

Abstract
Understanding the molecular mechanisms behind formation of melanoma, the deadliest form of skin cancer, is crucial for improved diagnosis and treatment. One key is to better understand the cross-talk between epidermal keratinocytes and pigment-producing melanocytes. Here, using a bigenic mouse model system combining mutant oncogenic NRASQ61K (constitutively active RAS) or mutant activated CDK4R24C/R24C (prevents binding of CDK4 by kinase inhibitor p16INK4A) with an epidermis-specific knockout of the nuclear retinoid X receptor alpha (RXRαep−/−) results in increased melanoma formation after chronic ultraviolet-B (UVB) irradiation compared to control mice with functional RXRα. Melanomas from both groups of bigenic RXRαep−/− mice are larger in size with higher proliferative capacity, and exhibit enhanced angiogenic properties and increased expression of malignant melanoma markers. Analysis of tumor adjacent normal skin from these mice revealed altered expression of several biomarkers indicative of enhanced melanoma susceptibility, including reduced expression of tumor suppressor p53 and loss of PTEN, with concomitant increase in activated AKT. Loss of epidermal RXRα in combination with UVB significantly enhances invasion of melanocytic cells to draining lymph nodes in bigenic mice expressing oncogenic NRASQ61K compared to controls with functional RXRα. These results suggest a crucial role of keratinocytic RXRα to suppress formation of UVB-induced melanomas and their progression to malignant cancers in the context of driver mutations such as activated CDK4R24C/R24C or oncogenic NRASQ61K.
Introduction
Malignant melanoma is the deadliest form of skin cancer (American Cancer Society, 2013), and exposure to ultraviolet (UV) radiation is an important etiological risk factor [1]. Therefore, understanding the molecular mechanisms behind UV-induced melanoma formation is crucial for determining new pathways that can be manipulated for diagnosis and therapeutic targeting. Retinoid-X-Receptors (RXRs) α, β, and γ are members of the nuclear hormone receptor (NR) superfamily, and have a complex and dynamic role in regulation of cellular processes. RXRs function as a ubiquitous DNA-binding transcription factor via ligand binding [2,3] and promiscuous heterodimerization with other NRs [2,4]. By interacting with several transcriptional coactivators and/or corepressors, RXRs can regulate gene expression via multiple signaling pathways [2]. Previously we established that RXRα ablation in keratinocytes (cells comprising skin epidermis) alters paracrine signals to the melanocytes (pigment producing cells in the skin) and can enhance melanomagenesis [5,6]. Epidermis-specific Rxrα knockout in a mouse model is achieved using Cre-LoxP recombination, with Cre gene expression driven by the Keratin 14 (K14) promoter [7], which is expressed specifically in stratified squamous epithelium [8]. These Rxrαep−/− mice show increased melanocyte proliferation and defective DNA damage repair following acute ultraviolet-B (UVB) irradiation [5], and have increased melanocytic tumor formation resulting from chemical carcinogenesis [6,9]. Expression of several mitogenic factors are upregulated in keratinocytes lacking RXRα, including Endothelin-1 (EDN1), Stem Cell Factor (SCF), Microphthalmia-associated Transcription Factor (MITF), and Hepatocyte Growth Factor (HGF) [5,6,10], which stimulate melanocyte activation/proliferation in vitro [5,10]. We also found that combining epidermis-specific RXRα knockout with an activating Cyclin-Dependent Kinase 4 (CDK4) mutation (R24C) in a bigenic mouse model further enhanced chemical carcinogen-induced melanomagenesis, suggesting a cooperative effect between RXRα signaling and a key oncogenic driver [6]. Analyses of human melanocytic lesions collected at different stages of disease progression revealed a progressive loss of RXRα protein as tumors progress from benign nevi to in situ and metastatic melanomas [6].
In this study we aimed to investigate a mechanistic role for keratinocytic RXRα in UVB-induced melanomagenesis, which is a more biologically-relevant model than chemical-induced carcinogenesis. Rxrαep−/− mice were combined with either oncogenic Neuroblastoma RAS Viral Oncogene Homolog (NRASQ61K) or activating Cdk4R24C/R24C mutations to elucidate the role of RXRα loss in melanoma formation when combined with aberrant signaling pathways. The CDK4 pathway (p16-cyclin D-CDK4/6-retinoblastoma protein pathway) is reported to be dysregulated in 90% of human melanomas [11], while NRAS gene mutations are detected in 15–20% [12].
We observed that keratinocytic Rxrα ablation combined with either Cdk4R24C/R24C or NRASQ61K mutations resulted in increased number/size of UVB-induced melanocytic tumors compared to control Cdk4R24C/R24C or NRASQ61K mice with functional RXRα (RxrαL2/L2). Melanocytic tumors from both groups of bigenic Rxrαep−/− mice were more proliferative and showed increased labeling for malignant melanoma and tumor angiogenesis markers. The tumors also had altered expression of several genes implicated in mouse cancer compared to corresponding RxrαL2/L2 control samples, which corroborate several observed phenotypic changes. We have also observed increased invasion of melanocytic cells into draining lymph nodes bigenic Rxrαep−/− mice expressing oncogenic NRASQ61K, compared to controls with functional RXRα. Additionally, the tumor adjacent normal (TAN) skin from both groups of UVB-treated bigenic Rxrαep−/− mice showed dysregulated expression of several melanoma biomarkers. Particularly, we observed reduced Phosphatase and Tensin Homolog (PTEN) protein and concomitant increase in the phosphorylated, active form of its downstream effector Protein Kinase B (commonly called AKT), in addition to reduced expression of Tumor Suppressor p53. These results suggest that loss of keratinocytic RXRα both promotes and enhances formation of UVB-induced malignant melanomas in combinations with distinct driver mutations. Therefore epidermal RXRα expression may represent a new potential biomarker for diagnosis and treatment of UVB-induced melanomas in humans.
Materials and Methods
Mice
Generation of Rxrαep−/− [7], Cdk4R24C/R24C [13], and Tyr-NRASQ61K [14] mice have been described previously. See Supplementary Figure S1 for breeding/genotyping strategies for Rxrαep−/− | Cdk4R24C/R24C and Rxrαep−/− | Tyr-NRASQ61K bigenic mice. PCR primers used for genotyping are listed in Supplementary Table S2. Mice were housed in our approved University Animal Facility with 12-h light cycles, food/water were provided ad libitum, and institutional approval was granted for all experiments via an Animal Care and Use Protocol (ACUP).
UVR treatment of mice
Age- and sex-matched (female) P2 mice were exposed to a single dose of 800 mJ/cm2 of UVB light from a bank of four Philips FS-40 UV sunlamps [5], in order to stimulate a large outmigration of melanocytes from hair follicles into the extrafollicular epidermis and dermis [5] and promote melanomagenesis [15] prior to chronic treatment. Upon weaning (21 days postnatal), dorsal skin was shaved weekly and exposed to chronic doses of 320 mJ/cm2, 3X weekly for 30 weeks [16]. For negative controls, parallel groups of mice were shaved but not UVB-treated. At the conclusion of 30 weeks, melanocytic lesions were quantitated, and biopsies of lesions and tumor adjacent normal (TAN) skin were collected for analyses. This experiment was repeated independently twice with at least five mice per group.
Histological analyses
All analyses were performed on 5 μm formalin-fixed paraffin (FFPE) sections. Hematoxylin and eosin (H&E) staining was performed as previously described [9]. Fontana-Masson and combined eosinophil/mast cell stain (CEM) procedures were done using commercial kits (American MasterTech) according to manufacturer’s protocol. Prior to CEM staining, melanin pigment was bleached by treating slides with 10% H2O2 in 1X PBS for 25 minutes at 60°C. See Supplementary Methods for detailed protocols.
Immunohistochemistry
All chromogenic and fluorescent IHC studies were performed on 5 μm FFPE sections. All slides were treated with 10% H2O2 in 1X PBS as described above to remove melanin pigment. Antibodies used are detailed in Supplementary Table 1. Sections on the same slide labeled without primary antibody was used as negative controls (for examples see Supplementary Figure S3A), and all experiments were performed in triplicates. See Supplementary Methods for detailed protocols.
Imaging and quantitation of histological experiments
Brightfield images were captured with a Leica DME light microscope using the Leica Application Suite software, version 3.3.1. Fluorescent images were captured using a Zeiss AXIO Imager.Z1 with a digital AxioCam HRm and processed using AxioVision 4.8 and Adobe Photoshop. Quantifications of cell labeling were performed using ImageJ software (NIH), multiple random fields (at least 12) were imaged from several replicate mice in all groups and counted. The slides were analyzed independently in a double-blinded manner by two investigators and significance was determined using a Student’s two-tailed t-test as calculated by GraphPad Prism software.
Laser Capture Microdissection (LCM) and RNA isolation from melanoma tissue
Melanocytic lesions embedded in OCT (fresh, not fixed) were sectioned at 14 μm. Blocks were kept on dry ice prior to sectioning. One section was affixed per glass slide, and slides were immediately placed on dry ice, stored at −80°C long term, and kept on dry ice until immediately before LCM. Sections were dehydrated through sequential washes of cold alcohol followed by cold xylene. LCM was performed using a Zeiss PALM MicroBeam laser microdissection system; tissue was collected from five individual sections from each lesion and pooled. RNA was isolated using an Arcturus Pico Pure RNA Isolation Kit according to manufacturer’s protocol.
RT-qPCR analysis of gene expression in melanoma tissue isolated by LCM
Equal amounts of RNA from five distinct lesions from each group (each from a different animal) were pooled and converted to cDNA using a RT2 PreAMP cDNA Synthesis Kit (SA Biosciences/Qiagen) according to manufacturer’s protocol. cDNAs were applied to an SA Biosciences/Qiagen RT2 Profiler PCR Array (Mouse Cancer, PAMM-033Z). Triplicate arrays were run for each group, and data analyses performed using SA Biosciences web-based software.
Immunoblotting analyses
Immunoblotting analyses were performed according to standard protocols (described in [5,17]). After incubation with appropriate secondary antibody, signals were detected using immunochemiluminescent reagents (GE Healthcare, Piscataway, NJ). Equal protein loading in each lane was confirmed with a β-actin antibody (#A300-491, Bethyl). See Supplementary Methods for detailed protocol.
Results
Loss of keratinocytic Rxrα expression in cooperation with activated Cdk4 or oncogenic NRAS mutations results in increased UVB-induced melanoma formation
Our previous studies indicated that loss of keratinocytic RXRα protein has a role in melanocyte homeostasis and melanomagenesis, as observed in mouse models with epidermis-specific knockout of Rxrα (Rxrαep−/−) [5,6,9] and human melanoma samples [6]. Therefore we hypothesized that loss of keratinocytic Rxrα expression would result in enhanced malignant melanoma formation from chronic UVB-irradiation in bigenic mouse models, combined with homozygous activated Cdk4R24C/R24C or heterozygous melanocyte-specific expression of oncogenic human NRAS (Tyr-NRASQ61K).
To that end, Rxrαep−/− mice were bred to either Cdk4R24C/R24C or Tyr-NRASQ61K mice to generate Rxrαep−/− | Cdk4R24C/R24C and Rxrαep−/− | Tyr-NRASQ61K bigenic mice, respectively (see Materials and Methods, Supplementary Figure S1). RxrαL2/L2 | Cdk4R24C/R24C and RxrαL2/L2 | Tyr-NRASQ61K mice (floxed Rxrα mice containing LoxP sites flanking exon 4) were used as controls for wild-type Rxrα function. Here onwards, the homozygous Cdk4R24C/R24C mutation will be simply referred to as Cdk4R24C. Cohorts of neonatal (P2) mice from each group were irradiated with a single large dose of UVB, followed by 3X weekly doses of UVB (2X minimal erythemal dose) for 30 weeks and monitored periodically for formation of melanocytic tumors (Figure 1A) [16]. The initial UVB dose served to stimulate outmigration of melanocytes into the extrafollicular epidermis and dermis prior to chronic treatments [5]. Additionally, since HGF is upregulated in Rxrαep−/− | Cdk4R24C skin [6], we found it prudent to apply a large neonatal dose to promote melanomagenesis; it has been previously found that HGF/Scatter Factor (SF) overexpressing mice are prone to later melanoma development when receiving a large dose of UVB as neonates, but not as adults [15].
Figure 1. Macroscopic and histological characterization of melanocytic tumors from bigenic Rxrαep−/− mice combined with Tyr-NRASQ61K or Cdk4R24C mutations.

(A) Scheme for chronic UVB treatment of mice. (B, F) Tyr-NRASQ61K or (C, G) Cdk4R24C mice with epidermal-specific Rxrα ablation have more growths than mice with functional Rxrα; Rxrαep−/− | Cdk4R24C mice also have an increase in large melanocytic (>2 mm) lesions. Lesions indicated by arrows. (D, E) H&E staining of melanocytic lesions. (D) Tyr-NRASQ61K or (E) Cdk4R24C mice with epidermal-specific Rxrα ablation have more densely pigmented lesions with enhanced penetration into epidermal basal layer (inset). Scale bar = 50 μm. *** = p ≤ 0.001
Phenotypically, overall skin coloration of adult homozygous Cdk4R24C mice was distinct from those expressing oncogenic Tyr-NRASQ61K. The Tyr-NRASQ61K mice have heavily pigmented skin throughout (Figure 1B, Supplementary Figure S2E) compared to skin of Cdk4R24C mice (Figure 1C, Supplementary Figure S2E), making it difficult to quantitate smaller melanocytic lesions. Only larger (>2 mm) lesions in Rxrαep−/− | Tyr-NRASQ61K mice could be quantitated, as they were raised at that size due to the presence of a follicular cyst underneath. Dermal cysts are another phenotype of epidermal Rxrα ablation [7], resulting from degenerate hair follicles, and were also found within the lesions from Rxrαep−/− | Cdk4R24C mice.
Both bigenic Rxrαep−/− mouse lines developed higher numbers of melanocytic lesions, most of which were larger in size (>2 mm), compared to mice with functional Rxrα expression (Figure 1B, C, F, G). The Rxrαep−/− lesions were also more densely pigmented (Figure 1D, E) and showed enhanced penetration into the epidermal basal layer (Figure 1D, E (inset)). Histopathological analyses confirmed that the melanocytic lesions (>2 mm) from both bigenic mouse lines are melanomas with hallmarks of round and/or spindle cell tumors. Cohorts of RxrαL2/L2 and Rxrαep−/− mice without Cdk4R24C or Tyr-NRASQ61K mutations were treated identically with chronic UVB and did not develop any melanocytic lesions after end of treatment (Supplementary Figure S2D). Additionally, bigenic mice from each group were shaved weekly without any UVB treatment to monitor spontaneous melanoma formation and did not develop any lesions (Supplementary Figure S2E). Non-UVB treated bigenic Rxrαep−/− mice do develop small melanocytic nevi (<1 mm) that do not increase in size over the life of the mouse (Supplementary Figure S2E). Altogether, our results suggest that loss of epidermal Rxrα expression in combination with oncogenic NRAS or activated CDK4 enhances UVB-induced melanomagenesis.
Increased proliferation, malignant conversion, and enhanced angiogenesis in melanomas from mice lacking keratinocytic Rxrα expression
We employed immunohistochemical (IHC) analyses in order to characterize the melanomas formed after chronic UVB exposure. We aimed to elucidate the differences between melanomas from bigenic Rxrαep−/− | Tyr-NRASQ61K and Rxrαep−/− | Cdk4R24C mice and their corresponding RxrαL2/L2 controls. Fluorescent IHC was performed on sections from melanocytic lesions. To characterize immune cell infiltration, IHC was performed to label for macrophages (Supplementary Figure S4 A–D) and T-cells (Supplementary Figure S4 E–H). A significant increase in infiltration of Macrophage-1 antigen (MAC1)-positive macrophages was observed specifically in melanomas from Rxrαep−/− | Cdk4R24C mice compared to their control (Supplementary Figure S4 C, D). IHC for Cluster of Differentiation (CD4)-positive T-cells (Supplementary Figure S4 E–H) and combined eosinophil-mast cell (CEM) staining (Supplementary Figure S5) did not reveal any changes in either bigenic group. These results suggest specific cooperative effects between epidermal Rxrα loss and activated Cdk4 mutation to alter UVB-induced macrophage response.
Next we co-labeled the lesions for proliferation marker Proliferating Cell Nuclear Antigen (PCNA) and melanocyte-specific marker Tyrosinase-Related Protein 1 (TYRP1), an enzyme involved in melanin synthesis [18]. We observed a significantly higher percentage of PCNA/TYRP1 co-labeled cells in lesions from bigenic Rxrαep−/− mice compared to their controls (Figure 2A, B and insets). Rxrαep−/− | Cdk4R24C lesions also displayed significantly higher overall numbers of TYRP1+ melanocytic cells (Figure 2B). An antibody cocktail against malignant melanoma (HMB45 + MART1) [19] showed markedly higher staining in melanomas from bigenic Rxrαep−/− mice (Figure 2C, D). We then labeled for tumor angiogenesis marker Cluster of Differentiation 31 (CD31) [20]. Although melanomas from Rxrαep−/− | Tyr-NRASQ61K mice exhibited a similar presence of CD31+ cells compared to controls, there was a higher incidence of larger, more complex CD31+ vasculature (Figure 2E, right panel). Similarly, we observed more intense staining of CD31 in Rxrαep−/− | Cdk4R24C melanocytic lesions than in the control group (Figure 2F). Altogether, these results suggest that loss of Rxrα expression in the epidermis contributes to enhanced proliferation, malignant conversion, and angiogenesis in UVB-induced melanomas in the context of oncogenic signaling or dysregulated cell-cycle regulators.
Figure 2. Melanocytic tumors from bigenic Rxrαep−/− mice combined with Tyr-NRASQ61K or Cdk4R24C mutations have enhanced proliferative, angiogenic, and malignant properties.

(A, B) Fluorescent IHC for proliferation marker PCNA (red) and melanocyte marker TYRP1 (green). A trend of increased overall TYRP1+ cells and significant increases in PCNA+/TYRP1+ cells was observed in lesions from Rxrαep−/− mice compared to RxrαL2/L2 controls in combination with either Tyr-NRASQ61K (A) or Cdk4R24C (B) mutations. (C, D) IHC using antibody cocktail against malignant melanoma (red). Overall, more positive staining was observed in lesions from bigenic Rxrαep−/− mice compared to RxrαL2/L2 controls. (E, F) IHC for tumor angiogenesis marker CD31 (red). Overall, more prominent staining was observed in lesions from bigenic Rxrαep−/− mice compared to controls. Particularly in mice carrying the Tyr-NRASQ61K mutation, loss of epidermal Rxrα results in lesions with large multicellular CD31+ blood vessels (E, right panel). E=Epidermis, D= Dermis. Scale bars = 50 μm.
Chronic UVB-irradiation results in increased melanoma invasion to draining lymph nodes in Tyr-NRASQ61K mice lacking keratinocytic Rxrα expression
Next we wanted to determine the effects of keratinocytic Rxrα loss on metastasis of UVB-induced melanomas. Upon conclusion of the 30 week chronic UVB treatment period, draining inguinal lymph nodes (LNs) were excised from the mice. Fontana-Masson (FM) staining for melanin pigment showed increased number of pigment-containing cells in LNs from mice expressing Tyr-NRASQ61K compared to those expressing the activated Cdk4R24C gene (Figure 3A). Interestingly, no difference in overall pigmentation was observed in Rxrαep−/− LNs compared to their corresponding RxrαL2/L2 controls in either group of bigenic mice (Figure 3A). To specifically characterize the presence of melanocytic cells in the LNs, we performed chromogenic IHC for TYRP1 on LN sections from mutant and control mice. We observed a much higher presence of TYRP1+ positive cells in the Tyr-NRASQ61K group compared to the Cdk4R24C group (Figure 3B, C), and a marked increase in TYRP1+ cells in LNs from the Rxrαep−/−| Tyr-NRASQ61K mice compared to their RxrαL2/L2 controls (Figure 3B,C). Epidermal Rxrα ablation did not result in significant changes to LN invasion in the Cdk4R24C group. These results suggest that loss of epidermal Rxrα expression cooperates with oncogenic NRAS to form UVB-induced melanomas with enhanced risk of metastasis of melanocytic cells to the distal LNs.
Figure 3. Enhanced invasion of TYRP1+ pigment-producing cells is observed in draining lymph nodes of Tyr-NRASQ61K mice compared to Cdk4R24C mice, which is further increased by absence of keratinocytic RXRα.
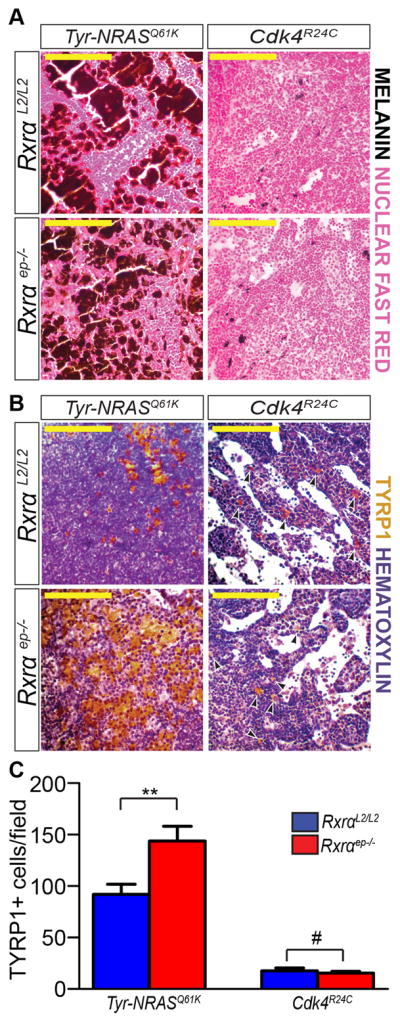
(A) General stain for melanin pigment (black staining) as determined by Fontana Masson assay. Nuclear Fast Red (pink) was used as a nuclear counterstain. More pigmentation is observed in LNs from the Tyr-NRASQ61K background as opposed to those expressing Cdk4R24C, but no difference in either background is observed between Rxrαep−/− and RxrαL2/L2 controls. (B, C) Chromogenic IHC for melanocyte-specific marker TYRP1 (brown). More positive staining overall is observed in Tyr-NRASQ61K LNs as opposed to the Cdk4R24C LNs, and a significant increase in melanocytic cells is observed in Tyr-NRASQ61K| Rxrαep−/− mice relative to its RxrαL2/L2 control. Black arrows indicate positive cells in Cdk4R24C group (B, right column). Hematoxylin (purple) was used as a nuclear counterstain. ** = p ≤ 0.01, # = no statistical significance. Scale bars = 100 μm.
Overlapping and distinct sets of genes related to cancer progression are dysregulated in melanomas from Rxrαep−/− | Tyr-NRASQ61K and Rxrαep−/− | Cdk4R24C bigenic mice
In order to determine gene signature changes that might cause the observed phenotypic differences between melanomas from bigenic Rxrαep−/− mice compared to RxrαL2/L2 controls, we utilized LCM to specifically isolate RNA from pigmented melanoma tissue (Figure 4A). Converted cDNA was then applied to an RT-qPCR array of genes implicated in mouse cancer progression (Qiagen/SA Biosciences PAMM-033Z) (Supplementary Figures S6, S7). Using a 1.7 fold cutoff, melanomas from both Rxrαep−/−|Tyr-NRASQ61K and Rxrαep−/− | Cdk4R24C mice exhibited similar upregulation of four gene transcripts relative to corresponding controls (Figure 4B). Three are involved in cell cycle regulation/proliferation: Marker of proliferation Mki67, Aurora Kinase A (Aurka, required for G2-M transition [21]) and Cell Division Cycle Protein 20 (Cdc20, activates anaphase promoting complex [22]). Additionally, both Rxrαep−/− | Tyr-NRASQ61K and Rxrαep−/−|Cdk4R24C melanomas had upregulated angiopoietin 1 (Angpt1) expression, which contributes to blood vessel formation/maturation [23]. Interestingly, a single gene was appreciably dysregulated specifically in the Rxrαep−/− | Tyr-NRASQ61K melanomas but not in the Rxrαep−/− | Cdk4R24C melanomas; Tankyrase-2 (Tnks2), associated with telomere maintenance processes [24], was upregulated (Figure 4B). Suppression of TNKS2 has also been previously associated with reduced colony formation, migration, and invasion of cervical cancer cells [25]. Additionally, a diverse subset of genes was dysregulated only in Rxrαep−/− | Cdk4R24C melanomas but not in the Rxrαep−/− | Tyr-NRASQ61K lesions (Figure 4B, C). They are involved in regulating key cellular processes including microtubule dynamics (Stathmin-1 (Stmn1) [26]), angiogenesis (Serpin Peptidase Inhibitor Clade F (Serpinf1) [27], Basic Fibroblast Growth Factor (Fgf2) [28], and Placental Growth Factor (Pgf) [29]), cell adhesion (Occludin (Ocln) [30]), apoptosis (Bcl-2-like Protein 11 (Bcl2l11) [31], Caspase-2 (Casp2) [32], and Fas Ligand (Fasl) [33]), chromatin remodeling (Alpha Thalassemia/Mental Retardation Syndrome X-Linked (Atrx) [34]), DNA Repair (X-ray repair cross-complementing protein 4 (Xrcc4) [35]), lipid synthesis/metabolism (Lipoprotein lipase (Lpl) [36]), growth factor signaling (Pgf [29]) and immune response (Chemokine (C-C Motif) Ligand 2 (Ccl2) [37], Fas ligand (Fasl) [33]), as well as ubiquitous transcription factors Goosecoid (Gsc) [38] and T-Box 2 (Tbx2) [39] (Figure 4B, C). Overall the above results suggest that epidermal ablation of Rxrα expression results in melanomas with altered gene signatures related to promoting proliferation and angiogenesis, and specific cooperation of epidermal Rxrα loss with activated CDK4 dysregulates a broad additional set of genes with diverse functions with regard to enhancing melanomagenesis.
Figure 4. Melanocytic tumors from bigenic Rxrαep−/− mice show both overlapping and distinct dysregulated expression of several genes implicated in melanoma progression.

(A) Laser Capture Microdissection (LCM) was performed to isolate RNA from melanoma tissue and analyze gene expression. (B) mRNA expression of several genes implicated in cancer were found to be dysregulated (>1.7 fold) in melanocytic lesions from Rxrαep−/− mice compared to RxrαL2/L2 controls, as determined by an RT-qPCR array (SA Biosciences PAMM-033Z). A subset of these genes were dysregulated in Rxrαep−/− mice from both the Tyr-NRASQ61K and Cdk4R24C groups, Tnks2 was dysregulated only in Rxrαep−/− | Tyr-NRASQ61K while several others were changed only in the Rxrαep−/− | Cdk4R24C group. RNA from five mice was pooled and three replicate RT-qPCR arrays were run per group. * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001 (C) Categories of gene functions represented by significantly dysregulated genes determined by RT-qPCR arrays shown in (B).
Dysregulated melanoma signaling in Tumor Adjacent Normal (TAN) skin from chronic UVB-irradiated mice lacking keratinocytic Rxrα expression
Thus far our data suggests that loss of keratinocytic RXRα has a role in enhancing progression of melanocytic tumors to malignant, metastatic lesions. As bigenic Rxrαep−/− mice formed a higher number of chronic UVB-induced melanocytic lesions relative to RxrαL2/L2 controls, we next aimed to determine if keratinocytic Rxrα loss also leads to phenotypic changes in the skin that enhance susceptibility to UVB-induced melanomas. To that end, we performed IHC and immunoblotting analyses on whole skin biopsies to analyze expression of known biomarkers of melanoma susceptibility in tumor adjacent normal (TAN) skin from chronic UVB-irradiated bigenic Rxrαep−/− mice and compared them to their controls. Morphologically, TAN skin from all groups was similar to non-UVB treated skin, except with dramatic thickening of the epidermal layer (Supplementary Figure S2 A,B,C) and increased pigmentation, particularly in the dermal layer (Supplementary Figure S2 A,B). Skin from non-UVB and UVB-treated Cdk4R24C mice was also prone to sebaceous gland hyperproliferation (Supplementary Figure S2 A,B).
We found that ablation of epidermal Rxrα expression resulted in reduced protein expression of tumor suppressor PTEN observed by both chromogenic IHC (Figure 5A–D) and immunoblotting (Figure 5E, F). As expected, we also observed a concomitant increase in Ser473 phosphorylation of its downstream effector AKT without change in total AKT levels (Figure 5G–L). In both bigenic Rxrαep−/− mouse lines we also observed reduced p53 expression in TAN skin, both by IHC (Figure 6A–D) and immunoblotting (Figure 6E–F). By Immunoblotting we observed upregulation of Cyclin D1 in TAN skin (Figure 6G,H); this protein has been found to be upregulated due to p53 inactivation [40]. Skin samples collected from non-UVB treated skin showed reduced staining intensity of PTEN, p-AKT, and p53; no discernable difference in expression pattern between RxrαL2/L2 and bigenic Rxrαep−/− mice was observed (Supplementary Figure S3).
Figure 5. Chronically UVB-irradiated tumor-adjacent normal (TAN) skin from bigenic Rxrαep−/− mice exhibit loss of PTEN and increase in phosphorylated AKT.

(A–F) Loss of PTEN expression in TAN skin of bigenic Rxrαep−/− compared to RxrαL2/L2 controls, as determined by IHC (A–D) and (E, F) immunoblotting. (G–L) Increase in phosphorylated AKT (Ser 473) in epidermis and dermis of bigenic Rxrαep−/− mice as determined by (G–J) IHC and (K, L) immunoblotting. Two biological replicates for each group are shown for all immunoblots. E=Epidermis, D= Dermis. Scale bars = 100 μm. # = no statistical significance, * = p ≤ 0.05, *** = p ≤ 0.001, **** = p ≤ 0.0001.
Figure 6. Chronically UVB-irradiated tumor-adjacent normal (TAN) skin from bigenic Rxrαep−/− mice exhibit loss of p53 and upregulation of Cyclin D1.

(A–F) Loss of p53 expression in TAN skin of bigenic Rxrαep−/− mice as compared to RxrαL2/L2 controls, as determined by IHC (A–D) and (E,F) immunoblotting. (G,H) Upregulation in Cyclin D1 in TAN skin of bigenic Rxrαep−/− mice, as shown by immunoblotting. Three biological replicates for each group are shown for all immunoblots. E=Epidermis, D= Dermis. Scale bars = 100 μm. # = no statistical significance, *** = p ≤ 0.001, **** = p ≤ 0.0001.
IHC revealed that expression of total CDK4 protein in TAN skin was modestly upregulated in both bigenic Rxrαep−/− mouse lines (Supplementary Figure S8 A–D). Expression of transcription factor E2F1, which has been associated with increased melanoma invasiveness [41], was upregulated only in Rxrαep−/−| Cdk4R24C TAN skin but not in that of Rxrαep−/− |Tyr-NRASQ61K mice (Supplementary Figure S8 G, H). Phosphorylated-ERK (p-ERK p44/42) was decreased in bigenic Rxrαep−/− TAN skin as determined by immunoblotting (Supplementary Figure S8 E, F). Increased p-ERK has been correlated with malignant melanoma progression [42], though reduced ERK activation has been reported in an epithelial subtype of melanomas [43]. Altogether, these results suggest that loss of keratinocytic Rxrα expression and chronic UVB exposure results in alteration of several key signaling pathways in the melanoma microenvironment that enhance melanoma susceptibility.
Discussion
We have previously established a role of keratinocytic RXRα in acute UV-induced melanocyte proliferation and in melanomagenesis induced by chemical carcinogenesis [5,6,9]. As exposure to UV is a major risk factor [1], we wanted to investigate mechanism(s) underlying loss of epidermal RXRα during UVB-induced melanomagenesis. Here we used Rxrαep−/− mice combined with oncogenic mutant NRAS (Q61K) or activated Cdk4 (R24C) to elucidate the role of keratinocytic RXRα protein in promoting chronic UVB-induced melanoma formation and determine cooperativity with signaling pathways linked to melanoma driver mutations.
As hypothesized, Rxrαep−/− mice developed more and larger UVB-induced melanocytic tumors in combination with either Tyr-NRASQ61K or Cdk4R24C mutations compared to their corresponding RxrαL2/L2 controls. Interestingly, we observed increased penetration of pigmented cells into the epidermal basal layer, possibly due to increased secretion of several mitogenic paracrine factors from the keratinocytes as we have reported previously [5,6]. Mice with epidermal Rxrα knockout alone did not develop UVB-induced melanomas, nor did age-matched bigenic mice that were not treated with UVB. Altogether, these results suggest that keratinocytic Rxrα loss is an etiologic factor for enhanced UVB-induced melanomagenesis in the context of a melanoma-susceptible background in the form of NRASQ61K or Cdk4R24C mutations.
Our LCM gene expression studies corroborate what we observed via immunohistochemistry. mRNA transcripts for several coordinators of cell cycle/proliferation were found to be upregulated in melanocytic tumors from both Rxrαep−/− bigenic mouse lines. We also found enhanced angiogenic properties in tumors from the bigenic Rxrαep−/− mice. Angiogenesis regulator Angpt1 was upregulated in tumors from both Rxrαep−/− bigenic mouse lines. Three additional genes (Serpinf1, Fgf, Pgf) with reported roles in angiogenesis were also altered specifically in Rxrαep−/− | Cdk4R24C tumors. We observed a higher degree of malignancy in melanocytic tumors from both bigenic Rxrαep−/− mouse lines, as well as increased invasion of TYRP1-expressing melanoma cells to draining lymph nodes in Rxrαep−/− | Tyr-NRASQ61K mice. Similar observations of enhanced proliferative, angiogenic, and malignant/metastatic properties were previously made in lesions from DMBA-TPA treated Rxrαep−/− | Cdk4R24C mice [6]. All of these results underscore an important role for RXRα in mediating melanocyte proliferation, homeostasis, and angiogenesis and suggest that loss of keratinocytic RXRα contributes to progression of UVB-induced melanocytic lesions to malignant and invasive tumors. That is further supported by our earlier observations in human melanomas, which demonstrated progressive loss of RXRα protein levels in adjacent epidermis as lesions progressed from benign nevi to metastatic melanomas [6].
Besides several overlapping observations in Rxrαep−/− | Tyr-NRASQ61K and Rxrαep−/− | Cdk4R24C melanocytic tumors, we observed changes specific to one group or the other, suggesting that in addition to a general role of keratinocytic RXRα in mediating melanomagenesis, specific cooperative effects result from combined loss of epidermal RXRα with activated CDK4 or oncogenic NRAS. A single gene, Tnks2, was upregulated specifically in in Rxrαep−/− | Tyr-NRASQ61K melanomas. Tnks2 is upregulated by microRNA miR20a [25], and TNKS2 suppression has been associated with reduced colony formation, migration, and invasion of cervical cancer cells [25]. It’s possible that upregulation specifically in the Rxrαep−/− | Tyr-NRASQ61K tumors contributes to the enhanced infiltration of melanocytic cells to LNs observed in this model. The role of Tnks2, and potentially miR20a, need to be investigated further going forward. Additionally, we found a diverse set of genes implicated in cancer formation dysregulated only in Rxrαep−/− | Cdk4R24C melanocytic lesions and not in the Tyr-NRASQ61K group. Those genes are involved in regulating a multitude of key cellular processes, including ubiquitous transcription factor function, which themselves may control a wide array of downstream processes affecting melanoma formation/progression. CDK4 initiates events leading to accumulation of transcription factor E2F1 and promotes entry into S-phase of the cell cycle [44]. E2F1 has been associated with increased melanoma invasion [41] and used as a biomarker toward sensitivity to MDM2 inhibitors [45]. We observed modest upregulations of total CDK4 protein in Rxrαep−/− TAN skin from both of our bigenic mouse models compared to RxrαL2/L2 controls, suggesting that loss of keratinocytic RXRα contributes to enhanced CDK4 expression. As the Rxrαep−/−| Cdk4R24C mice are homozygous for the mutant Cdk4R24C allele, any upregulated CDK4 expression in these animals is exclusively the mutant, activated CDK4R24C protein, resulting in increased expression levels of activated CDK4 (Cdk4R24C) above that of RxrαL2/L2| Cdk4R24C control mice, thereby further enhancing alterations of other downstream targets. Selective enhancement of E2F1 expression in Rxrαep−/− | Cdk4R24C TAN skin, and not in mice expressing oncogenic NRAS, corroborates that notion. Our observation of increased MAC1+ macrophage infiltration exclusively in Rxrαep−/− | Cdk4R24C lesions is also supported by the LCM gene expression studies, where expression of the macrophage attractant Ccl2 was upregulated in Rxrαep−/− | Cdk4R24C lesions relative to RxrαL2/L2 control, and this was not observed in melanomas from Rxrαep−/− | Tyr-NRASQ61K mice. CCL2 is a ligand of receptor CCR2, and melanocytes have been reported to secrete CCR2 ligands to attract CCR2+ macrophages into the skin following UVB irradiation [37]. These macrophages secrete interferon-y which promotes post-UV survival of melanocytes [37]. Therefore, specific cooperative effects may result from combined loss of epidermal RXRα and activated CDK4, contributing to enhanced melanoma progression. The increased immune responses and enhanced macrophage infiltration in the Rxrαep−/− | Cdk4R24C mice may also prevent and/or eliminate the highly proliferative and aggressive melanoma cells from invading and infiltrating to the adjacent lymph nodes. Additional studies are necessary to elucidate the specific cooperative effects of combined epidermal RXRα loss and activated CDK4 in melanomagenesis.
Thus far our results suggest that loss of keratinocytic RXRα can enhance progression of UVB-induced melanocytic lesions to malignant, metastatic tumors in the context of activating Cdk4R24C or oncogenic Tyr-NRASQ61K mutations. We observed reduced expression of tumor suppressor p53 and an upregulation of Cyclin D1 in bigenic Rxrαep−/− TAN skin. p53 has been reported to cooperate with both NRAS [40,46] or CDK4 [47,48] mutations to promote melanoma formation. Enhanced expression of Cyclin D1 has been correlated with progression of primary melanoma [42,49] and is upregulated due to p53 inactivation [40].
Similarly, functional loss of PTEN expression occurs in 30–60% of melanomas [50]. PTEN expression inactivation can occur through deletion/mutation [50], or alternatively via aberrant epigenetic silencing [50]. This makes it difficult to accurately estimate the impact of PTEN inactivation as no detectable mutation in the gene itself may be present [50]. PTEN functions as a tumor suppressor by degrading second messenger lipids produced by Phosphoinositide 3-kinase (PI3K). Without functional PTEN these signals result in excessive AKT activation, suppress apoptosis, and promote tumorigenesis [50]. PTEN normally functions in homeostatic balance with AKT, and when functionally suppressed results in excessive AKT phosphorylation [50]. Our observed loss of PTEN and concomitant increase in levels of activated, phosphorylated AKT protein in Rxrαep−/− TAN skin, further corroborates those previous reports. Since the above results are obtained from non-tumorigenic skin, they strongly suggest that loss of epidermal RXRα in combination with chronic UVB exposure creates a microenvironment in the skin that is susceptible to melanoma formation in the context of multiple signaling pathways mediated by driver mutations such as oncogenic NRAS or activating CDK4 mutations.
Overall, our results suggest that loss of RXRα in the epidermis alters the skin microenvironment in a manner that both promotes formation and enhances progression of malignant and metastatic UVB-induced melanomas when combined with upregulated oncogenic NRAS or activated CDK4 signaling pathways. Further studies will be necessary to establish RXRα as a clinical diagnostic marker and a therapeutic target for preventing melanoma progression and metastasis in humans.
Supplementary Material
Figure 7. Cooperativity between loss of epidermal RXRα expression and activated CDK4 or oncogenic NRAS during UVB induced melanoma progression in bigenic mice.

Schematic representation of the results of epidermal RXRα ablation in combination with Cdk4R24C or Tyr-NRASQ61K oncogenic mutations and chronic UVB exposure. TAN = “Tumor Adjacent Normal.” Similarities/differences between the two bigenic Rxrαep−/− mouse lines are represented by a Venn diagram. Relative to control Cdk4R24C or Tyr-NRASQ61K mice with functional Rxrα expression, melanomas from bigenic Rxrαep−/− mice have increased proliferation, malignant conversion, and tumor angiogenesis. The TAN skin from the bigenic Rxrαep−/− mice show altered expression of several known melanoma biomarkers relative to RxrαL2/L2 controls, including reduced expression of PTEN/concomitant increase in phosphorylated AKT, reduced p53 expression, and upregulation of Cyclin D1. On their own, Rxrαep−/− | Tyr-NRASQ61K mice had heavily pigmented skin overall and higher numbers of melanocytic cells invading draining LNs compared to the Cdk4R24C group, which is enhanced further by keratinocytic Rxrα loss. Increased macrophage infiltration and a distinct, increased E2F1 expression, and a diverse subset of dysregulated genes were observed specifically in Rxrαep−/− | Cdk4R24C lesions.
Implications.
These findings suggest that RXRα may serve as a clinical diagnostic marker and therapeutic target in melanoma progression and metastasis.
Acknowledgments
We thank all Indra lab members and the OSU College of Pharmacy, specifically Dr. Mark Zabriskie and Dr. Gary Delander for their continuous support and encouragement. We are grateful for services provided by the Cell and Tissue Analysis Facilities, specifically C. Samuel Bradford for help with LCM, and Services Core of the Environmental Health Sciences Center (EHSC) at OSU, grant number P30 ES00210, National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH). Research reported in this publication was supported by the NIEHS of the NIH under award numbers ES016629-01A1 as well as the National Cancer Institute (NCI) of the NIH under award number T32CA106195.
Footnotes
Conflict of Interest (COI): No financial relationships/conditions/circumstances that present a potential conflict of interest exist.
References
- 1.Markovic SN, Erickson LA, Rao RD, McWilliams RR, Kottschade LA, et al. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Elsevier; 2007. pp. 364–380. [DOI] [PubMed] [Google Scholar]
- 2.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 3.Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, et al. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 4.Leid M, Kastner P, Chambon P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Coleman DJ, Bajaj G, Liang X, Ganguli-Indra G, et al. RXRalpha ablation in epidermal keratinocytes enhances UVR-induced DNA damage, apoptosis, and proliferation of keratinocytes and melanocytes. J Invest Dermatol. 2011;131:177–187. doi: 10.1038/jid.2010.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyter S, Bajaj G, Liang X, Barbacid M, Ganguli-Indra G, et al. Loss of nuclear receptor RXRalpha in epidermal keratinocytes promotes the formation of Cdk4-activated invasive melanomas. Pigment Cell Melanoma Res. 2010;23:635–648. doi: 10.1111/j.1755-148X.2010.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li M, Chiba H, Warot X, Messaddeq N, Gerard C, et al. RXR-alpha ablation in skin keratinocytes results in alopecia and epidermal alterations. Development. 2001;128:675–688. doi: 10.1242/dev.128.5.675. [DOI] [PubMed] [Google Scholar]
- 8.Nelson WG, Sun T-T. The 50-and 58-kdalton keratin classes as molecular markers for stratified squamous epithelia: cell culture studies. J Cell Biol. 1983;97:244–251. doi: 10.1083/jcb.97.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Indra AK, Castaneda E, Antal MC, Jiang M, Messaddeq N, et al. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidermal keratinocytes. J Invest Dermatol. 2007;127:1250–1260. doi: 10.1038/sj.jid.5700672. [DOI] [PubMed] [Google Scholar]
- 10.Hyter S, Coleman DJ, Ganguli-Indra G, Merrill GF, Ma S, et al. Endothelin-1 is a transcriptional target of p53 in epidermal keratinocytes and regulates ultraviolet-induced melanocyte homeostasis. Pigment Cell Melanoma Res. 2013;26:247–258. doi: 10.1111/pcmr.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19:5320–5328. doi: 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- 12.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–1510. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 14.Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, et al. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005;65:4005–4011. doi: 10.1158/0008-5472.CAN-04-2970. [DOI] [PubMed] [Google Scholar]
- 15.Noonan FP, Recio JA, Takayama H, Duray P, Anver MR, et al. Neonatal sunburn and melanoma in mice. Nature. 2001;413:271–272. doi: 10.1038/35095108. [DOI] [PubMed] [Google Scholar]
- 16.Newkirk KM, Parent AE, Fossey SL, Choi C, Chandler HL, et al. Snai2 expression enhances ultraviolet radiation-induced skin carcinogenesis. Am J Pathol. 2007;171:1629–1639. doi: 10.2353/ajpath.2007.070221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang X, Bhattacharya S, Bajaj G, Guha G, Wang Z, et al. Delayed cutaneous wound healing and aberrant expression of hair follicle stem cell markers in mice selectively lacking Ctip2 in epidermis. PLoS One. 2012;7:e29999. doi: 10.1371/journal.pone.0029999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi T, Urabe K, Winder A, Jimenez-Cervantes C, Imokawa G, et al. Tyrosinase related protein 1 (TRP1) functions as a DHICA oxidase in melanin biosynthesis. EMBO J. 1994;13:5818–5825. doi: 10.1002/j.1460-2075.1994.tb06925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki F, Okamoto H, Matsumura Y, Tanaka K, Kunisada T, et al. Development of a new mouse model (xeroderma pigmentosum a-deficient, stem cell factor-transgenic) of ultraviolet B-induced melanoma. J Invest Dermatol. 2005;125:521–525. doi: 10.1111/j.0022-202X.2005.23753.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang D, Stockard CR, Harkins L, Lott P, Salih C, et al. Immunohistochemistry in the evaluation of neovascularization in tumor xenografts. Biotech Histochem. 2008;83:179–189. doi: 10.1080/10520290802451085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci. 2004;117:2523–2531. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- 22.Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, et al. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 24.Sbodio JI, Lodish HF, Chi NW. Tankyrase-2 oligomerizes with tankyrase-1 and binds to both TRF1 (telomere-repeat-binding factor 1) and IRAP (insulin-responsive aminopeptidase) Biochem J. 2002;361:451–459. doi: 10.1042/0264-6021:3610451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang H-W, Wang F, Wei Q, Zhao Y-F, Liu M, et al. miR-20a promotes migration and invasion by regulating TNKS2 in human cervical cancer cells. FEBS Lett. 2012;586:897–904. doi: 10.1016/j.febslet.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Karst AM, Levanon K, Duraisamy S, Liu JF, Hirsch MS, et al. Stathmin 1, a marker of PI3K pathway activation and regulator of microtubule dynamics, is expressed in early pelvic serous carcinomas. Gynecol Oncol. 2011;123:5–12. doi: 10.1016/j.ygyno.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren JG, Jie C, Talbot C. How PEDF prevents angiogenesis: a hypothesized pathway. Med Hypotheses. 2005;64:74–78. doi: 10.1016/j.mehy.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–583. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 30.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 31.Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–1679. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 33.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 34.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A. 2003;100:10635–10640. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 36.Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. doi: 10.1194/jlr.r200015-jlr200. [DOI] [PubMed] [Google Scholar]
- 37.Zaidi MR, Davis S, Noonan FP, Graff-Cherry C, Hawley TS, et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469:548–553. doi: 10.1038/nature09666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conway SJ. Novel expression of the goosecoid transcription factor in the embryonic mouse heart. Mech Dev. 1999;81:187–191. doi: 10.1016/s0925-4773(98)00235-4. [DOI] [PubMed] [Google Scholar]
- 39.Christoffels VM, Hoogaars WM, Tessari A, Clout DE, Moorman AF, et al. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev Dyn. 2004;229:763–770. doi: 10.1002/dvdy.10487. [DOI] [PubMed] [Google Scholar]
- 40.Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, et al. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001;21:2144–2153. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alla V, Engelmann D, Niemetz A, Pahnke J, Schmidt A, et al. E2F1 in melanoma progression and metastasis. J Natl Cancer Inst. 2010;102:127–133. doi: 10.1093/jnci/djp458. [DOI] [PubMed] [Google Scholar]
- 42.Oba J, Nakahara T, Abe T, Hagihara A, Moroi Y, et al. Expression of c-Kit, p-ERK and cyclin D1 in malignant melanoma: an immunohistochemical study and analysis of prognostic value. J Dermatol Sci. 2011;62:116–123. doi: 10.1016/j.jdermsci.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–1512. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- 44.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 45.Verhaegen M, Checinska A, Riblett MB, Wang S, Soengas MS. E2F1-dependent oncogenic addiction of melanoma cells to MDM2. Oncogene. 2012;31:828–841. doi: 10.1038/onc.2011.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dovey M, White RM, Zon LI. Oncogenic NRAS cooperates with p53 loss to generate melanoma in zebrafish. Zebrafish. 2009;6:397–404. doi: 10.1089/zeb.2009.0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muthusamy V, Hobbs C, Nogueira C, Cordon-Cardo C, McKee PH, et al. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer. 2006;45:447–454. doi: 10.1002/gcc.20310. [DOI] [PubMed] [Google Scholar]
- 48.Yang G, Rajadurai A, Tsao H. Recurrent patterns of dual RB and p53 pathway inactivation in melanoma. J Invest Dermatol. 2005;125:1242–1251. doi: 10.1111/j.0022-202X.2005.23931.x. [DOI] [PubMed] [Google Scholar]
- 49.Ramirez JA, Guitart J, Rao MS, Diaz LK. Cyclin D1 expression in melanocytic lesions of the skin. Ann Diagn Pathol. 2005;9:185–188. doi: 10.1016/j.anndiagpath.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 50.Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, et al. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–2890. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.