

Abstract
Current evidence suggests that neonatal immunity is functionally distinct from adults. Though toll like receptor (TLR) signaling through the adaptor protein, MyD88, has been shown to be critical for survival to sepsis in adults, little is known about the role of MyD88 or TRIF in neonatal sepsis. We demonstrate that TRIF−/− but not MyD88−/− neonates are highly susceptible to Escherichia coli peritonitis and bacteremia. This was associated with decreased innate immune recruitment and function. Importantly, we found that the reverse was true in adults, that MyD88−/−, but not TRIF−/− or wild-type adults are susceptible to E coli peritonitis and bacteremia. In addition, we demonstrate that TRIF but not MyD88 signaling is critical for the TLR4 protective adjuvant effect we have previously demonstrated. These data suggest a differential requirement for the survival of neonates versus adults to gram negative infection, and that modulation of TRIF in neonates can be used to augment survival to neonatal sepsis.
Keywords: mice, infection, neutrophil, cytokine
Introduction
One million newborn children die each year from sepsis or severe infection (1, 2). Mortality rates among these babies range from 10–40% depending on birth weight and age at onset of sepsis, but are especially high in very low birth weight infants (VLBW) (3, 4). In addition, despite progress in outcomes with postpartum group B streptococcus infections and sepsis, gram negative sepsis from Escherichia coli continues to be a significant problem in the neonatal intensive care units (5, 6).
It is now generally accepted that the functional status of neonatal innate and adaptive immunity may be one of the reasons why this population fares so poorly during infection (2). Numerous studies have examined the epidemiology of neonatal sepsis and the molecular markers associated with neonatal sepsis (3, 4, 7, 8), but few studies have characterized the underlying immunological responses and pathophysiology of neonatal sepsis. Though many studies in adult models of infection and sepsis exist, examining the relative requirements of innate immunity and potential modulators of innate immune response, few studies have established these responses or mechanisms in the neonate and whether they are similar or different than in the adult.
Of these modulators of innate immune activation, Toll like receptor (TLR) signaling is arguably one of the most important. TLR signaling proceeds through one of three different pathways: myeloid differentiation primary response gene 88 (MyD88) alone, leading predominantly to the production of proinflammatory cytokines/chemokines, TIR-domain containing adapter protein-inducing interferon-β (TRIF), alone leading to the production of both type I interferons, as well as the production of proinflammatory cytokines and chemokines, or in the unique case of TLR4 in which both MyD88 and TRIF pathways are activated (9). Several adult studies have identified MyD88 signaling as critical for survival to gram negative infection or sepsis in adults, as well as important for the production of reactive oxygen species in neutrophils (10–14). The role of TRIF signaling in adult gram negative infection is somewhat controversial; the response to some gram negative infections such as Burholderia infection is thought to be MyD88 dependent, whereas TRIF signaling was shown to be critical for survival to Yersinia enterocolitica (14, 15). The role of these TLR adaptor signaling proteins in the survival of the neonate to gram negative infection is unknown.
In this report, we demonstrate that TRIF−/− neonates are more susceptible to E coli sepsis compared to wild type (WT) and MyD88−/− neonates. This is associated with decreased recruitment of peritoneal neutrophils and macrophages at 12 hours post E coli infection, decreased neutrophil and macrophage reactive oxygen species production, as well as increased peritoneal and blood bacterial counts in TRIF−/− compared to WT neonates. Importantly, we also demonstrate that MyD88−/−, but not TRIF−/−, young adults were more susceptible to E coli gram negative infection. In addition, we have previously shown that pretreatment of murine neonates with toll-like receptor (TLR) 4 ligand, lipopolysaccharide (LPS) (16), is able to significantly improve survival to subsequent polymicrobial sepsis (2). Although this effect was associated with increased activation/recruitment of innate immune effector cells, the mechanism was unknown. In this report, we demonstrate that TRIF, but not MyD88 signaling, is critical for the observed TLR4 adjuvant effect.
Methods
Mice
Six to eight week old male and female C57BL/6J (wild type (WT)) and TRIF−/− (C57Bl/6J-Ticam1Lps2/J) mice were purchased from the Jackson Laboratories (Bar Harbor, ME) and used for adult breeders and adult experiments. MyD88−/− mice (B6.129P2-Myd88tm1Defr) were obtained from S Akira, initially maintained at Rhode Island Hospital and Brown University, and transferred to UF for breeding and colony maintenance. Interferon type 1 receptor null mice (B6.129S2-Ifnar1tm1Agt/J; IFNAR−/−) breeders were purchased from Jackson Laboratories. These mice had been backcrossed onto a B6 background for six generations, and inbred on the B6 background for an additional 12 generations. Adult mice were mated in a harem schema with one male to two females. Neonatal mice were studied between four and seven days of age. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Florida, College of Medicine.
Cecal slurry (CS) model
Polymicrobial sepsis was induced as previously described (17). Briefly, cecal contents of adult C57BL/6J mice were suspended in 5% dextrose solution (D5W) at a final concentration of 80 mg/mL and injected intraperitoneally (ip) into five to seven day old neonates. An LD70 (1.3 mg fecal contents/g body weight) of cecal slurry, as determined from previous experiments, was used (17).
Escherichia coli gram negative sepsis
After institutional review board permission was obtained to review patient records, a clinical isolate of E coli was found from a blood culture drawn from a neonate (less than 30 days of age) with E. coli urosepsis. This isolate was then subsequently serotyped as O11:H18 by Pennsylvania State University laboratories and used in the murine studies described in this manuscript. E. coli O11:H18 was grown in trypticase soy agar broth and plates (Fisher Scientific). Neonatal C57BL/6 mice five to seven days old received ip an LD70 of E. coli (2 × 103 CFUs) in 50 µL of phosphate buffered saline. Total volume of injected solution was less than 50 µL. Neonates were either followed for survival for seven days or euthanized and tissues harvested at the time points indicated.
Colony forming unit calculation from blood and peritoneal washes
Blood harvested via intracardiac puncture or peritoneal washes were collected from neonates or adults, 24 hours post E coli inoculation. Peritoneal washes were harvested from neonates or adults via intraperitoneal injection of 500 µL or 5 mL of PBS at 4° C, respectively. Peritoneal washes and blood was then diluted and plated on blood agar plates (Fisher scientific) (18–20).
Blockade of type 1 interferon signaling
To block type 1 interferon signaling, anti-IFNAR1 or isotype (100 µg/animal) (Mouse IgG1) (Leinco technologies, St. Louis, MO) was administered intraperitoneally to neonatal C57Bl/6J mice. Amount of antibody used was gleaned from Sotolongo et al (15).
Pretreatment with a TLR4 agonist (bacterial LPS)
Neonatal C57BL/6 mice four to six days old received ip administration of lipopolysaccharide (16) (1 µg/g BW; ultrapure via ion exchange chromatography E. coli O26:B6) (Sigma-Aldrich, St Louis, MO) in physiologic saline 24 hours before challenge with cecal slurry. Control animals received saline only ip 24 hours before cecal slurry challenge. Total volume of injected solution was less than 50 µL.
Peritoneal, sera, and media cytokine concentrations
Blood and peritoneal washes were harvested from neonates at the time points described following either LPS, cecal slurry or E coli administration. As reports have shown that heparin can bind to CXCL10 and possibly reduce its detection in blood, serum was used for these assays (21, 22). Blood was collected via intracardiac puncture and centrifuged to collect the supernatant (serum). To collect peritoneal washes, the peritoneal cavity was instilled with 500 µL of cold phosphate-buffered saline (PBS) (Cellgro, Manassas, VA). The peritoneal cavity was then opened over a sterile tissue culture dish and the peritoneal wash collected. Peritoneal washes were then centrifuged to remove debris and the supernatants were stored at −80° C. Media was collected from in vitro LPS stimulation experiments and centrifuged to remove cellular debris. The supernatant was then used for cytokine detection assays. Sera, peritoneal washes, and media were then measured for IL-1β, IL-6, IL-10, IL-12p70, KC, IFNγ, TNFα, MCP-1, MIP-1α, CXCL10 concentrations via Multiplex Luminex® Assay (Miltenyi Bitoec, Auburn, CA).
Flow cytometry
Single cell suspensions were characterized using anti-Ly6G APC, anti-CD11b-PECy7, and/or anti-F4/80 APC-Alexfluor-750 (Ebioscience, San Diego, CA). All antibodies were purchased from Becton-Dickinson (BD) (Franklin Lakes, NJ) unless otherwise indicated. Samples were acquired and analyzed on an LSR II flow cytometer (BD) and analyzed via FACSDiva™ (BD) or FloJo™ (Treestar Inc, Ashland, OR) software. At least 1 × 104 live cells (SYTOX Blue−; Invitrogen, Carlsbad, CA) were collected for analysis.
Isolation of blood, bone marrow, and splenocytes for phenotypic analysis
Blood was obtained via intracardiac puncture of isoflurane anesthetized neonatal mice into a heparinized syringe. Spleens were harvested from neonates and then dissociated through a 70 µm sterile filter. To harvest bone marrow, both femurs and tibias from individual animals were collected and flushed with phosphate-buffered saline (PBS) (Cellgro, Manassas, VA). Blood, splenocyte, and bone marrow suspensions were then subjected to erythrocyte lysis using ammonium chloride lysis solution. Cell suspensions were then stained for neutrophil (Ly6G+,CD11b+) or macrophage (F4/80+,CD11b+) cell surface markers and analyzed via flow cytometry. A representative scattergram for staining of neutrophil (Ly6G+,CD11b+) or macrophage (F4/80+,CD11b+) cell surface markers from neonatal splenocytes is presented in Supplemental Figure 1.
Harvest of peritoneal washes
Peritoneal cell isolation was carried out in a similar manner as described previously (2). Due to the fact that peritoneal lavages from individual animals contained relatively few cells, peritoneal washes from three to five animals were pooled for analysis and considered to be one sample. For each time point or condition analyzed, a total of three to four peritoneal samples were collected per experiment. To determine absolute numbers of innate immune effector cells, the percentage of macrophages or neutrophils within the total sample population was determined via flow cytometry, multiplied by the total sample cell number, and then divided by the total number of mice in the pooled sample. Absolute numbers represent total macrophages or neutrophils per mouse.
Isolation of splenic macrophages and neutrophils for functional analyses
Spleens were disassociated through a 70 µm sterile filter into RPMI 1640 media (Cellgro) with 10% heat inactivated low endotoxin fetal bovine serum 0.5% and penicillin/streptomycin (Cellgro). Suspensions were then overlayed on top of a Histopaque 1119® (Sigma Aldrich, St Louis, MO) density gradient and centrifuged. Mixed cell suspensions were then washed with media alone, and individual cell populations were assayed for phagocytosis or reactive oxygen species (23) production by flow cytometry.
Reduction of cytochrome c assay
In Hanks’ balanced salt solution (Sigma) containing 145 µmol/L cytochrome c (Sigma), 1 × 105 cells isolated from peritoneal washes of WT or TRIF−/− neonates 24 hours post E coli infeciton were stimulated with or without phorbol myristic acid (PMA; 98 nmol/L) (Sigma) at 37°C. Reduction of cytochrome c was measured at 550 nm at 1-min intervals for 45 min. The maximal rate of reduction (Vmax) was calculated by linear regression analysis. Purified superoxide dismutase 1 (0.5 units/mL) was used to confirm specificity of superoxide production.
In vitro stimulation with recombinant CXCL10
Splenocytes isolated by Histopaque 1119® (Sigma-Aldrich) gradient were stimulated with or without recombinant murine CXCL10 at 100 ng/mL for four hours (R&D Systems, Minneapolis, MN). After incubation with CXCL10, cells were washed with PBS and subsequently assayed for phagocytic function or ROS production and phenotypically characterized via flow cytometry.
Functional analysis of splenic macrophages and neutrophils or peritoneal macrophages or neutrophils
For phagocytosis assays, 1 × 105 splenocytes isolated via density gradient or peritoneal cell suspensions were incubated with 1 × 108 fluorescent polystyrene microspheres (Fluospheres; Invitrogen, Carlsbad, CA) for 30 min at 37°C. Cells were then stained and analyzed by flow cytometry, as described above. After cells were gated on either neutrophil (Ly-6G+,CD11b+) or macrophage (F4/80+,CD11b+) populations, FITC+ cells were considered to be phagocytic cells. To assay for ROS production, 2 × 106 splenocytes isolated via density gradient were first stained for cell surface markers and labeled with dihydrorhodamine 1,2,3 (Invitrogen). Cells were then stimulated with 1 µM of phorbol-12-myristate-13-acetate (PMA) (Sigma -Aldrich) at 37°C and aliquots were evaluated by flow cytometry at various points over a 30 minute period using a LSR II flow cytometer (BD). A minimum of 1 × 104 live, non-debris cells were collected for analysis.
Statistical analyses
Continuous variables were tested for normality and equality of variances. Differences in survival were determined by Fisher’s exact test, whereas differences in continuous variables among groups were evaluated by either one-way ANOVA with either Dunn’s or Tukey post-hoc analysis or Student’s t-test. In those cases where normality failed, either a Mann-Whitney U test using the Hodges–Lehmann (HL) estimator or a Kruskal-Wallace ANOVA on ranks was also performed. In the latter case, a Dunn’s post hoc analysis was performed. However, non- parametric tests are not without their limits. In particular, for small sample sizes the power of the Mann-Whitney U versus the Student’s t-test is considerably less. In all cases, however, significance was determined at the 95% confidence level
Results
TRIF−/− neonates, but not adults, are susceptible to gram negative sepsis
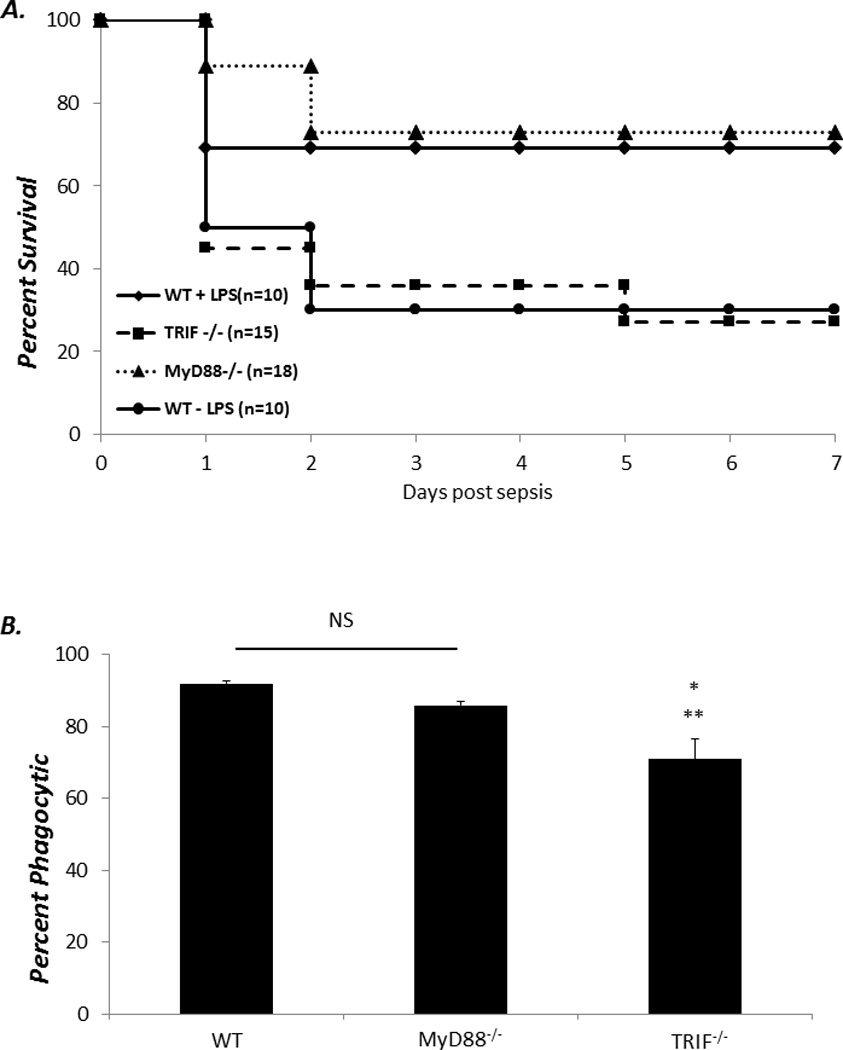
Though the loss of MyD88 has been associated with susceptibility to gram negative and polymicrobial infection in adults, the role of MyD88 in neonatal gram negative infection and sepsis is unclear (13, 14, 24, 25). To address this, TRIF−/−, MyD88−/−, and WT neonates were challenged with 1 × 103 (LD20–30) CFUs of an E coli isolate (serotype O11:H18) from a neonate with urosepsis and followed for survival. As demonstrated in Figure 1A, TRIF−/− neonates were more susceptible to E coli challenge compared to both MyD88−/− and WT neonates. This was associated with increased peritoneal and blood CFUs (Figure 1B). To see if previous observations regarding the adult requirement for MyD88 for survival to gram negative infection held true in our model system, we administered 1 × 106 E coli (LD20–30) ip into adult MyD88−/−, TRIF−/−, and WT mice. Not surprisingly, MyD88−/− adults were dramatically more susceptible to E coli challenge compared to TRIF−/− or WT adults (Figure 1C). This increased susceptibility of MyD88−/− adults to E coli was associated with increased bacteremia and peritoneal bacterial counts (Figure 1D). These data suggest intrinsic differences in the TLR signaling adaptor protein pathways that are important for the clearance of gram negative infection between neonates and adults.
Figure 1. TRIF−/− neonates, but not adults, are more susceptible to gram negative infection compared to MyD88−/− or WT neonates.
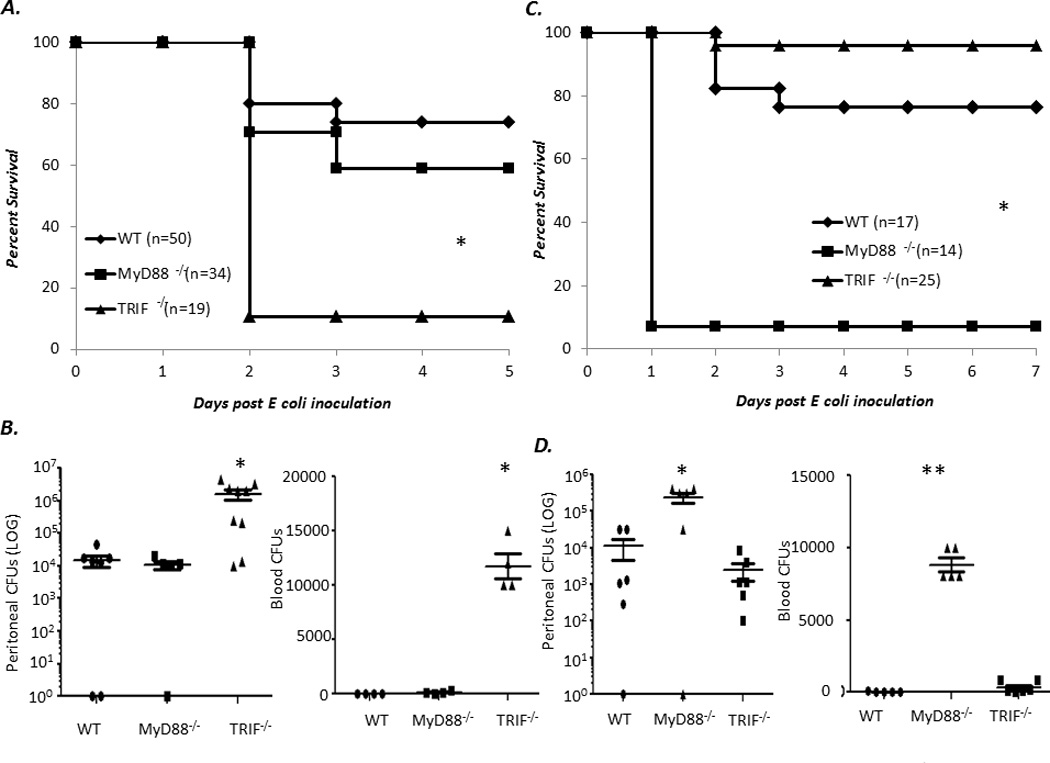
(A) WT, TRIF−/−, and MyD88−/− neonates were inoculated ip. with E coli and subsequently followed for survival (Fisher’s exact *p< 0.001). (B) Peritoneal washes or blood were harvested 24 hours post E coli inoculation and plated on blood agar, and colony forming units (CFUs) counted (n=5–7 animals per group) (Data were analyzed before log transformation by both one way ANOVA and Kruskal-Wallace ANOVA; in both cases, data were significantly different * p<0.05) (C) Six to twelve week old WT, TRIF−/−, and MyD88−/− were inoculated ip. with E coli and followed for survival (Fisher’s exact p<0.001) (D) Peritoneal washes and blood were collected from MyD88−/−, TRIF−/− and WT adults 24 hours post E coli infection and plated on blood agar, and colony forming units (CFUs) counted (Data were analyzed before log transformation by both one way ANOVA and Kruskal-Wallace ANOVA * p<0.009 ** p<0.001) (n=5 animals per group).
TRIF−/− neonates produce decreased local inflammatory cytokines and chemokines early, and increased inflammatory cytokines and chemokines late in response to E coli gram negative sepsis
As the downstream products of TLR signaling are inflammatory mediators, we next examined whether there was any difference in the production of key cytokines or chemokines in TRIF−/− neonates compared to WT or MyD88−/− neonates that could potentially explain this increased susceptibility to gram negative sepsis. Peritoneal washes from TRIF−/−, MyD88−/−, and WT neonates were harvested at 0, 6, 12, and 24 hours post E coli administration and assayed for the concentration of IL-10, IL-12, IFNγ, IL-1β, TNFα, IL-6, CXCL10, MCP-1, MIP-1α, and KC via Luminex® multiplex cytokine arrays. There were significant reductions in the peritoneal concentration of IL-6, KC, and MIP-1α, and CXCL10 in TRIF−/− neonates compared to MyD88−/− or WT neonates at 12 hrs following E coli inoculation (Figure 2). There were also differences in TNFα concentrations between both MyD88−/− and TRIF−/− neonates compared to WT neonates in response to E coli infection. Concentrations of several cytokines (IL-1β, IL-6 ,MIP1α, TNFα) were significantly elevated in the TRIF−/− neonates, but not the wild-type or the MyD88−/− at 24 hours. These increased concentrations were associated with markedly increased bacterial counts seen in the blood and peritoneal fluid in TRIF−/− mice at 24 hours (Figure 1B) and dramatic mortality over the next 24 hours.
Figure 2. Decreased peritoneal inflammatory mediators in TRIF−/− neonates compared to MyD88−/− or WT neonates.
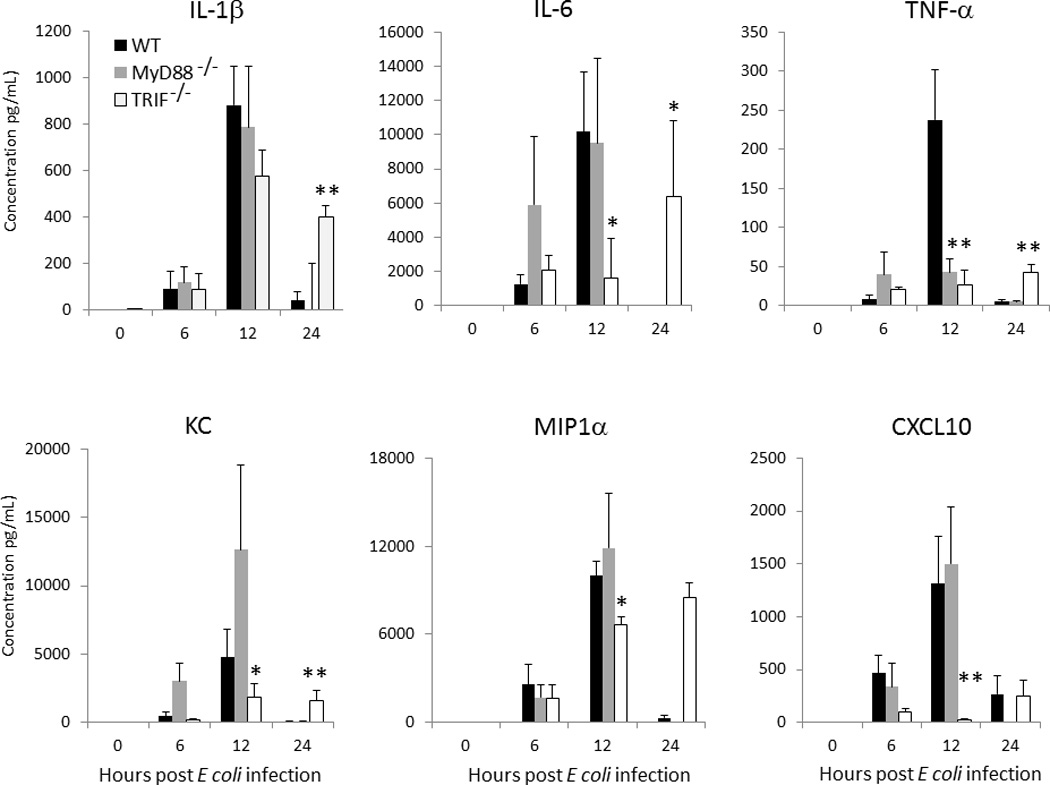
Peritoneal washes were obtained from TRIF−/−, MyD88−/−, and WT neonates at 0, 6, 12, and 24 hours post E. coli administration and assayed for concentration (Values represent mean ± SD; Data were analyzed by either one way ANOVA or Kruskal-Wallace ANOVA * p<0.05 ** p<0.001) (n=4–5 animals per group).
Increased susceptibility of neonates to gram negative infection is not dependent on type -1 interferons
A recent report by Sotolongo and colleagues demonstrated that the loss of type-1 interferons increased the susceptibility of adults to gram negative infection and was associated with defects in innate immune phagocytosis (15). To address whether type 1 interferons played a role in the survival of neonates to E coli, two approaches were used. First, we administered anti-IFNAR1 or isotype antibody to neonatal mice six hours before E coli challenge and then subsequently followed them for survival. No difference in survival was noted between isotype and anti-IFNAR antibody mice suggesting that the increased susceptibility of TRIF−/− neonates was type 1 interferon independent (Figure 3A). Second, we repeated the studies in interferon type 1 receptor knockout mice (IFNAR−/−) and wild-type controls. Both groups of mice were administered an LD50 of E coli and survival was evaluated over the next seven days. Once again, no difference in survival was noted (Figure 3B).
Figure 3. Susceptibility of neonates to gram negative infection is not dependent on Type-1 interferon signaling.
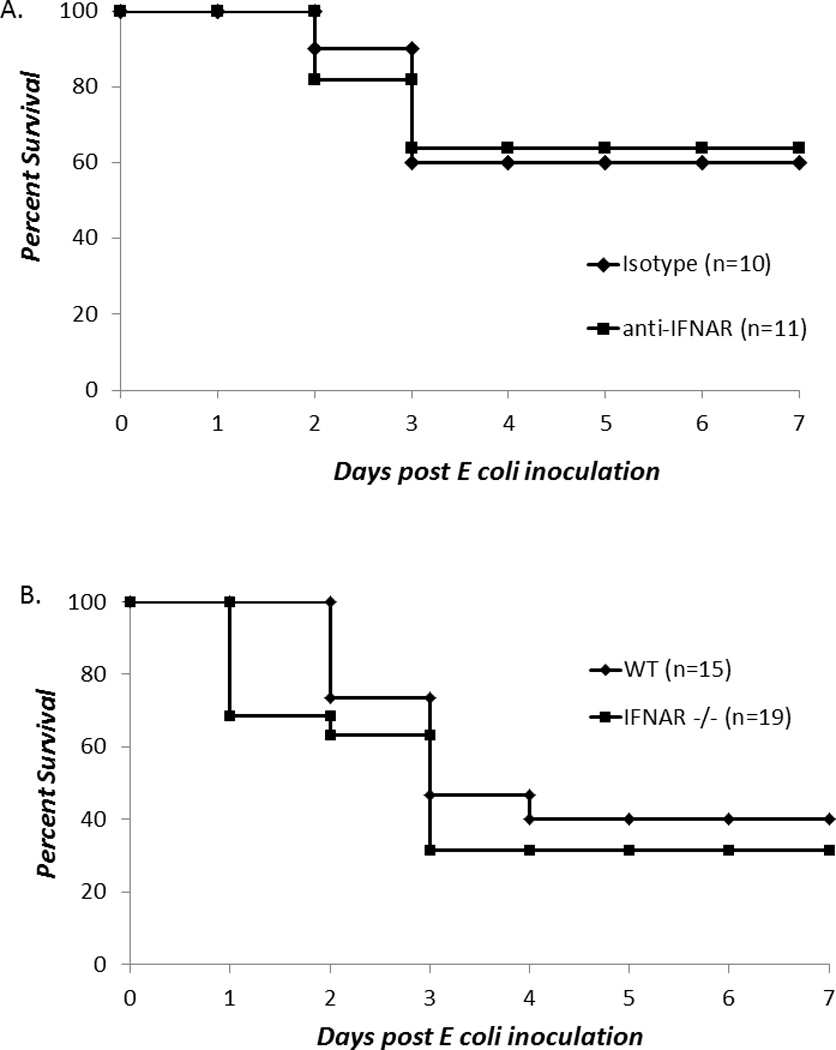
(A) Neonatal mice were administered either anti-IFNAR1 or isotype antibody ip six hours before E coli challenge and followed for 5–7 days for survival (Fisher’s exact; no significant differences in survival were noted). (B) Neonatal WT or IFNAR−/− mice were challenged with E coli and followed for 5–7 days for survival (Fisher’s exact; no significant differences in survival were noted).
Decreased early recruitment of peritoneal neutrophils and macrophages and decreased late ROS production in TRIF−/− neonates in response to E coli neonatal sepsis
To understand why TRIF−/− neonates were so susceptible to E coli sepsis compared to WT neonates, we harvested peritoneal washes from TRIF−/− and WT neonates at 12 and 24 hours post E coli infection. TRIF−/− neonates had significantly fewer peritoneal neutrophils and macrophages compared to WT neonates at 12 hours post E coli administration (Figure 4A). A representative scattergram of splenic neutrophil (Ly6G+,CD11b+) or macrophage (Ly6G−,F4/80+,CD11b+) populations is presented in Supplemental Figure 1. While no differences existed in the phagocytic function of these innate immune effector cells, TRIF−/− neonatal neutrophils and macrophages produced significantly less ROS than WT neonates at 24 hours post E. coli infection at baseline (Figure 4B). To further support this, we performed a cytochrome c reduction assay on peritoneal washes isolated from neonates 24 hours following E coli. TRIF−/− mice had significantly slower reduction of cytochrome c, and as a corollary decreased ROS production, compared to peritoneal washes isolated from WT neonates (Figure 4C) (p<0.001). Together these data suggest that TRIF−/− neonates have impaired innate immune neutrophil and macrophage function compared to WT neonates that subsequently impairs clearance of gram negative infection.
Figure 4. Despite increased numbers and phagocytic peritoneal neutrophils and macrophages, TRIF−/− neonates have decreased ROS production 24 hrs post E. coli infection.
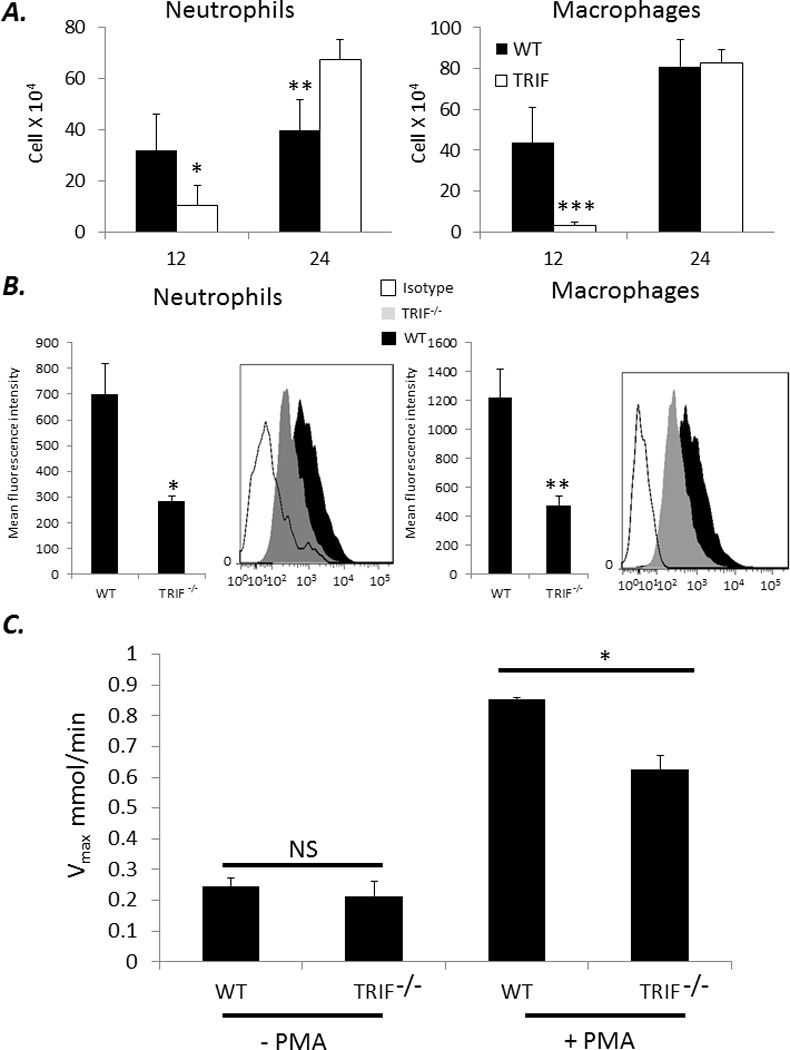
(A) Peritoneal washes were harvested from WT and TRIF−/− neonates at 12 and 24 hours post E coli inoculation and assayed for the presence of neutrophils and macrophages via flow cytometry (Values represent mean ± SD ; Student t-test or Mann Whitney U * p<0.04 ** p<0.01 ***p<0.001) (n=4–5 animals per group). (B) Peritoneal washes from WT and TRIF−/− neonates 24 hours post E. coli were phenotyped for the presence of neutrophils and macrophages and subsequently stained for DHR 123 expression via flow cytometry (Values represent mean ± SD; *p=0.016 ** p<0.001) (n=4–5 animals per group). (C) Twenty four hours following E coli infection, peritoneal washes from WT or TRIF−/− were isolated and stimulated with or without PMA. Maximal rate of cytochrome c reduction (V max) calculated (Values represent mean ± SD; Student t-test p<0.001) (n=5 animals per group).
Deletion of toll like receptor signaling does not affect survival to polymicrobial neonatal sepsis
The data above suggests that TRIF−/− neonatal mice are more susceptible to classic gram negative infections such as E coli; however, this does not address the response in a mixed polymicrobial infection that exist in conditions such as necrotizing enterocolitis, an important cause of mortality in premature infants (26). Therefore to address this, we utilized a cecal slurry model of peritonitis and sepsis to generate polymicrobial “hit”, previously reported by our group (2, 18). Surprisingly, no differences in survival were seen between TRIF−/−, MyD88−/−, or WT animals in response to either an LD40 or LD70 of cecal slurry (Figure 5). This data suggests there are other signaling pathways that are able to compensate for the lack of TLR signaling following polymicrobial infection.
Figure 5. Deletion of TLR signaling does not affect survival to polymicrobial neonatal sepsis.
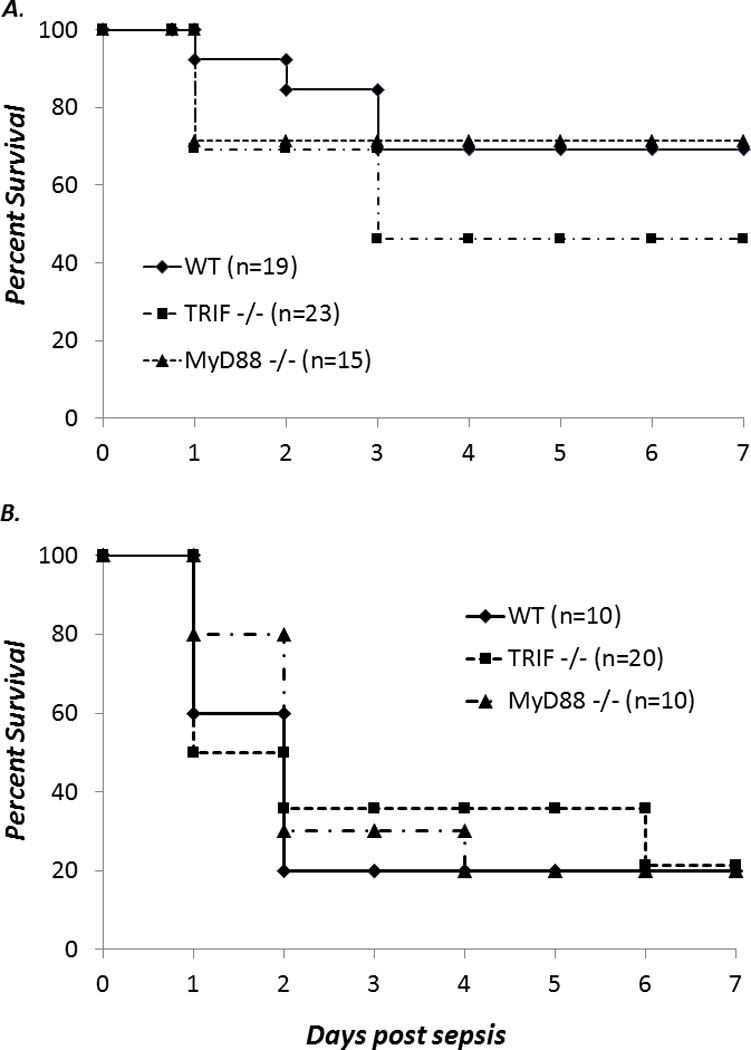
WT, TRIF−/−, or MyD88−/− neonates were treated with either an LD40 (A) or an LD70 (B) dose of cecal slurry and then followed for 7 days for survival (Fisher’s exact NS).
TRIF−/− neonates are not protected against polymicrobial sepsis in response to TLR4 adjuvant protection
We have previously shown that WT neonates pretreated with LPS are protected against polymicrobial sepsis compared to neonates pretreated with saline (2). Despite the data above, demonstrating that there is no difference in mortality in response to polymicrobial sepsis, this does not address the adjuvant effect of LPS in this setting. To address this, WT, TRIF−/−, and MyD88−/− neonates were pretreated with LPS (1 µg/g BW) 24 hours before polymicrobial sepsis challenge, and subsequently followed for survival. As demonstrated in Figure 6A, in contrast to both MyD88−/− and WT neonates, TRIF−/− neonates were not protected against cecal slurry (CS)-induced polymicrobial sepsis. These data suggests that the loss of TRIF not only impacts the inflammatory responses and functional maturity of innate immune effectors cells recruited to the peritoneum, but also is critical for the TLR4 adjuvant protective effect that we have previously demonstrated (2).
Figure 6. TRIF−/− neonates are not protected against polymicrobial sepsis with TLR4 agonist pretreatment.
(A) WT, TRIF−/−, and MyD88−/− neonates were treated with either LPS or saline 24 hours before CS challenge and followed for survival (Fisher’s exact *p<0.01) (B) Phagocytic uptake of polystyrene FITC labeled beads in neutrophils following 24 hour stimulation with LPS (Values represent mean ± SD; One way ANOVA * p <0.001; ** p< 0.004) (n=4–5 animals per group).
Both the phagocytic function and production of neutrophil extracellular traps have been demonstrated to be decreased or impaired in neonatal neutrophils relative to adults (2, 27, 28). To investigate if TLR signaling through TRIF or MyD88 dependent pathways played any role in the neonatal neutrophil function, splenocytes were harvested from neonates and stimulated with LPS (100 ng/mL) for 24 hours. Following staining for neutrophil cell surface markers, cell suspensions were then either incubated with either FITC-labeled polystyrene beads or evaluated for ROS production via phorbol ester stimulation. Though no differences were noted between neutrophils from MyD88−/−, TRIF−/−, and WT neonates with regards to ROS respiratory burst following PMA stimulation (data not shown), neutrophils from TRIF−/− animals had significantly decreased phagocytic function relative to neutrophils from both MyD88−/− and WT animals in response to LPS (p<0.001) (Figure 6B).
One of the reasons why neutrophil phagocytic function may be impaired in TRIF−/− neonates may be due to poor inflammatory mediator production, which can have a paracrine effect on innate immune effector function (18, 29). To assess if the inflammatory cytokine production by inflammatory cell populations was also impaired in TRIF−/− neonates, media supernatant from neonatal splenocytes stimulated with LPS (100 ng/mL) for 24 hours and assayed via Luminex™ for the concentration of cyto/chemokines, including IL-1β, TNFα, IFNγ, and CXCL10. While both MyD88−/− and TRIF−/− produced significantly less IL-6, TRIF−/− neonates produced significantly less IL-1β and CXCL10 (Figure 7A). These data suggest that deletion of TRIF, but not MyD88 in the neonate, significantly impairs both neutrophil phagocytic function and cellular inflammatory responses to LPS.
Figure 7.
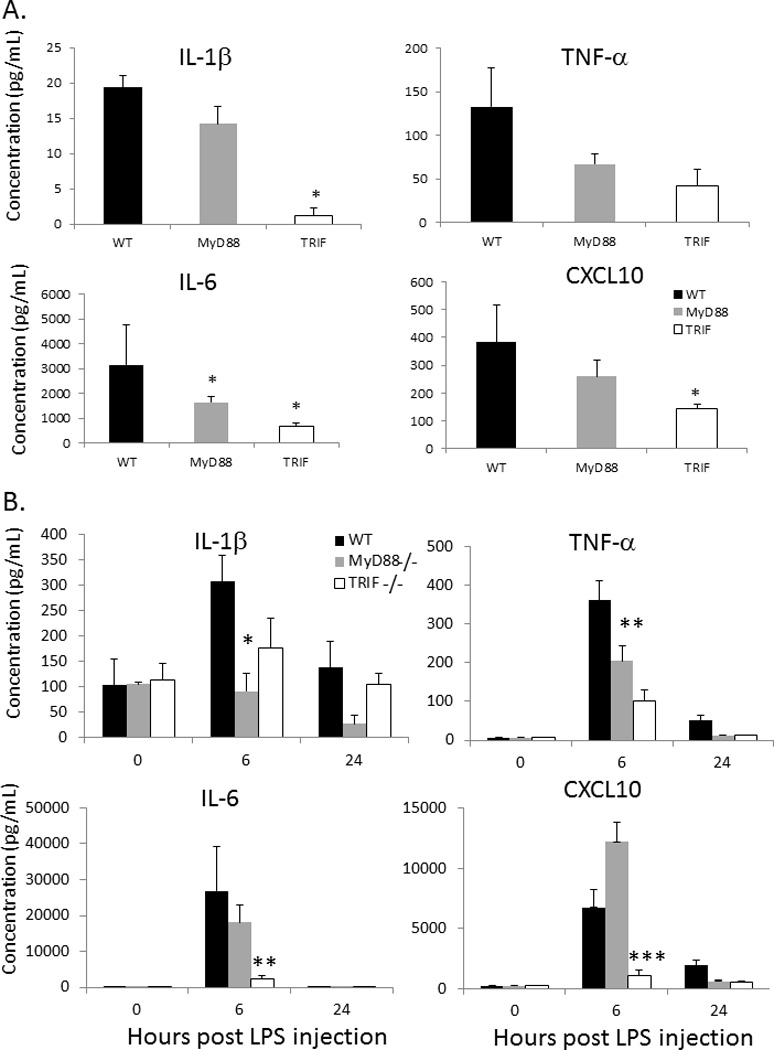
(A) Cytokine production from splenocytes isolated from WT, TRIF−/−, and MyD88−/− neonates following 24 hour stimulation in vitro with LPS (100ng/mL)(* p<0.05) (n=3–4 animals per group). (B) Sera cytokine concentrations from WT, TRIF−/−, and MyD88−/− neonates at 0, 6, and 24 hours post LPS administration (Values represent mean ± SD; Data were analyzed by either one way ANOVA or Kruskal-Wallace ANOVA *p<0.05; **p<0.01; ***p<0.024) (n=4–5 animals per group)
To determine if this observed decrease in cytokine/chemokine production is recapitulated in vivo, WT, MyD88−/− and TRIF−/− neonates were injected with LPS at a dose of 1 µg/g body weight (BW) and serum was harvested at 0, 6, and 24 hours post injection. Interestingly in contrast to the in vitro data, MyD88−/− neonates produced significantly less IL-1β and TNFα compared to WT neonates, whereas TRIF−/− neonates produced significantly less TNFα, IL-6, and CXCL10 (Figure 7B). This data further supports the data above and suggests that TRIF plays a critical role in the endotoxin-mediated inflammatory responses in the neonate.
Reversal of poor phagocytic function of TRIF−/− neonatal granulocytes in the presence of CXCL10
We have previously demonstrated that CXCL10 is important for neonatal and adult neutrophil phagocytosis. As CXCL10 was significantly decreased following both in vitro stimulation and in vivo administration of LPS in TRIF−/−, but not MyD88−/− or WT neonates, and TRIF−/− granulocytes had significantly decreased phagocytic function compared to WT or MyD88 −/− neonates following LPS stimulation, we treated TRIF−/− and WT neonatal splenocytes with or without 100 ng/mL of recombinant murine CXCL10 in the presence of LPS. As demonstrated in Figure 8, incubation of TRIF−/− splenocytes with CXCL10 partially but significantly reversed the phagocytic function of TRIF−/− neutrophils, 84% with CXCL10 vs 70% without CXCL10, following LPS stimulation (p< 0.05). No differences were noted between MyD88−/− and WT neutrophil phagocytic function in the presence of LPS (see Figure 8). Though we were not are able to demonstrate complete reversal of the phagocytic defect in TRIF−/− mice via CXCL10, this data supports the importance of TRIF signaling and the production of TRIF dependent inflammatory mediators in innate immune function.
Figure 8.
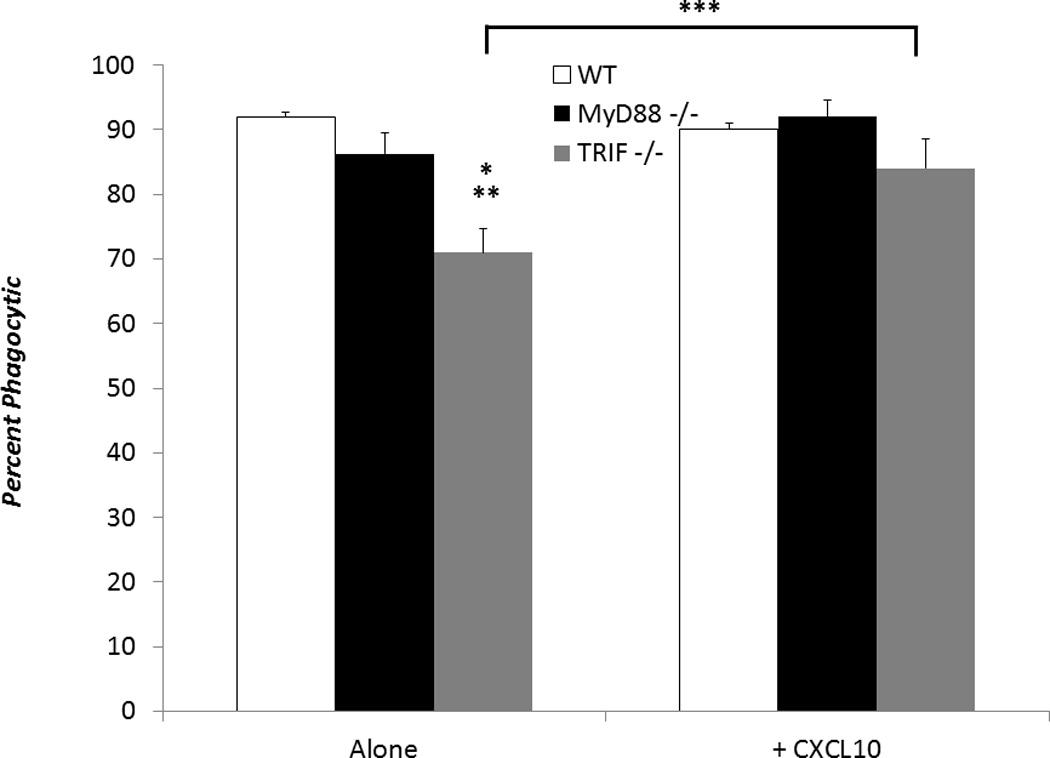
Splenocytes from WT, MyD88−/−, or TRIF−/− neonates were stimulated with LPS (100 ng/mL). TRIF−/− splenocytes were also either stimulated with or without CXCL10 (100 ng/mL) (Values represent mean ± SD; Data were analyzed by either one way ANOVA * WT vs TRIF−/− p<0.01; ** MyD88−/− vs TRIF−/− p<0.05; Student t-test *** TRIF−/− vs TRIF−/− + CXCL10 p< 0.05 ) (n=4-5 animals per group).
Discussion
Many studies have focused on the role of MyD88 in response to polymicrobial or gram negative sepsis in adults (10–14, 24, 25). Further, these studies have linked or suggested a critical role for TLR4-MyD88 signaling in the survival of adult mice to both polymicrobial and gram negative sepsis. Though Akira’s group has identified the importance of MyD88 and TRIF signaling in the lethality of adults to endotoxemia, again the loss of MyD88 appears to offer more protection to lethal endotoxin challenge in young adults than the loss of TRIF in these studies (30, 31). TLR signaling in neonates, however, is not as well characterized although a recent study by Andrade and colleagues has demonstrated that blockade of TLR2 signaling following group B streptococcus infection improves neutrophil recruitment and improves survival to gram positive neonatal sepsis (32).
Our data suggests that TLR4-TRIF signaling in the neonate, in contrast to the young adult, is critical for response to TLR4 agonists in neonates. We demonstrate that the loss of TRIF in neonates impairs the early production of several inflammatory cytokines and chemokines in response to E coli infection. Mortality is markedly increased in these animals and is associated with systemic bacteremia and an ongoing, exaggerated late inflammatory cytokine response. In addition, following either in vitro stimulation with LPS or in vivo administration, our data shows a reduced phagocytic function in neutrophils from TRIF−/− mice compared to neutrophils from WT or MyD88−/− animals. Previous work from our laboratory has also demonstrated a critical role for CXCL10 in the phagocytosis and survival of neonates to polymicrobial challenge (18). In the current study, we show that the loss of TRIF in neonates impairs both in vitro and in vivo CXCL10 production, as well as the phagocytic response of neutrophils to LPS. Further, in the current study, we also demonstrate that TRIF−/− neonates are not protected following LPS adjuvant pretreatment to polymicrobial sepsis. Similarly, our recent work shows that if the CXCL10 response to LPS is blocked, the protective adjuvant effect of LPS is lost in response to polymicrobial sepsis (18). These findings suggest a novel critical role for a TRIF dependent inflammation in response to LPS and gram negative infection. Importantly, we also demonstrate that the same TRIF signaling is not as critical in adults, and suggest a reliance of neonates on TRIF signaling in contrast to adults who appear to be more dependent upon MyD88 for response and survival to gram negative infection.
While analysis of cytokines at individual time points can be difficult to interpret, we also attempted to compare the cytokine production of WT, TRIF−/−, and MyD88−/− neonates at 0, 12, and 24 hours post E coli inoculation. Of the presented cytokines/chemokines, IL-1β, TNFα, KC and MIP-1α, were all found to be significantly (p<0.05) higher in the TRIF knockout animals compared to either MyD88−/− or WT animals at 24 hours post E coli inoculation (Figure 2). In addition, we found that the numbers of neutrophils were significantly greater in the TRIF KO animals compared to WT animals at 24 hours post E coli infection (Figure 4A). However, as shown in Figure 1C, TRIF−/− neonates have significantly more bacterial CFUs than either WT or MyD88−/− animals at 24 hours post E coli inoculation. We postulate that the increases in peritoneal neutrophils and cytokines/chemokines that were measured are more a reflection of the overwhelming E coli dependent sepsis that is occurring in the TRIF−/− animals at 24 hours post inoculation compared to the MyD88−/− or WT animals.
Although a recent report by Sotolongo and colleagues has demonstrated that TRIF signaling is important for the adult survival to gram negative infection, it is important to note that their model of infection utilized the gram negative enteropathogen Yersinia enterocolitica, not Escherichia coli, which may indeed have different immunopathological processes and requirements for clearance despite both being gram negative organisms (15). In addition, susceptibility of MyD88−/− adults to gram negative infection was not examined in the report but was rather inferred from data investigating phagocytic function of MyD88−/−, TRIF−/−, TLR4−/−, and WT macrophages. Finally, the authors stressed and demonstrated the importance of TRIF-dependent type I interferon production in the survival of adult mice to gram negative infection. In a previous report, we have demonstrated that loss of type I Interferon signaling is not important for survival of neonates to gram negative infection (2). Though it was in a polymicrobial model of neonatal sepsis, most of the enteric organisms within the cecum that are administered through cecal slurry challenge are gram negative (> 99%).
These data highlight important differences between the neonatal and adult responses to bacterial products and gram negative infection. Previous data have suggested that neonates rely more heavily on a functioning innate immune system for protective immunity than do young adults2. This report moves that observation forward, and demonstrates for the first time the novel finding that TRIF but not MyD88 signaling is essential for innate immune activation and is required for survival of neonates to gram negative infection. This is important, as much of the focus on early signaling pathways of innate immunity to microbial infection has been on MyD88, as it is the primary adaptor protein responsible for downstream signaling for the majority of TLRs (30). In addition, these data suggests an ontological difference between adults and neonates that reflect differential requirement for TRIF-dependent inflammation during the neonatal period and redirect focus on therapeutics designed around TRIF-dependent inflammation and innate immune activation to improve outcome in these particularly susceptible patients.
Supplementary Material
Acknowledgments
Supported in part by GM-40586 and GM-81923, awarded by the National Institute of General Medical Sciences (NIGMS), P.H.S. AGC and KMK-S were supported by the Ruth Kirschstein NRSA training grant (T32 GM-08721) in burns and trauma, awarded by NIGMS. AGC was also supported by an individual NRSA training grant (F32 GM-093665), awarded by NIGMS.
References
- 1.Lawn JE, Wilczynska-Ketende K, Cousens SN. Estimating the causes of 4 million neonatal deaths in the year 2000. Int J Epidemiol. 2006;35:706–718. doi: 10.1093/ije/dyl043. [DOI] [PubMed] [Google Scholar]
- 2.Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, Ungaro R, Levy O, Moldawer LL. Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood. 2008;112:1750–1758. doi: 10.1182/blood-2008-01-130500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kermorvant-Duchemin E, Laborie S, Rabilloud M, Lapillonne A, Claris O. Outcome and prognostic factors in neonates with septic shock. Pediatr Crit Care Med. 2008;9:186–191. doi: 10.1097/PCC.0b013e31816689a8. [DOI] [PubMed] [Google Scholar]
- 4.Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. 2003;167:695–701. doi: 10.1164/rccm.200207-682OC. [DOI] [PubMed] [Google Scholar]
- 5.Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, Bizzarro MJ, Goldberg RN, Frantz ID, 3rd, Hale EC, Shankaran S, Kennedy K, Carlo WA, Watterberg KL, Bell EF, Walsh MC, Schibler K, Laptook AR, Shane AL, Schrag SJ, Das A, Higgins RD N. Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research. Early onset neonatal sepsis: the burden of group B Streptococcal and E. coli disease continues. Pediatrics. 2011;127:817–826. doi: 10.1542/peds.2010-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson JE, Oh W, Bauer CR, Korones SB, Shankaran S, Laptook AR, Stevenson DK, Papile LA, Poole WK. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics. 2002;110:285–291. doi: 10.1542/peds.110.2.285. [DOI] [PubMed] [Google Scholar]
- 7.Bizzarro MJ, Dembry LM, Baltimore RS, Gallagher PG. Changing patterns in neonatal Escherichia coli sepsis and ampicillin resistance in the era of intrapartum antibiotic prophylaxis. Pediatrics. 2008;121:689–696. doi: 10.1542/peds.2007-2171. [DOI] [PubMed] [Google Scholar]
- 8.Lukacs SL, Schoendorf KC, Schuchat A. Trends in sepsis-related neonatal mortality in the United States, 1985–1998. Pediatr Infect Dis J. 2004;23:599–603. doi: 10.1097/01.inf.0000131633.74921.90. [DOI] [PubMed] [Google Scholar]
- 9.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 10.Lima HR, Gelani V, Fernandes AP, Gasparoto TH, Torres SA, Santos CF, Garlet GP, da Silva JS, Campanelli AP. The essential role of toll like receptor-4 in the control of Aggregatibacter actinomycetemcomitans infection in mice. J Clin Periodontol. 37:248–254. doi: 10.1111/j.1600-051X.2009.01531.x. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien GC, Wang JH, Redmond HP. Bacterial lipoprotein induces resistance to Gram-negative sepsis in TLR4-deficient mice via enhanced bacterial clearance. J Immunol. 2005;174:1020–1026. doi: 10.4049/jimmunol.174.2.1020. [DOI] [PubMed] [Google Scholar]
- 12.Anand RJ, Kohler JW, Cavallo JA, Li J, Dubowski T, Hackam DJ. Toll-like receptor 4 plays a role in macrophage phagocytosis during peritoneal sepsis. J Pediatr Surg. 2007;42:927–932. doi: 10.1016/j.jpedsurg.2007.01.023. discussion 933. [DOI] [PubMed] [Google Scholar]
- 13.Ko HJ, Yang JY, Shim DH, Yang H, Park SM, Curtiss R, 3rd, Kweon MN. Innate immunity mediated by MyD88 signal is not essential for induction of lipopolysaccharide-specific B cell responses but is indispensable for protection against Salmonella enterica serovar Typhimurium infection. J Immunol. 2009;182:2305–2312. doi: 10.4049/jimmunol.0801980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiersinga WJ, Wieland CW, Roelofs JJ, van der Poll T. MyD88 dependent signaling contributes to protective host defense against Burkholderia pseudomallei. PLoS One. 2008;3:e3494. doi: 10.1371/journal.pone.0003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sotolongo J, Espana C, Echeverry A, Siefker D, Altman N, Zaias J, Santaolalla R, Ruiz J, Schesser K, Adkins B, Fukata M. Host innate recognition of an intestinal bacterial pathogen induces TRIF-dependent protective immunity. J Exp Med. 2011;208:2705–2716. doi: 10.1084/jem.20110547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, Kennedy KA, Poindexter BB, Finer NN, Ehrenkranz RA, Duara S, Sanchez PJ, O'Shea TM, Goldberg RN, Van Meurs KP, Faix RG, Phelps DL, Frantz ID, 3rd, Watterberg KL, Saha S, Das A, Higgins RD N. Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–456. doi: 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wynn JL, Scumpia PO, Delano MJ, O'Malley KA, Ungaro R, Abouhamze A, Moldawer KL. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28:675–683. doi: 10.1097/SHK.0b013e3180556d09. [DOI] [PubMed] [Google Scholar]
- 18.Cuenca AG, Wynn JL, Kelly-Scumpia KM, Scumpia PO, Vila L, Delano MJ, Mathews CE, Wallet SM, Reeves WH, Behrns KE, Nacionales DC, Efron PA, Kunkel SL, Moldawer LL. Critical role for CXCL10 (IP-10)/CXCR3 signaling in the murine neonatal response to sepsis. Infect Immun. 2011 doi: 10.1128/IAI.01291-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delano MJ, Thayer T, Gabrilovich S, Kelly-Scumpia KM, Winfield RD, Scumpia PO, Cuenca AG, Warner E, Wallet SM, Wallet MA, O'Malley KA, Ramphal R, Clare-Salzer M, Efron PA, Mathews CE, Moldawer LL. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J Immunol. 2011;186:195–202. doi: 10.4049/jimmunol.1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delano MJ, Kelly-Scumpia KM, Thayer TC, Winfield RD, Scumpia PO, Cuenca AG, Harrington PB, O'Malley KA, Warner E, Gabrilovich S, Mathews CE, Laface D, Heyworth PG, Ramphal R, Strieter RM, Moldawer LL, Efron PA. Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J Immunol. 2011;187:911–918. doi: 10.4049/jimmunol.1100588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campanella GS, Lee EM, Sun J, Luster AD. CXCR3 and heparin binding sites of the chemokine IP-10 (CXCL10) J Biol Chem. 2003;278:17066–17074. doi: 10.1074/jbc.M212077200. [DOI] [PubMed] [Google Scholar]
- 22.Ranjbaran H, Wang Y, Manes TD, Yakimov AO, Akhtar S, Kluger MS, Pober JS, Tellides G. Heparin displaces interferon-gamma-inducible chemokines (IP-10, I-TAC, and Mig) sequestered in the vasculature and inhibits the transendothelial migration and arterial recruitment of T cells. Circulation. 2006;114:1293–1300. doi: 10.1161/CIRCULATIONAHA.106.631457. [DOI] [PubMed] [Google Scholar]
- 23.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ, Jr C.W.E. i. S. S. s. g. Recombinant human protein. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 24.Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J Immunol. 2005;175:5596–5600. doi: 10.4049/jimmunol.175.9.5596. [DOI] [PubMed] [Google Scholar]
- 25.Peck-Palmer OM, Unsinger J, Chang KC, Davis CG, McDunn JE, Hotchkiss RS. Deletion of MyD88 markedly attenuates sepsis-induced T and B lymphocyte apoptosis but worsens survival. J Leukoc Biol. 2008;83:1009–1018. doi: 10.1189/jlb.0807528. [DOI] [PubMed] [Google Scholar]
- 26.Cuenca AG, Wynn JL, Moldawer LL, Levy O. Role of innate immunity in neonatal infection. American journal of perinatology. 2013;30:105–112. doi: 10.1055/s-0032-1333412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, Chandler NB, Rodesch CK, Albertine KH, Petti CA, Weyrich AS, Zimmerman GA. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood. 2009;113:6419–6427. doi: 10.1182/blood-2008-07-171629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad M, Fleit HB, Golightly MG, La Gamma EF. In vivo effect of recombinant human granulocyte colony-stimulating factor on phagocytic function and oxidative burst activity in septic neutropenic neonates. Biol Neonate. 2004;86:48–54. doi: 10.1159/000077585. [DOI] [PubMed] [Google Scholar]
- 29.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2011;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 31.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 32.Andrade EB, Alves J, Madureira P, Oliveira L, Ribeiro A, Cordeiro-da-Silva A, Correia-Neves M, Trieu-Cuot P, Ferreira P. TLR2-induced IL-10 production impairs neutrophil recruitment to infected tissues during neonatal bacterial sepsis. Journal of immunology. 2013;191:4759–4768. doi: 10.4049/jimmunol.1301752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.