We did not observe a difference in MAP2 staining (Fig 1e, f). By 14 days following injury, we no longer observed a significant macroscopic tissue sparing effect (Supplemental Fig 1). There was more variability at this time point, mainly as the result of an outlier in the vehicle-treated group, which showed almost no lesion. We did not observe a significant difference in NF68 or MAP2 immunoreactivity at 14 days post-injury (data not shown).
3.2. GM-CSF alters the immune response following CCI
Microglia respond within minutes to damage to the brain (Davalos et al., 2005, Nimmerjahn et al., 2005). The number of activated microglia peak in the brain at seven days post-injury (Loane et al., 2014). Other studies have found the expression of M1 markers (classical activation) to peak around seven days post-injury and remain elevated until at least day 14 while M2 marker (alternative activation) peaked early and decreased to baseline levels by day 14 (Wang et al., 2013).
To examine whether GM-CSF’s neuroprotective effects are associated with changes in the glial microenvironment and immunological function, we first examined staining for the glial-specific markers Iba1 and GFAP in sections taken from GM-CSF- or vehicle-treated mice. Following CCI, a persistent and robust microglia/macrophage response at the lesion site and corresponding area on the contralateral cortex were detected, with the lesion site having significantly more Iba1 immunoreactivity compared to the corresponding contralateral site (Fig 2a; p < 0.005). GM-CSF administration reduced Iba1 immunoreactivity in both areas relative to vehicle-treated animals (Fig 2a; p < 0.0001). Upon closer examination, Iba1-labeled cells from the GM-CSF-treated animals displayed a different morphology than the vehicle-treated animals (Fig 2b). Sections from vehicle-treated animals (Fig 2b, top panel) had a higher frequency of Iba1+ cells with larger cell bodies and shorter processes, a characteristic of classically-activated microglia/macrophages (Loane et al., 2014). However, the occurrence of Iba1+ cells with ramified morphology, characterized by cells with small circular and centrally located soma surrounded radially by long and highly branched system of processes, was more pronounced in GM-CSF-treated mice (Fig 2b, bottom panel). At 14 days following CCI, Iba1 immunoreactivity was increased in both the ipsilateral and contralateral hemispheres (Supplemental Fig 2). As was observed at 7 days, GM-CSF administration by 14 days reduced Iba1 immunoreactivity in both hemispheres (p < 0.001), although the effect was not as pronounced compared to 7 days.
Fig. 2. GM-CSF treatment reduces Iba1+ staining in the lesion area seven days following CCI.
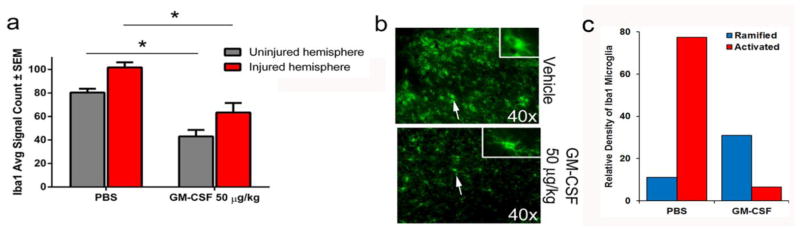
a) CCI induces increased Iba1+ staining in the contralateral and ipsilateral hemisphere that is reduced by GM-CSF administration (p < 0.0001). b) GM-CSF alters Iba1+ cell morphology. Cells from animals treated with vehicle (n = 6) have round bodies (top panel) and shortened processes whereas cells from GM-CSF treated animals (n = 7) have smaller cell bodies and elongated processes (bottom panel). The insets are images of Iba1 stained cells taken with a 63x objective to highlight these differences. DAPI staining was removed from images to improve clarity of Iba1 staining.
By day 7 post-injury, GFAP immunoreactivity in the lesion area of both vehicle- and GM-CSF-treated animals were increased compared to the respective uninjured contralateral hemispheres (Fig 3a), but GM-CSF administration did not affect expression compared to vehicle (Fig 3b). In contrast to day seven, GM-CSF administration for 14 days significantly (p < 0.05) reduced GFAP immunoreactivity in the lesion area compared to that of vehicle controls suggesting a reduction in astrocytosis (Supplemental Fig 3).
Fig. 3. GM-CSF treatment has no effect on GFAP staining in the lesion area seven days following CCI.
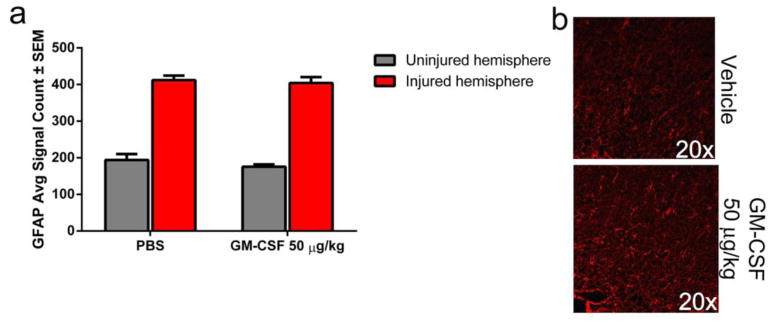
a) GFAP immunoreactivity is significantly increased in the lesion area compared to the contralateral hemisphere. However, there is no significant difference between vehicle and GM-CSF treatment. b) Representative photomicrographs of GFAP immunoreactivity in the lesion area of vehicle (n = 6) and GM-CSF treated (n = 7) animals. DAPI staining was removed from images to improve clarity of GFAP staining.
Cervical lymph nodes and spleens were harvested to determine Treg populations in these organs. Qualitatively, we observed that these organs were enlarged in GM-CSF treated animals compared to vehicle-treated animals or shams. We did not detect a significant difference in the percentages of CD4+ T cell populations in either the cervical lymph nodes or the spleen due to GM-CSF administration (Fig 4a), similar to a study recently published by our group (Kosloski et al., 2013). We observed a trend for increased Treg populations in the cervical lymph nodes of GM-CSF treated animals, but did not reach statistical significance (p = 0.06; Fig 4b). Splenic Treg populations were not significantly different when derived from sham-, vehicle-, or GM-CSF-treated groups.
Fig. 4. Peripheral immune response seven days after GM-CSF treatment.

a) There was no significant differences in frequency of CD4+ T cells in either the cervical lymph nodes or spleens of sham (n = 4), vehicle-treated (n = 5) or GM-CSF-treated (n = 5) animals. b) Treg cells (CD4+CD25+FoxP3+) showed increased numbers in cervical lymph nodes but did not reach statistical significance.
As with the seven day animals, we did not detect a significant difference in CD4+ cell populations in either the cervical lymph nodes or the spleen due to GM-CSF administration by day 14 post CCI (Supplemental Fig 4a). The trend that we observed for increased Treg population in the cervical lymph nodes at 7 days was no longer observed by 14 days (Supplemental Fig 4b). However, in splenocytes, there was a strong trend towards a decrease in Treg populations, though it did not reach statistical significance.
3.3. Evolution of inflammatory response following TBI
To elucidate the mechanism of GM-CSF mediated neuroprotective effects, we investigated expression changes of inflammatory genes adjacent to the lesion area that may be reflective of innate and adaptive immune modulation. Due to the inflammatory response that can be initiated by the surgical preparation required for the CCI (Cole et al., 2011, Lagraoui et al., 2012), comparisons between biologically significant (> 2-fold) mRNA expression changes were made for naïve versus injured animals and injured, vehicle-treated animals versus GM-CSF-treated animals following two, seven, or fourteen days post-injury (Supplemental Fig 5). Two days following injury, mRNA expression was altered for 59 genes (19 upregulated, 40 downregulated; Table 1) out of the 84 analyzed compared to naïve animals. Upregulated genes included tumor necrosis factor alpha (Tnf), GM-CSF administration significantly affected mRNA expression for chemokine ligand 2 (Ccl2), Il13, Il17b, and tumor necrosis factor (ligand) superfamily, member 10, (Tnfsf10) (Supplemental Fig 5, Day 2).
Table 1.
Regulation of inflammatory mediators two days following TBI.
| Upregulated | |||
|---|---|---|---|
| Gene | Gene Name | GO Molecular Function (Mi et al., 2013) | Fold |
| Aimp1 | Aminoacyl tRNA synthase complex-interacting multifunctional protein 1 | - | 3.4967 |
| Ccl12 | C-C motif chemokine 12 | Chemokine activity | 25.3867 |
| Ccl2 | C-C motif chemokine 2 | Chemokine activity | 21.2737 |
| Ccl3 | C-C motif chemokine 3 | Chemokine activity | 9.3082 |
| Ccl4 | C-C motif chemokine 4 | Chemokine activity | 11.4995 |
| Ccl6 | C-C motif chemokine 6 | Chemokine activity | 4.7027 |
| Ccl7 | C-C motif chemokine 7 | Chemokine activity | 7.7195 |
| Ccl8 | C-C motif chemokine 8 | Chemokine activity | 2.447 |
| Ccr1 | C-C chemokine receptor type 1 | G-protein coupled receptor activity | 2.8749 |
| Ccr3 | C-C chemokine receptor type 3 | G-protein coupled receptor activity | 2.4175 |
| Ccr5 | C-C chemokine receptor type 5 | G-protein coupled receptor activity | 2.3311 |
| Cxcl10 | C-X-C motif chemokine 10 | Chemokine activity | 8.96 |
| Il10rb | Interleukin 10 receptor 2 (precursor) | Receptor activity | 4.4568 |
| Il11 | Interleukin 11 | - | 5.3647 |
| Il1b | Interleukin 1 beta | Cytokine receptor binding | 3.1569 |
| Il1rn | Interleukin 1 receptor antagonist protein | Cytokine receptor binding | 2.7721 |
| Il2rg | Cytokine receptor common subunit gamma | Cytokine receptor activity | 2.4811 |
| Spp1 | Osteopontin | Cytokine activity | 38.6126 |
| Tnf | Tumor necrosis factor | Tumor necrosis factor receptor binding | 2.3432 |
| Downregulated | |||
| Gene | Gene Name | GO Classification | Fold |
| Ccl1 | C-C motif chemokine 1 | Chemokine activity | 0.1293 |
| Ccl17 | Protein Ccl17 | Chemokine activity | 0.3987 |
| Ccl20 | C-C motif chemokine 20 | Chemokine activity | 0.1282 |
| Ccl22 | C-C motif chemokine 22 | Chemokine activity | 0.4121 |
| Ccl24 | C-C motif chemokine 24 | Chemokine activity | 0.1293 |
| Ccr10 | C-C chemokine receptor type 10 | G-protein coupled receptor activity | 0.3987 |
| Ccr4 | C-C chemokine receptor type 4 | G-protein coupled receptor activity | 0.132 |
| Ccr6 | C-C chemokine receptor type 6 | G-protein coupled receptor activity | 0.1659 |
| Ccr8 | C-C chemokine receptor type 8 | G-protein coupled receptor activity | 0.1247 |
| Cd40lg | CD40 ligand | Tumor necrosis factor receptor binding | 0.1293 |
| Csf2 | Granulocyte-macrophage colony-stimulating factor | Cytokine activity | 0.1345 |
| Csf3 | Granulocyte colony-stimulating factor | Cytokine activity | 0.2677 |
| Cxcl1 | Growth-regulated alpha protein | Chemokine activity | 0.3172 |
| Cxcl11 | C-X-C motif chemokine 11 | Chemokine activity | 0.1352 |
| Cxcl12 | Stromal cell-derived factor 1 | -- | 0.447 |
| Cxcl13 | C-X-C motif chemokine 13 | Chemokine activity | 0.1482 |
| Cxcl15 | C-X-C motif chemokine 15 | Chemokine activity | 0.0534 |
| Cxcl9 | C-X-C motif chemokine 9 | Chemokine activity | 0.1293 |
| Cxcr3 | C-X-C chemokine receptor type 3 | G-protein coupled receptor activity | 0.1592 |
| Cxcr5 | C-X-C chemokine receptor type 5 | G-protein coupled receptor activity | 0.2608 |
| Fasl | Fas ligand | 0.1293 | |
| Ifng | Interferon gamma | Cytokine receptor binding | 0.1329 |
| Il13 | Interleukin-13 | Cytokine receptor binding | 0.4652 |
| Il15 | Interleukin-15 | Cytokine receptor binding | 0.4022 |
| Il17a | Interleukin-17A | Cytokine receptor binding | 0.1293 |
| Il17b | Interleukin-17B | -- | 0.475 |
| Il17f | Interleukin-17f | Cytokine receptor binding | 0.251 |
| Il21 | Interleukin-21 | -- | 0.1535 |
| Il27 | Interleukin-27 subunit alpha | -- | 0.2286 |
| Il2rb | Interleukin-2 receptor subunit beta | Receptor activity | 0.4288 |
| Il3 | Interleukin-3 | Interleukin-3 receptor binding | 0.1341 |
| Il5ra | Interleukin-5 receptor subunit alpha | Cytokine receptor activity | 0.1912 |
| Il7 | Interleukin-7 | -- | 0.3085 |
| Lta | Lymphotoxin-alpha | Tumor necrosis factor receptor binding | 0.1809 |
| Mif | Macrophage migration inhibitory factor | -- | 0.458 |
| Tnfsf10 | Tumor necrosis factor ligand superfamily member 10 | Tumor necrosis factor receptor binding | 0.3746 |
| Tnfsf11 | Tumor necrosis factor ligand superfamily member 11 | Tumor necrosis factor receptor binding | 0.4371 |
| Tnfsf13 | Tumor necrosis factor ligand superfamily member 13 | -- | 0.3313 |
| Tnfsf4 | Tumor necrosis factor ligand superfamily member 4 | Tumor necrosis factor receptor binding | 0.1293 |
| Vegfa | Vascular endothelial growth factor A | Growth factor activity | 0.2649 |
However, by seven days after injury, mRNA expression was also altered for 59 genes out of 84 (Table 2). Interestingly as compared to two days, 58 genes were upregulated (Chemokines Ccl1, Ccl11, Ccl12, Ccl17, Ccl2, Ccl20, Ccl22, Ccl24, Ccl3, Ccl4, Ccl5, Ccl6, Ccl7, Ccl8, Ccl9, Cxcl10, Cxcl11, Cxcl13, Cxcl5; chemokine receptors Ccr1, Ccr10, Ccr2, Ccr3, Ccr5, Ccr6, Cxcr3, Cxcr5; interleukins Il11, Il15, Il16, Il17a, Il17b, Il1a, Il1b, Il27, Il3, Il4, Il7; interleukin receptors Il10ra, Il10rb, Il2rb, Il2rg, Il5ra; cytokines Aimp1, Bmp2, Csf2, Csf3, Fasl, Ltb, Nampt, Pf4, Spp1, Tnf, Tnfsf10, Tnfsf11, Tnfsf13b, Tnfsf4, Vegfa; and cytokine receptor Tnfrsf11b) while Ccl19 was the sole downregulated gene. At this time point, there is a significant inflammatory response that is attenuated by GM-CSF treatment (Fig 5 and Supplemental Fig 5, Day 7). GM-CSF administration altered expression of 68 genes compared to vehicle-treated animals, with 66 being downregulated (Chemokines Ccl1, Ccl11, Ccl12, Ccl17, Ccl2, Ccl20, Ccl22, Ccl24, Ccl3, Ccl4, Ccl6, Ccl7, Ccl8, Ccl9, Cxcl1, Cxcl11, Cxcl13, Cxcl15, Cxcl5, Cxcl9; chemokine receptors Ccr1, Ccr10, Ccr2, Ccr3, Ccr4, Ccr5, Ccr6, Ccr8, Cxcr2, Cxcr3, Cxcr5; interleukins Il11, Il13, Il16, Il17a, Il17b, Il17f, Il1b, Il1rn, Il21, Il27, Il3, Il4, Il5, Il7; interleukin receptors Il10ra, Il2rb, Il2rg, Il5ra; cytokines Aimp1, Bmp2, Cd40lg, Csf2, Csf3, Fasl, lfng, Lta, Ltb, Nampt, Osm, Spp1, Tnf, Tnfsf11, Tnfsf13b, Tnfsf4, Vegfa), while only mRNA for chemokine (C-X3-C motif) ligand 1 (Cx3cl1) and IL-6 signal transducer (Il6st, Gp130) genes were upregulated.
Table 2.
Regulation of inflammatory mediators seven days following TBI.
| Upregulated | |||
|---|---|---|---|
| Gene | Gene Name | GO Molecular Function (Mi et al., 2013) | Fold |
| Aimp1 | Aminoacyl tRNA synthase complex-interacting multifunctional protein 1 | -- | 149.0859 |
| Bmp2 | Bone morphogenetic protein 2 | Growth factor activity | 132.7437 |
| Ccl1 | C-C motif chemokine 1 | Chemokine activity | 26.7846 |
| Ccl11 | C-C motif chemokine 11 | Chemokine activity | 10.8528 |
| Ccl12 | C-C motif chemokine 12 | Chemokine activity | 119.2903 |
| Ccl17 | C-C motif chemokine 17 | Chemokine activity | 9.4698 |
| Ccl2 | C-C motif chemokine 2 | Chemokine activity | 190.6787 |
| Ccl20 | C-C motif chemokine 20 | Chemokine activity | 5.1934 |
| Ccl22 | C-C motif chemokine 22 | Chemokine activity | 97.737 |
| Ccl24 | C-C motif chemokine 24 | Chemokine activity | 15.1369 |
| Ccl3 | C-C motif chemokine 3 | Chemokine activity | 286.3561 |
| Ccl4 | C-C motif chemokine 4 | Chemokine activity | 89.7288 |
| Ccl5 | C-C motif chemokine 5 | Chemokine activity | 2.8663 |
| Ccl6 | C-C motif chemokine 6 | Chemokine activity | 19.6075 |
| Ccl7 | C-C motif chemokine 7 | Chemokine activity | 9.6186 |
| Ccl8 | C-C motif chemokine 8 | Chemokine activity | 6.4382 |
| Ccl9 | C-C motif chemokine 9 | Chemokine activity | 8.9125 |
| Ccr1 | C-C chemokine receptor type 1 | G-protein coupled receptor activity | 20.6299 |
| Ccr10 | C-C chemokine receptor type 10 | G-protein coupled receptor activity | 2.8089 |
| Ccr2 | C-C chemokine receptor type 2 | G-protein coupled receptor activity | 13.4466 |
| Ccr3 | C-C chemokine receptor type 3 | G-protein coupled receptor activity | 8.4806 |
| Ccr5 | C-C chemokine receptor type 5 | G-protein coupled receptor activity | 19.5509 |
| Ccr6 | C-C chemokine receptor type 6 | G-protein coupled receptor activity | 40.5979 |
| Csf2 | Granulocyte-macrophage colony-stimulating factor | Cytokine activity | 10.459 |
| Csf3 | Granulocyte colony-stimulating factor | Cytokine activity | 3.9495 |
| Cxcl10 | C-X-C motif chemokine 10 | Chemokine activity | 26.3245 |
| Cxcl11 | C-X-C motif chemokine 11 | Chemokine activity | 9.2535 |
| Cxcl13 | C-X-C motif chemokine 13 | Chemokine activity | 13.6107 |
| Cxcl5 | C-X-C motif chemokine 5 | Chemokine activity | 11.2096 |
| Cxcr3 | C-X-C chemokine receptor type 3 | G-protein coupled receptor activity | 6.4234 |
| Cxcr5 | C-X-C chemokine receptor type 5 | G-protein coupled receptor activity | 2.5024 |
| Fasl | Tumor necrosis factor ligand superfamily member 6 | Tumor necrosis factor receptor binding cytokine activity | 2.7321 |
| Il10ra | Interleukin-10 receptor subunit alpha | Receptor activity | 3.5946 |
| Il10rb | Interleukin-10 receptor subunit beta | Receptor activity | 5.1515 |
| Il11neuro | Interleukin-11 | -- | 6.5206 |
| Il15 | Interleukin-15 | Cytokine receptor binding | 2.0861 |
| Il16 | Interleukin-16 | Cytokine receptor binding | 3.1583 |
| Il17a | Interleukin-17 alpha | Cytokine receptor binding | 4.6054 |
| Il17b | Interleukin-17 beta | Cytokine receptor binding | 10.226 |
| Il1a | Interleukin-1 alpha | Cytokine receptor binding | 3.1456 |
| Il1b | Interleukin-1 beta | Cytokine receptor binding | 2.5301 |
| Il27 | Interleukin-27 | -- | 4.357 |
| Il2rb | Interleukin-2 receptor subunit beta | Receptor activity | 2.2307 |
| Il2rg | Interleukin-2 receptor subunit gamma | Receptor activity | 10.4831 |
| Il3 | Interleukin-3 | Cytokine receptor binding Growth factor activity |
9.0631 |
| Il4 | Interleukin-4 | Cytokine receptor binding | 5.3147 |
| Il5ra | Interleukin-5 receptor subunit alpha | Cytokine receptor activity | 4.8793 |
| Il7 | Interleukin-7 | Cytokine receptor binding | 9.1103 |
| Ltb | Lymphotoxin-beta | Tumor necrosis factor binding Cytokine activity |
3.5166 |
| Nampt | Nicotinamide phosphoribosyltransferase | Cytokine activity | 2.9656 |
| Pf4 | Platelet factor-4 | Chemokine activity | 3.6893 |
| Spp1 | Osteopontin | Cytokine activity | 9.9578 |
| Tnf | Tumor Necrosis Factor | -- | 5.8395 |
| Tnfrsf11b | Tumor necrosis factor receptor superfamily member 11b | Tumor necrosis factor receptor activity Cytokine activity |
2.0058 |
| Tnfsf10 | Tumor necrosis factor ligand superfamily member 10 | Tumor necrosis factor receptor activity | 4.9876 |
| Tnfsf11 | Tumor necrosis factor ligand superfamily member 11 | Tumor necrosis factor receptor activity | 5.7624 |
| Tnfsf13b | Tumor necrosis factor ligand superfamily member 13b | -- | 13.1091 |
| Tnfsf4 | Tumor necrosis factor superfamily member 4 | Tumor necrosis factor receptor activity | 2.7959 |
| Downregulated | |||
| Gene | Gene Name | GO Molecular Function | Fold |
| Ccl19 | C-C motif chemokine 19 | Chemokine activity | −2.3161 |
Fig. 5. Network maps visualizing the functions predicted to be perturbed at seven-days after CCI.

Network maps were generated using IPA core analysis in order to visualize and predict altered biological functioning of immune cells seven days post-insult in naïve compared to CCI/vehicle-treated mice and in CCI mice treated with vehicle compared to GM-CSF. As apparent, a stark difference was observed for processes that included the top 10 biological functions predicted to be altered. These were used for generating the network maps and include lymphocyte migration, cell movement of leukocytes, leukocyte migration, cell movement of mononuclear leukocytes, inflammatory response, homing, homing of cells, chemotaxis, T cell migration, chemotaxis of cells. For each map, pink represents increased expression (> 2-fold), green represents decreased expression (> 2-fold), and grey indicates that the factor was measured but not biologically significant. In addition, orange represents predicted activation, blue represents predicted inhibition, grey indicates a relationship without substantial evidence for activation or inhibition, and yellow represents a conflicted prediction.
These results were corroborated using the Ingenuity Pathway Analysis (IPA) core analysis tool, which identified a differential prediction for the activation of a great number of immune processes. The top 10 immune processes (by p-value) that were predicted to be altered are cell movement of mononuclear leukocytes, chemotaxis, chemotaxis of cells, homing, homing of cells, inflammatory response, lymphocyte migration, leukocyte migration, cell movement of leukocytes, and T cell migration (Fig. 5).
By 14 days post-injury, there were still significant alterations in the inflammatory response as evidenced by 68 genes that were altered compared to naïve animals (Table 3, Supplemental Fig 5, Day 14). Of those 66 genes, 53 were downregulated (Ccl1, Ccl11, Ccl20, Ccl22, Ccl24, Ccl9, Cx3cl1, Cxcl1, Cxcl11, Cxcl15, Cxcl9, Ccr10, Ccr3, Ccr4, Ccr6, Ccr8, Cxcr2, Cxcr3, Cxcr5, Il11, Il13, Il16, Il17a, Il17b, Il17f, Il1rn, Il21, Il27, Il3, Il4, Il5, Il7, Il10ra, Il1r1, Il2rb, Il5ra, Il6ra, CD40lg, Csf1, Csf2, Csf3, Fasl, lfng, Lta, Ltb, Osm, Pf4, Tnf, Tnfsf11, Tnfsf13, Tnfsf13b, Tnfsf4, Vegfa) and 13 upregulated (Ccl12, Ccl2, Ccl4, Ccl5, Ccl6, Ccl7, Ccl8, Cxcl10, Cxcl13, Cxcl5, Il10rb, Mif, Spp1). GM-CSF treatment largely did not have an effect on the inflammatory profile compared to vehicle-treated animals at this time point with 15 genes being downregulated (Ccl12, Ccl20, Cxcl11, Cxcl13, Cxcl5, Cxcl9, Il13, Il17a, Il21, Il3, Il5, Csf3, Fasl, lfng, Tnfsf4) and 10 upregulated (Cx3cl1, Ccr3, Cxcr2, Il16, Il10ra Il2rb, Il2rg, Il6ra, Ltb, Tnfsf1).
Table 3.
Regulation of inflammatory mediators fourteen days following TBI.
| Upregulated | |||
|---|---|---|---|
| Gene | Gene Name | GO Molecular Function (Mi et al., 2013) | Fold |
| Aimp1 | Aminoacyl tRNA synthase complex-interacting multifunctional protein 1 | -- | 4.3289 |
| Ccl12 | C-C motif chemokine 12 | Chemokine activity | 18.8828 |
| Ccl2 | C-C motif chemokine 2 | Chemokine activity | 6.4935 |
| Ccl4 | C-C motif chemokine 4 | Chemokine activity | 2.1557 |
| Ccl5 | C-C motif chemokine 5 | Chemokine activity | 2.8151 |
| Ccl6 | C-C motif chemokine 6 | Chemokine activity | 5.148 |
| Ccl7 | C-C motif chemokine 7 | Chemokine activity | 3.0381 |
| Ccl8 | C-C motif chemokine 8 | Chemokine activity | 3.5983 |
| Cxcl10 | C-X-C motif chemokine 10 | Chemokine activity | 24.8011 |
| Cxcl13 | C-X-C motif chemokine 13 | Chemokine activity | 3.9014 |
| Cxcl5 | C-X-C motif chemokine 5 | Chemokine activity | 4.5652 |
| Il10rb | Interleukin-10 receptor subunit beta | Receptor activity | 2.8992 |
| Mif | Macrophage migration inhibitory factor | -- | 2.0512 |
| Spp1 | Osteopontin | Cytokine activity | 3.7751 |
| Downregulated | |||
| Gene | Gene Name | GO Molecular Function | Fold |
| Ccl1 | C-C motif chemokine 1 | Chemokine activity | −4.6139 |
| Ccl11 | C-C motif chemokine 11 | Chemokine activity | −2.0792 |
| Ccl20 | C-C motif chemokine 20 | Chemokine activity | −4.6892 |
| Ccl22 | C-C motif chemokine 22 | Chemokine activity | −2.5289 |
| Ccl24 | C-C motif chemokine 24 | Chemokine activity | −4.6139 |
| Ccl9 | C-C motif chemokine 9 | Chemokine activity | −4.1703 |
| Ccr10 | C-C chemokine receptor type 10 | G-protein coupled receptor activity | −6.1731 |
| Ccr3 | C-C chemokine receptor type 3 | G-protein coupled receptor activity | −2.0165 |
| Ccr4 | C-C chemokine receptor type 4 | G-protein coupled receptor activity | −4.6139 |
| Ccr6 | C-C chemokine receptor type 6 | G-protein coupled receptor activity | −3.5373 |
| Ccr8 | C-C chemokine receptor type 8 | G-protein coupled receptor activity | −3.3815 |
| Cd40lg | CD40 ligand | Tumor necrosis factor receptor binding | −4.4158 |
| Csf1 | Macrophage colony-stimulating factor | Cytokine activity | −4.3525 |
| Csf2 | Granulocyte-macrophage colony-stimulating factor | Cytokine activity | −4.4774 |
| Csf3 | Granulocyte colony-stimulating factor | Cytokine activity | −5.9012 |
| Cx3cl1 | Fractalkine | Chemokine activity | −3.8843 |
| Cxcl1 | C-X-C motif chemokine 1 | Chemokine activity | −3.8 |
| Cxcl11 | C-X-C motif chemokine 11 | Chemokine activity | −4.3853 |
| Cxcl15 | C-X-C motif chemokine 15 | Chemokine activity | −11.8023 |
| Cxcl9 | C-X-C motif chemokine 9 | Chemokine activity | −2.3016 |
| Cxcr2 | C-X-C chemokine receptor type 2 | G-protein coupled receptor activity | −4.0446 |
| Cxcr3 | C-X-C chemokine receptor type 3 | G-protein coupled receptor activity | −2.7818 |
| Cxcr5 | C-X-C chemokine receptor type 5 | G-protein coupled receptor activity | −4.6139 |
| Fasl | Tumor necrosis factor ligand superfamily member 6 | Tumor necrosis factor receptor binding cytokine activity | −4.3853 |
| Ifng | Interferon gamma | Cytokine receptor binding | −2.9353 |
| Il10ra | Interleukin-10 receptor subunit alpha | Receptor activity | −5.774 |
| Il11 | Interleukin-11 | -- | −3.9935 |
| Il13 | Interleukin-13 | Cytokine receptor binding | −4.5242 |
| Il16 | Interleukin-16 | Cytokine receptor binding | −6.132 |
| Il17a | Interleukin-17 alpha | Cytokine receptor binding | −3.4486 |
| Il17b | Interleukin-17 beta | -- | −2.1463 |
| Il17f | Interleukin-17f | Cytokine receptor binding | −4.7601 |
| Il1r1 | Interleukin-1 | Cytokine receptor binding | −2.9319 |
| Il1rn | Interleukin-1 | Cytokine receptor binding | −2.0553 |
| Il21 | Interleukin-21 | -- | −4.1106 |
| Il27 | Interleukin-27 | -- | −3.0652 |
| Il2rb | Interleukin-2 receptor subunit beta | Receptor activity | −5.9732 |
| Il3 | Interleukin-3 | Cytokine receptor binding | −4.6139 |
| Il4 | Interleukin-4 | Cytokine receptor binding | −7.8317 |
| Il5 | Interleukin-5 | Cytokine receptor binding | −5.206 |
| Il5ra | Interleukin-5 receptor subunit alpha | Cytokine receptor activity | −9.754 |
| Il6ra | Interleukin-6 receptor subunit beta | Cytokine receptor activity | −5.2452 |
| Il7 | Interleukin-7 | Cytokine receptor binding | −4.796 |
| Lta | Lymphotoxin-alpha | Tumor necrosis factor binding Cytokine activity |
−4.5557 |
| Ltb | Lymphotoxin-beta | Tumor necrosis factor binding Cytokine activity |
−2.4541 |
| Osm | Oncostatin-M | Cytokine receptor binding | −3.4886 |
| Pf4 | Platelet factor-4 | Chemokine activity | −2.3554 |
| Tnf | Tumor Necrosis Factor | -- | −3.5888 |
| Tnfrsf11b | Tumor necrosis factor ligand superfamily member 11b | Tumor necrosis factor receptor activity Cytokine activity |
−2.3378 |
| Tnfsf11 | Tumor necrosis factor ligand superfamily member 11 | Tumor necrosis factor receptor activity | −2.9472 |
| Tnfsf13 | Tumor necrosis factor ligand superfamily member 13 | Tumor necrosis factor receptor activity | −9.7766 |
| Tnfsf13b | Tumor necrosis factor ligand superfamily member 13b | -- | −2.0235 |
| Tnfsf4 | Tumor necrosis factor ligand superfamily member 4 | Tumor necrosis factor receptor activity | −4.6139 |
| Vegfa | Vascular endothelial growth factor A | Growth factor activity | −5.1224 |
4. Discussion
The results of the present study add to a growing body of literature that suggests GM-CSF has neuroprotective properties against models of neurological disease including PD (Kim et al., 2009, Kosloski et al., 2013, Mangano et al., 2011), stroke (Kong et al., 2009, Nakagawa et al., 2006, Schabitz et al., 2008), myasthenia gravis (Sheng et al., 2008, Sheng et al., 2011), and TBI (Nishihara et al., 2011, Shultz et al., 2014). Our work demonstrated that GM-CSF administration following experimental TBI produced a neuroprotective effect at seven days following injury while also reducing the microglia/macrophage burden. Administration of GM-CSF for seven days following TBI also attenuated many of the inflammatory processes occurring in the brain, possibly influencing the innate immune response as evidenced by a shift in microglia/macrophage morphology. By 14 days post-injury, many of these effects are not as pronounced. However, the results of the PCR array suggest that GM-CSF induces alterations in the innate immune response that require more in-depth investigation.
Although few studies have investigated the role of GM-CSF as a therapy for TBI, those that have been published support our current findings. To date, one study used GM-CSF administration in a model of induced TBI. Nishihara et al. administered 10 μg/kg of GM-CSF by subcutaneous injection to adult male Wistar rats beginning two days post-injury (stab model created by inserting a 26 gauge syringe needle into the brain with a stereotaxic device) and continuing injections daily for another seven days (Nishihara et al., 2011). When the animals were sacrificed two months following injury, there was not a significant difference in tissue loss between the saline-treated animals and the GM-CSF-treated animals. Unfortunately, this was the only outcome measure determined in this study. Recently, Shultz et al. performed a more thorough assessment of chronic outcomes for GM-CSF deficient mice (GM−/−) and wild-type (WT) mice following a lateral fluid percussion injury, a widely used and accepted model of TBI (Shultz et al., 2014). Three months after injury, GM−/− mice had behavioral alterations as evidenced by the Y-maze and elevated plus maze. These animals also had larger ventricle volumes on MRI scans and decreased GFAP immunoreactivity. However, neuronal cell counts were not significantly different than WT. These two studies support our findings where the immunomodulatory effects of GM-CSF appear to be chronic, though evidence of neuronal survival is not readily apparent. However, the impaired behavior of the GM−/− mice at three months after injury (Shultz et al., 2014) suggests long-term neuronal dysfunction which warrants additional study.
A significant finding of this study was the shift in microglia morphology from amoeboid-like in vehicle-treated animals to a ramified cell type in treated animals. This shift in morphology was associated with phenotype changes. Change from cytotoxic M1 to an anti-inflammatory M2 microglial state and under similar circumstances was reported (Loane et al., 2014). However, macrophage/microglia activation states are known to have multiple stages based on morphology marker expression, and cytokine secretion (Marshall et al., 2013, Raivich et al., 1999) and would require more in-depth investigation to fully investigate this possibility. It is also possible that different phenotypes may reside at different spatial locations in relation to the lesion.
It is also possible that GM-CSF-induced neuroprotection is mediated through the interaction with the GM-CSF receptor (GM-CSFR) in addition to its immunomodulatory actions. GM-CSFR is expressed in primary neuronal cultures and neuroblastoma cell lines and can prevent apoptosis resulting from camptothecin (Schabitz et al., 2008). Expression of GM-CSFR in vivo has been found on neurons throughout the human brain including regions and subregions of the cortex, basal ganglia, hippocampus, thalamus, hypothalamus, substantia nigra, cerebellum, and amygdala with expression being altered in many of these regions in Alzheimer’s disease (Ridwan et al., 2012). In the rat, these receptors have been found expressed in neurons of the somatosensory cortex, entorhinal cortex, CA2/CA3 region of the hippocampus, olfactory bulbs, lateral posterior thalamic nucleus, cerebellum, and mesencephalic tract (Schabitz et al., 2008). Co-localization of the receptor has also been observed in oligodendrocytes, but not astrocytes or microglia. GM-CSFR is upregulated in the rat cortex in the early post-ischemic period (< 2 days) before returning to control levels around 21 days. This finding also supports the use of GM-CSF in the early post-insult period.
Alternatively, GM-CSF is not the only factor that can influence Treg populations in the brain. Neurons and astrocytes can influence Treg populations through signaling of cytotoxic T lymphocyte antigen 4 (CTLA4) and transforming growth factor (TGF)-β (Liu et al., 2006, Trajkovic et al., 2004), suggesting a positive feedback loop for endogenous repair. Future studies will determine the extent to which Treg enter the brain following TBI.
Although we did not observe significant increase in percentages of the Treg population in spleen or cervical lymph nodes as a result of GM-CSF administration, it is still possible that this is the mechanism by which GM-CSF is eliciting a neuroprotective response. In a dose finding study that was recently published by our group, a 50 μg/kg dose of GM-CSF administered by ip injection for five days produced a significant increase in splenic Treg populations in normal C57Bl6/J mice (Kosloski et al., 2013). In this current analysis of Treg populations, we selected sham animals as a control instead of naïve and it is possible that the inflammatory response observed by others (Cole et al., 2011, Lagraoui et al., 2012) after craniotomy is more widespread than local changes observed in the area of the craniotomy.
Secondly, Jin et al. reported a differential regulation of T cell populations following CCI which has relevance with regards to our findings (Jin et al., 2012). In that study, T cells (defined as CD45+CD3+ by flow cytometric analyses) in the cervical lymph nodes were reduced at one day following injury and no different from baseline levels at three, seven, and 14 days before increasing by day 28. A similar pattern was observed in the spleen. In the brain, CD45+CD3+ T cells peaked around the third day after injury and decreased steadily thereafter the lowest post-TBI point at day 14. T cell numbers in the brain then increased at day 28. In the context of our study, it is possible that we missed the large shifts in T cell populations by performing our analyses at seven and 14 days post-injury.
Thirdly, the Ingenuity Pathway Analysis suggests that Treg populations and/or function are affected by drug treatment. It is not surprising that the majority of the gene products identified by Ingenuity Pathway Analysis were related to the inflammatory response, since genes related to that response predominate the chosen PCR kit. However, some of the gene products that were differentially regulated by GM-CSF are also known to have a role in microglia activation (CCL1 and CCL7), function of macrophages (CCR1, CCR6, and IL27) with some of these products overlapping with known mediators of encephalitis (CCR1, CCR6, and IL27 along with CCR4, TNFSF4, and CCR8). In conclusion, our study demonstrates the neuroprotective and immunomodulatory effects of GM-CSF when used as a treatment strategy for TBI. Future works will include behavioral analysis of treated animals, classification of microglia/macrophage activation states, and tests of neuronal integrity and function in the chronic post-traumatic period.
In conclusion, we demonstrated that in CCI injured mice, GM-CSF increased splenic regulatory T cell percentages coincident with cortical tissue sparing. This paralleled increased integrity of neurofilament staining. Lesion volumes were reduced by 15% in treated animal at 7 days after injury. Staining for ionized calcium binding adaptor molecule 1 (Iba1), a microglia marker, was observed at increased levels at or around the lesion on both 7 and 14 days. GM-CSF altered microglial morphology and reduced staining intensities by 25% of controls. Immunoreactivity for GFAP expression by astrocytes was not altered. Transcriptomics revealed a robust neuroprotective immune modulatory response 7 days after injury. Thus, in TBI wherein innate and adaptive immune systems respond to the injury facilitating early neural damage and subsequent time-dependent repair, these data demonstrated that such processes and injury-associated immune responses can be facilitated by modulating innate-, microglial-, and adaptive T cell-mediated immunity to induce a broad immune neuroprotective response.
Supplementary Material
Highlights.
GM-CSF treatment after CCI brain injury:
Induces immunomodulatory and neuroprotective responses,
Increases peripheral Treg frequencies,
Diminishes reactive microglial responses,
Increased integrity of the neuronal architecture, and
Diminishes lesion volumes while increasing cortical tissue sparing.
Acknowledgments
We thank Dr. Pawel Ciborowski, Department of Pharmacology and Experimental Neuroscience for support provided for the transcriptome analyses. Acquisition of flow cytometric data was performed by the UNMC Cell Analysis Facility. This work was supported by Carol Swarts Neuroscience Research Laboratory, the Frances and Louie Blumkin Foundation, Department of Defense Grant W81XWH11-1-0700, and National Institutes of Health grants (1P01DA028555, 2R01NS034239, 2R01NS36126, P01NS43985, and P01MH64570) to H.E.G. and a Centers of Biomedical Research Excellence grant (P20GM103480) to M.K. and H.E.G.
Footnotes
Authors’ Declaration. We declare the authors have no conflicts of interest. This manuscript contains original work and has not been concurrently published or submitted for publication elsewhere.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boulet T, Kelso ML, Othman SF. Long-term in vivo imaging of viscoelastic properties of the mouse brain after controlled cortical impact. J Neurotrauma. 2013;30:1512–20. doi: 10.1089/neu.2012.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsel PC, Forgey A, Grape FB, Hamann SS. Granulocyte macrophage colony-stimulating factor: current practice and novel approaches. Clin J Oncol Nurs. 2002;6:198–205. doi: 10.1188/02.CJON.198-205. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–48. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61:279–85. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- Clausen F, Hanell A, Bjork M, Hillered L, Mir AK, Gram H, et al. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30:385–96. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- Cole JT, Yarnell A, Kean WS, Gold E, Lewis B, Ren M, et al. Craniotomy: true sham for traumatic brain injury, or a sham of a sham? J Neurotrauma. 2011;28:359–69. doi: 10.1089/neu.2010.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Gorantla S, Gendelman HE, Knibbe J, Casale GP, Makarov E, et al. Loss of neuronal integrity during progressive HIV-1 infection of humanized mice. J Neurosci. 2011;31:3148–57. doi: 10.1523/JNEUROSCI.5473-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Appel SH. Neuroprotective activities of regulatory T cells. Trends Mol Med. 2011;17:687–8. doi: 10.1016/j.molmed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25:235–47. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One. 2012;7:e41892. doi: 10.1371/journal.pone.0041892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso ML, Scheff SW, Pauly JR, Loftin CD. Effects of genetic deficiency of cyclooxygenase-1 or cyclooxygenase-2 on functional and histological outcomes following traumatic brain injury in mice. BMC Neurosci. 2009;10:108. doi: 10.1186/1471-2202-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso ML, Wehner JM, Collins AC, Scheff SW, Pauly JR. The pathophysiology of traumatic brain injury in alpha7 nicotinic cholinergic receptor knockout mice. Brain Res. 2006;1083:204–10. doi: 10.1016/j.brainres.2006.01.127. [DOI] [PubMed] [Google Scholar]
- Kim NK, Choi BH, Huang X, Snyder BJ, Bukhari S, Kong TH, et al. Granulocyte-macrophage colony-stimulating factor promotes survival of dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced murine Parkinson’s disease model. Eur J Neurosci. 2009;29:891–900. doi: 10.1111/j.1460-9568.2009.06653.x. [DOI] [PubMed] [Google Scholar]
- Kong T, Choi JK, Park H, Choi BH, Snyder BJ, Bukhari S, et al. Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte-macrophage colony-stimulating factor in rats. Laboratory investigation. J Neurosurg. 2009;111:155–63. doi: 10.3171/2008.12.JNS08172. [DOI] [PubMed] [Google Scholar]
- Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol. 2013;265:1–10. doi: 10.1016/j.jneuroim.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kox M, Pompe JC, Pickkers P, Hoedemaekers CW, van Vugt AB, van der Hoeven JG. Increased vagal tone accounts for the observed immune paralysis in patients with traumatic brain injury. Neurology. 2008;70:480–5. doi: 10.1212/01.wnl.0000279479.69502.3e. [DOI] [PubMed] [Google Scholar]
- Lagraoui M, Latoche JR, Cartwright NG, Sukumar G, Dalgard CL, Schaefer BC. Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Front Neurol. 2012;3:155. doi: 10.3389/fneur.2012.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74:458–71. doi: 10.1002/ana.23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–9. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Na SY, Hammerling GJ, Garbi N, Karcher S, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013;33:17350–62. doi: 10.1523/JNEUROSCI.4901-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljefors M, Nilsson B, Mellstedt H, Frodin JE. Influence of varying doses of granulocyte-macrophage colony-stimulating factor on pharmacokinetics and antibody-dependent cellular cytotoxicity. Cancer Immunol Immunother. 2008;57:379–88. doi: 10.1007/s00262-007-0377-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–25. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangano EN, Peters S, Litteljohn D, So R, Bethune C, Bobyn J, et al. Granulocyte macrophage-colony stimulating factor protects against substantia nigra dopaminergic cell loss in an environmental toxin model of Parkinson’s disease. Neurobiol Dis. 2011;43:99–112. doi: 10.1016/j.nbd.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, Nixon K. Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiol Dis. 2013;54:239–51. doi: 10.1016/j.nbd.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Suga S, Kawase T, Toda M. Intracarotid injection of granulocyte-macrophage colony-stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res. 2006;1089:179–85. doi: 10.1016/j.brainres.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nishihara T, Ochi M, Sugimoto K, Takahashi H, Yano H, Kumon Y, et al. Subcutaneous injection containing IL-3 and GM-CSF ameliorates stab wound-induced brain injury in rats. Exp Neurol. 2011;229:507–16. doi: 10.1016/j.expneurol.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Marion DW, Botscheller ML, Swedlow PE, Styren SD, DeKosky ST. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J Neurochem. 1993;61:2015–24. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–83. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol. 2007;82:1083–94. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184:2261–71. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+CD25+ T cells. J Proteome Res. 2009;8:3497–511. doi: 10.1021/pr9001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridwan S, Bauer H, Frauenknecht K, von Pein H, Sommer CJ. Distribution of granulocyte-monocyte colony-stimulating factor and its receptor alpha-subunit in the adult human brain with specific reference to Alzheimer’s disease. J Neural Transm. 2012;119:1389–406. doi: 10.1007/s00702-012-0794-y. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Kruger C, Pitzer C, Weber D, Laage R, Gassler N, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF) J Cereb Blood Flow Metab. 2008;28:29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010;27:121–30. doi: 10.1089/neu.2009.1114. [DOI] [PubMed] [Google Scholar]
- Segev-Amzaleg N, Trudler D, Frenkel D. Preconditioning to mild oxidative stress mediates astroglial neuroprotection in an IL-10-dependent manner. Brain Behav Immun. 2013;30:176–85. doi: 10.1016/j.bbi.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Sheng JR, Li LC, Ganesh BB, Prabhakar BS, Meriggioli MN. Regulatory T cells induced by GM-CSF suppress ongoing experimental myasthenia gravis. Clin Immunol. 2008;128:172–80. doi: 10.1016/j.clim.2008.03.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JR, Muthusamy T, Prabhakar BS, Meriggioli MN. GM-CSF-induced regulatory T cells selectively inhibit anti-acetylcholine receptor-specific immune responses in experimental myasthenia gravis. J Neuroimmunol. 2011:240–241. 65–73. doi: 10.1016/j.jneuroim.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz SR, Tan XL, Wright DK, Liu SJ, Semple BD, Johnston L, et al. Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma. 2014;31:976–83. doi: 10.1089/neu.2013.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares HD, Hicks RR, Smith D, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15:8223–33. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic V, Vuckovic O, Stosic-Grujicic S, Miljkovic D, Popadic D, Markovic M, et al. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia. 2004;47:168–79. doi: 10.1002/glia.20046. [DOI] [PubMed] [Google Scholar]
- Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864–74. doi: 10.1038/jcbfm.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–42. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Adelson PD, Lifshitz J. Microglia: dismantling and rebuilding circuits after acute neurological injury. Metab Brain Dis. 2014 doi: 10.1007/s11011-014-9539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou T, Satake A, Ojha P, Kambayashi T. Cellular therapies supplement: the role of granulocyte macrophage colony-stimulating factor and dendritic cells in regulatory T-cell homeostasis and expansion. Transfusion (Paris) 2011;51(Suppl 4):160S–8S. doi: 10.1111/j.1537-2995.2011.03379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.