

Abstract
The attenuated yellow fever vaccine (YF-17D) was developed in the 1930’s, yet little is known about the protective mechanisms underlying its efficiency. In this study, we analyzed the relative contribution of cell-mediated and humoral immunity to the vaccine-induced protection in a murine model of YF-17D infection. Using different strains of knock-out mice, we found that CD4+ T cells, B cells and antibodies are required for full clinical protection of vaccinated mice, while CD8+ T cells are dispensable for long-term survival following intracerebral (i.c.) challenge. However, by analysing the immune response inside the infected CNS, we observed an accelerated T-cell influx into the brain following i.c. challenge of vaccinated mice, and this T-cell recruitment correlated with improved virus control in the brain. Using mice deficient in B cells we found that, in the absence of antibodies, YF vaccination can still induce some antiviral protection, and in vivo depletion of CD8+ T cells from these animals revealed a pivotal role for CD8+ T cells in controlling virus replication in the absence of a humoral response. Finally, we demonstrated that effector CD8+ T cells also contribute to viral control in the presence of circulating YF-specific antibodies. To our knowledge this is the first time that YF-specific CD8+ T cells have been demonstrated to possess antiviral activity in vivo.
Introduction
The yellow fever (YF) vaccine, based on the live-attenuated YF-17D virus, is one of the most effective vaccines ever made (1), and in the 80 years that have passed since its establishment it has been administered to over 600 million people globally. It was developed in the 1930’s by Max Theiler and associates, who experimentally attenuated the wild type (wt) Asibi strain of YF virus by more than 200 serial tissue culture passages through monkey, mouse embryonic tissue and chicken embryonic tissue (2,3). Vaccination with YF-17D virus results in an acute viral infection during which there is a transient viral replication that peaks approximately 5–7 days after virus inoculation, and subsequently dissipates. A single immunization is known to protect against infection in more than 90% of vaccinees (1,3), and neutralizing antibodies are thought to be the primary correlate of protection against infection with wt YF virus (4). However, YF-17D virus has also been demonstrated to be a potent inducer of cytotoxic T cell responses (5,6), suggesting a potential role also for cell-mediated immunity in the control of the natural infection.
The last decade has seen a growing interest in the YF vaccine because of its live viral nature, which offers the possibility to study the immune response to an acute viral infection in humans, and for its emerging potential as a recombinant vaccine vector (7–10). Moreover, the re-emergence of YF in some areas of the world in the last 20 years has contributed in bringing the YF-17D vaccine back to the attention of the scientific community.
An interesting feature of the YF-17D virus is its interaction with human DCs; a recent study has shown its ability to activate several DC subsets - such as myeloid and plasmacytoid DCs through engagement of TLR2, TLR7, TLR8 and TLR9, resulting in the production of a mixed Th1/Th2 cytokine profile (11). Moreover, Barba-Spaeth et al. demonstrated direct infection of both immature and mature DCs by YF-17D virus, leading to presentation of endogenous antigen and consequent CD8+ T cell activation; something that has been proposed as a mechanism contributing to the strong and long lasting immunity elicited by vaccination (12).
A number of studies have described in details the development of the human T cell response following vaccination with YF-17D virus, and characterized the phenotypical changes occurring during the transition from the effector to the memory phase (6,13,14). However, there is still very little known about the contribution of the virus-induced T cell response to the establishment and maintenance of protection from yellow fever infection. This is due, at least in part, to the intrinsic limitations of studying immune responses in humans, where only some features of the host response following vaccination can be analyzed. In this context, a small animal model may prove to be a valuable tool to examine in much greater detail the functional role of the different arms of the immune system in YF-17D induced immunity.
In this report, we describe a mouse model for infection with YF-17D virus and characterize the effector mechanisms underlying vaccine-induced protection in vivo. Most important, we show that, even though humoral immunity represents the principal effector arm of the adaptive immune response when it comes to preventing a lethal outcome of YF infection, effector CD8+ T cells also significantly contribute to viral control in the brain, which is the decisive site for virus replication in this model system. Collectively, our data demonstrate for the first time that CD8+ T cells may play an important role in controlling infection with YF virus.
Materials and Methods
Mice
Female C57BL/6 (wt B6) mice as well as MHC class I (β2m−/−) and MHC class II (Aβ−/−) deficient mice on a B6 background were obtained from Taconic Farms. B cell deficient (μMT) mice were either obtained directly from The Jackson Laboratory (Bar Harbor, ME) or the local progeny of such animals. Perforin (Prf) deficient and IFN-γ deficient mice were originally obtained from The Jackson Laboratory, and IFN-γ/Perforin double-deficient (IFN-γ/Prf) mice were generated locally through intercrossing of these strains, as previously described (15). Rag 1 deficient mice (B6.129S7-Rag1tm1Mom/J), CD40L deficient mice (B6.129P2-Cd40tm1Kik/J) and CXCR5 deficient mice (B6.129S2(Cg)-Cxcr5tm1Lipp/J) came directly from the Jackson Laboratory. All mice used in this study were 7 to10 weeks old and housed in a specific pathogen–free facility. All experiments were approved by the national animal ethics committee and performed in accordance with national guidelines.
Virus preparation and quantitation
YF-17D virus (Stamaril, Sanofi Pasteur; reconstituted as recommended by the manufacturer) was propagated in Vero cells (ATCC CCL-81) grown in DMEM containing 10 % FCS, glutamine and antibiotics (penicillin and streptomycin). Cell monolayers were seeded 24 hours earlier and infected with virus at a multiplicity of infection (MOI) of 0.001 in DMEM 2% FCS; infectious supernatants were harvested when cytopathic effect was more than 60% (day 6 p.i ), freeze-thawed and clarified of cell debris at 2000 rpm for 15 min at 4°C. Viral stocks were stored as single use aliquots at −80°C. An immune focus assay (IFA) was developed to quantitate infectious virus. In brief, virus stocks were serially diluted (10-fold) in DMEM and adsorbed for 1hour at 37°C onto Vero cell monolayers in 24-well plates; cells were then overlaid with media containing 0.9% methyl cellulose and incubated at 37°C for 3 days. Following fixation and permeabilization, a monoclonal antibody made against NS3 from YF-17D virus was used for detection of virus antigens within infected cells; foci of infection (analogous to plaques) were visualized using a horse radish peroxidase (HRP)-conjugated secondary antibody and counted. When virus titers in the brain were determined, the IFA was performed on homogenized and clarified 10% organ suspensions. The detection limit for virus in organs was 200 PFU/g organ.
Neutralization of YF-17D virus by sera from vaccinated mice
Appropriately diluted YF-17D virus (resulting in about 70–100 pfu per well) was incubated for 1 hour at 37°C with serial (2-fold) dilutions of mouse sera obtained from individual YF-vaccinated mice. The virus-serum mixture was subsequently added to Vero cells monolayers in 24-well plates and incubated for 2 hours at 37°C, after which the overlay media containing 0.9% methyl cellulose was added. After 3 days of incubation at 37°C, the overlay media was removed and, following fixation and permeabilization, cells were stained as described above (IFA). Following counting, the neutralizing antibody titer, defined as the highest serum dilution neutralizing more than 50% of the viral plaques, was determined.
Vaccination and challenge
Mice were vaccinated by either intravenous (i.v.) or subcutaneous (s.c.) injection of YF-17D virus using a dose of 105 pfu in 300 μl. For virus challenge, deeply anesthetized mice were injected intracerebrally (i.c.) with 104 pfu of YF-17D virus in a volume of 30 μl. In clinical protection studies, the mice were checked at least twice a day for two weeks following i.c. challenge and euthanized when irreversible signs of illness (body weight drop, ruffled fur, twitching) together with neurologic signs (hind leg paralysis and failure to get into an upright position within 10 seconds after being tilted) were observed. Mice developing sickness within the first 4 days post challenge were excluded from the experiments.
Serum transfer and adoptive transfer of splenocytes
Serum was harvested from C57BL/6 mice vaccinated with 105 pfu of YF-17D virus i.v. 28 days earlier, and each recipient received a dose of 1 ml serum, equally distributed between the i.v. and the intraperitoneal (i.p.) route on day −3 and additionally 0.3 ml i.v. on day −1 relative to virus challenge. Normal mouse serum was used for control.
For splenocyte preparation, spleens were collected from donor mice primed with 105 pfu of YF-17D 28 days earlier and transferred to Hanks balanced salt solution (HBSS). A single-cell suspension was obtained by gently pressing the spleens through a nylon mesh (mesh size 70 μm), followed by centrifugation and two washes in HBSS after which 7×107 cells were injected into recipient mice i.v.; YF-immune splenocytes (or naïve splenocytes ) were transferred to recipient animals 2 days prior to i.c. challenge.
Flow cytometry
Single cell suspensions of splenocytes were prepared as mentioned above. Frequencies of antigen-specific CD8+ T cells in the spleen were determined by intracellular cytokine staining performed after 5 hours of incubation with relevant peptides (0.1 μg/ml of NS3(268-275) or E(4-12) (16)) in the presence of monensin (3 μM) at 37°C in 5% CO2. After incubation, cells were stained with antibodies for cell surface markers using peridinin chlorophyll protein-Cy5.5-CD8, and APC-Cy7-CD44. Subsequently the cells were washed, permeabilzed, and stained for intracellular cytokine using APC-IFN-γ.
Infiltrating mononuclear cells were extracted from the CNS as previously described (17). In brief, brains were removed from deeply anesthetized mice following intracardial perfusion with 20 ml of ice-cold PBS, and single-cell suspensions were prepared by gently pressing the organs through a nylon mesh (mesh size 70 μm). After centrifugation in 37% Percoll (Sigma-Aldrich), the myelin layer was removed and cells were resuspended in RPMI containing 5% FCS, 2-Mercaptoethanol and antibiotics (penicillin and streptomycin). Following centrifugation, cells were resuspended in buffer containing rat serum and stained for cell surface markers using FITC-CD45.2, PE-CD3, peridinin chlorophyll protein-Cy5.5-CD8, PE-Cy7-CD4 and APC-Cy7-CD44. Frequencies of YF specific CD8+ T cells among CNS derived mononuclear cells were determined by tetramer staining using NS3(268-275) or E(4-12) PE and APC-conjugated tetramers in combination with Abs for PerCP-Cy5.5-CD8 and FITC-CD44; LCMV-specific tetramers was used to define the background. Samples were run on an LSRII flow cytometer (BD biosciences) and analyzed using FlowJo software (TreeStar).
Immunohistochemistry for CD4+ and CD8+ T cells
Brains were removed from deeply anesthetized and exsanguinated mice, frozen in liquid nitrogen and sectioned in a cryostat. Parallel brain sections (20 μm thick) were fixed 45 min in 4% paraformaldehyde for CD4 immunostaining or 4 min in 4% phosphate buffered formaline, pH 7.4, followed by 2 min in 50%, 100%, and 50% acetone, respectively, and finally air dried for CD8a immunostaining. Sections were then rinsed in 0.5 % triton X-100 diluted in 0.05M trizma base buffered saline (TBS+T), pH 7.4, and incubated with 10% fetal calf serum (FBS) in TBS+T before incubation overnight at 4°C with purified rat-anti-mouse CD4 IgG2a (2.5 μg/mL) (553647, BD Pharmingen) or purified rat-anti-mouse CD8a IgGa (7 μg/mL) (MCA1108GT, AbDSerotec) diluted in 10% FCS in TBS+T. Parallel sections were incubated with rat IgG2a isotype control (2.5–7 μg/mL) (BD Pharmingen). After a rinse in TBS+T, endogenous peroxidase activity was blocked with 1.9% H2O2 in TBS, and sections were rinsed in TBS+T before and after incubation with biotinylated goat anti-rat IgG antibody (5 μg/mL) (BA9400, Vector Laboratories Inc) and HRP-conjugated streptavidin (1:300) (RPN1231V, GE Healthcare), respectively, diluted in 10% FCS in TBS+T. After a final rinse in TBS+T, the colour signal was developed in 0.05% 3.30-diaminobenzidine tetrahydrochloride and 0.003% H2O2 in TBS. Finally, sections were coverslipped with Depex.
Isolation of total RNA for quantitative PCR (Q-PCR)
Brains from deeply anesthetized and exsanguinated mice were immediately removed, snap-frozen in liquid nitrogen, and stored at −80°C. Total RNA was extracted from the homogenized brains using the RNeasy midi kit (Qiagen).
Detection of mRNA in the brain by Q-PCR
One microgram of RNA was reverse transcribed to cDNA using RevertAid First strand cDNA synthesis kit (Fermentas, Thermo Scientific). For the qPCR reaction, a Brilliant II SYBR Green qPCR Mastermix was used according to the manufacturer’s instructions (Stratagene, AH diagnostics). The qPCR components included Brilliant II qPCR Mastermix, milliQ water, reverse transcribed cDNA, and the forward and reverse target gene primers (see table I for primer sequences). Target gene expression was normalized to expression of the reference gene PBGD (porphobilinogen). The Mx3005P Real-time qPCR instrument was used with the following program: Initial denaturation at 95°C (10 min), 40 cycles of denaturation at 95°C (30 sec), annealing at 60°C (60 sec), and extension at 72°C (60 sec). Each reaction was run in triplicate plus a no template control and a no RT control. Results were analyzed using Mx3005P system software. The relative expression ratio (R) in each sample was calculated using a mathematical model based on the amplification efficiency (18):
Table I.
Primer sequences for qPCR
| Gene | Forward primer 5′ → 3′ | Reverse primer 3′ → 5′ |
|---|---|---|
| CD8 | GTATCATGAATGTGAAGCCA | GAAGGACATCAACCACAGTC |
| CD4 | TTAATTAGAGGAGGTTCGCC | CCTGTTCTCCAGCTCACA |
| CD19 | GGAGGATAGTGGGGAGATG | TACAGCTGGGAACCAGAAG |
| CXCL10 | CGATGACGGGCCAGTGAGAATG | TCAACACGTGGGCAGGATAGGCT |
| IFN-γ | CGTCATTGAATCACACCTG | GGTTGTTGACCTCAAACTTG |
| PBGD | GTGAGTGTGTTGCACGATC | GGGTCATCTTCTGGACCAT |
An amplification efficiency (E) of 100% corresponds to a doubling of the PCR product for every cycle. E is calculated from the slope of a standard curve based on 5-fold dilutions for each primer pair used (E = 10(−1/slope)). Thus, Etarget corresponds to the target gene primers, and Ereference corresponds to the reference gene primers. Throughout this study, brains from YF-immunized C57BL/6 mice infected i.c. with YF-17D virus 7 days before removal were used as standard curve templates. ΔCP (control-sample) refers to the difference in threshold cycle (Ct) between day 7 p.i. (control) and 3, 5 or 7 p.i. (sample). Ct reflects the number of cycles it takes to reach a point in which the fluorescent signal is above the background fluorescence (18).
In vivo depletion of T cells
A combination of two monoclonal antibodies (YTS 169 and YTS 156) was used for in vivo depletion of CD8+ T cells from vaccinated mice prior to challenge, while the monoclonal antibody YTS 191was used for depletion of CD4+ T cells; hybridomas producing these monoclonals were kindly provided by S. Cobbold, Sir Williams Dunn School of Pathology, Oxford, UK (19). Mice to be depleted were injected i.p. with 100 μg of antibody one day prior to i.c challenge and at days 1 and 4 post challenge. The efficiency of the depletion was confirmed by flow cytometric analysis at day 7 post challenge.
Statistical evaluation
A nonparametric Mann-Whitney U test was used to compare quantitative data (*p < 0.05; **p < 0.01; ***p< 0.001; ****p<0.0001 unless otherwise stated). GraphPad Prism (version 6) software was used for statistical analysis.
Results
B cells are pivotal in the adaptive immune response to YF-17D vaccination, but other effector mechanisms mediate partial protection in B cell deficient mice
Mice are not susceptible to peripheral infection with YF-17D. However, it is possible to induce a lethal CNS disease by i.c. inoculation of the virus (20–22). In agreement with previous studies, we found that infection of wt B6 mice with 105 pfu of YF-17D virus via the i.v. route or the s.c. route results in sub-clinical infection. In a preliminary evaluation of acute i.c. challenge of wt B6 mice with 104 pfu of YF-17D virus, we noted initial signs of illness around day 6–7 p.c., progressing to severe lethal disease by day 8–9 p.c. in all the tested animals, demonstrating the stringency of this challenge model. Notably, when B6 mice were vaccinated with 105 pfu of YF-17D virus i.v. or s.c. 4 weeks prior to i.c. challenge, 100% protection was observed, demonstrating that YF-17D virus acts as a protective vaccine in mice as well as in humans (Fig. 1A). This allowed us to carry out vaccination-challenge experiments with the aim to investigate the requirement for specific cell subsets and effector molecules in mounting a protective immune response following YF-17D vaccination in mice.
Figure 1. Adaptive immune response to infection with YF-17D virus: role of B cells in protection.

A) Percent survival after i.v. infection, i.c. infection, and i.c infection 28 days after i.v vaccination of WT B6 mice. (n=10 in all groups). B) Percent survival after i.v. infection, i.c. infection, and i.c infection 28 days after i.v. vaccination of Rag 1 KO mice (x: n= 10; △: n=5; □: n=10). C) Percent survival after i.v. infection, i.c. infection, and i.c. infection 28 days after i.v. vaccination of B cell KO mice. (x: n= 35; △: n=5; □: n=35). D) Analysis of splenic CD8+ T cell responses of vaccinated WT and B cell KO mice at day 15 post i.c. challenge; representative plots (gated on CD8+ cells) showing the percentages of CD44+IFN-γ+ cells after peptide stimulation with NS3(268-275) and E(4-12); non-stimulated samples serve as controls.
To this end, we tested several strains of genetically modified mice with targeted immune defects. A complete vaccine failure (i.e. no survival when i.v. vaccinated animals were subsequently challenged i.c.) was observed in Rag1 deficient mice (Fig. 1B). This demonstrates not only the requirement for T cells and/or B cells in the vaccine-induced protection, but also suggests that, despite its attenuation, YF-17D is likely to be a cytolytic virus causing direct cell damage, although activation of innate immune mechanisms cannot be ruled out. Interestingly, even in the absence of B and T cells (Rag1 KO), primary i.v. infection with YF-17D was still sub-clinical, which given the apparently cytolytic nature and replicative capacity of YF-17D virus, strongly support the idea that virus control and clearance following peripheral infection/vaccination can be accomplished entirely by innate host mechanisms (23,24).
When analyzing μMT mice lacking B cells, we found that, although B cell deficiency resulted in a high mortality, some vaccinated μMT mice could still mount a protective immune response and survive long term after i.c. viral challenge (23% survival) (Fig. 1C). Given the pivotal role of effector CD8+ T cells in the control of many other acute murine and human viral infections, we speculated that the protection observed in the absence of antibody responses might be mediated by this cell subset.
Some years back, Van der Most et al. have identified two YF virus-derived, MHC class I-restricted T cell epitopes in B6 mice: a dominant H-2Kb-restricted epitope mapped to the NS3 protein, and an H-2Db-restricted subdominant epitope found in the viral E protein (16). Consequently, we studied the CD8+ T cell responses specific for these two murine epitopes in the spleen of B cell deficient mice that had survived an i.c. viral challenge, and compared our findings to those in surviving wt B6 mice. Notably, B cell deficient mice displayed an increased YF specific CD8+ T cell response compared to matched wt mice, as demonstrated by a higher frequency of IFN-γ producing CD44+ T cells responding to stimulation with both peptides (6.35% vs 2.16% for the NS3 epitope; 6.1% vs 1.69% for the E epitope) at day 15 post challenge (p.c.) (Fig. 1D). This was not due to an intrinsically increased capacity of B cell deficient mice to generate an augmented CD8+ T cell response, as we have previously observed that primary epitope-specific CD8+ T cell responses in these mice tend to be of a lesser magnitude compared to those mounted in wt B6 mice ((25,26) and data not shown). These findings suggest that B cells are crucial for the YF induced immune response in wt mice; however, in the absence of B cells and/or antibodies, other effector mechanisms, likely involving CD8+ T cells, may mediate a partial protection.
CD8+ T cell responses appear dispensable for long term survival in vivo
The importance of T cells in the immune response to viral infections is well documented; in particular, CD8+ T cells synthesize a variety of effector molecules that may contribute to resistance against intracellular pathogens. Perforin (prf) is a key effector molecule for CD8+ T cell-mediated cytolysis of virus-infected cells; effector CD8+ T cells can also release IFN-γ, among other cytokines, which can inhibit viral replication directly and can act as an immunomodulatory and immunostimulatory entity.
As a first approach to study the contribution of CD8+ T cell responses in the YF infection model, we tested MHC class I deficient mice (β2 microglobulin−/−) for their ability to mount a protective immune response following vaccination. We found that absence of CD8+ T cells did not impair vaccine-induced protection; thus, we observed no mortality after i.c. challenge of previously vaccinated MHC I KO mice (Table II). Next, we analyzed vaccine-induced protection in mice with targeted deficiencies in IFN-γ and prf; similarly to MHC I deficient mice, both IFN-γ KO mice and prf KO mice displayed 100% survival following vaccination (Table II). Only the combined deficiency of these molecules (IFN-γ/prf double KO) resulted in a very modest impairment in vaccine-induced protection (20% mortality) (Table II). These data suggest that, in otherwise immunologically intact mice, CD8+ T cell responses are not essential for clinical protection following vaccination with YF-17D in mice.
Table II.
Role of CD8+ T cells in YF-17D virus-induced protectiona
| Strain | i.v.% mortality (dead/total)b | i.c.% mortality (dead/total)c | i.v. prior to i.c.% mortality (dead/total)d |
|---|---|---|---|
| WT (B6) | 0% (0/10) | 100% (10/10) | 0% (0/10) |
| IFN-γ KO | 0% (0/5) | 100% (5/5) | 0% (0/5) |
| Prf KO | 0% (0/5) | 100% (5/5) | 0% (0/5) |
| IFN-γ/Prf KO | 0% (0/15) | 100% (5/5) | 20% (3/15) |
| MHC-I KO | 0% (0/15) | N.D. | 0% (0/15) |
Percent mortality of WT B6 mice, IFN-γ KO mice, perforin (Prf) KO mice, IFN-γ and Prf double KO mice and MHC class I KO mice after infection with YF-17D virus via different routes (N.D.= not determined).
Mice were infected with 105 pfu of YF-17D virus intravenously (i.v.).
Mice were infected with 104 pfu of YF-17D virus intracerebrally (i.c.).
Mice were infected i.v. with 105 pfu of YF-17D virus and 28 days later challenged i.c. with 104 pfu of YF-17D virus.
CD4 T cells and CD40L are required for protection against a lethal outcome of YF-17D i.c. infection in previously vaccinated mice
CD4+ T cells can contribute to the immune response to a viral infection in very different ways. Antigen-specific CD4+ T cells can migrate to sites of infection and carry out direct effector functions, or they can persist in secondary lymphoid organs and provide help for expansion and differentiation of other effector cells. The role of Th cells in the formation of high-affinity neutralizing antibody responses by B cells is well established, and follicular helper CD4+ T cells (Tfh) have been identified as the key CD4+ subset that mediates this function and promotes germinal center reactions (27). Expression of CXCR5 is one of the defining hallmarks of T follicular helper cells (28). CD40–CD40L interaction is also an essential signal for B cell activation, germinal center formation and differentiation and isotype switching to IgG molecules. Having established a critical role for B cell responses in YF-17D-induced resistance to lethal outcome of i.c. challenge, we wanted to investigate the requirement for CD4+ T cell help in the observed protection.
Using mice deficient in MHC class II, we could demonstrate that CD4+ T cells were critical for the vaccine-induced protective response, as none of the vaccinated mice survived subsequent viral challenge (100% mortality) (Table III). Similarly, lack of CD40L expression completely abolished vaccine-induced protection (Table III), suggesting that production of IgG is essential for long-term protection. We also sought to determine whether CXCR5+ T follicular helper cells would be critical for promoting YF-specific B cell responses. Surprisingly, lack of CXCR5 only very modestly reduced the overall survival following i.c. challenge of YF-vaccinated mice; mortality in CXCR5 KO mice was only 15% (Table III). Assuming a key role for antibodies in vaccine-induced protection, this finding suggests that sufficient neutralizing antibodies may be produced even without substantial germinal center formation (29).
Table III.
CD4+ T cell contribution to YF-17D virus-induced protectiona
| Strain | i.v.% mortality (dead/total)b | i.c.% mortality (dead/total)c | i.v. prior to i.c.% mortality (dead/total)d |
|---|---|---|---|
| WT (B6) | 0% (0/15) | 100% (5/5) | 0% (0/15) |
| MHC-II KO | 0% (0/15) | 100% (5/5) | 100% (15/15) |
| CD40L KO | 0% (0/20) | N.D. | 100% (20/20) |
| CXCR5 KO | 0% (0/20) | N.D. | 15% (3/20) |
Percent mortality of WT B6 mice, MHC-II KO mice, CD40L KO mice and CXCR5 KO mice after infection with YF-17D virus via different routes (N.D.= not determined).
Mice were infected with 105 pfu of YF-17D virus intravenously (i.v.).
Mice were infected with 104 pfu of YF-17D virus intracerebrally (i.c.).
Mice were infected i.v. with 105 pfu of YF-17D virus and 28 days later challenged i.c. with 104 pfu of YF-17D virus.
Transfer of YF immune serum or YF-primed splenocytes confers protection from YF-17D challenge
The strikingly impaired protective immunity of YF-vaccinated B cell KO mice, CD40L KO mice and MHC class II KO mice, pointed towards a major role for humoral immunity in the vaccine induced protective response. However, these findings did not unequivocally prove that antibodies were the major effector molecules in mediating survival, as all of the above KO strains have been previously characterized as carrying defects in CD8+ T cells antiviral immunity as well (30–33). Consequently, to confirm the role of antibodies in protection, we wanted to test whether serum from immunized mice contained neutralizing antibodies and whether we could passively transfer immunity to subsequent i.c. challenge through the transfusion of YF immune serum.
Analyzing sera from vaccinated mice, we found high levels of neutralizing antibodies in serum of wt mice 28 days after vaccination, while no neutralizing activity was observed in serum from naive animals or from similarly vaccinated B cell deficient mice (Fig. 2). Consequently, donor wt B6 mice were vaccinated i.v. with 105 pfu of YF-17D virus, and immune serum was harvested 28 days post vaccination and transfused into naïve recipients by i.v. and i.p. injection. Pooled serum obtained from naïve mice was also transferred to a group of recipients as a control. Recipient mice were challenged with 104 pfu YF-17D virus i.c. 3 days after serum transfer (Fig. 3A). Transfusion of YF immune serum resulted in 100% protection from virus challenge, while no protection was observed in groups of mice that had been sham injected (PBS) or transfused with serum obtained from naïve animals (Fig. 3A). Thus, these results confirmed a pivotal role for antibodies in the immunity to YF.
Figure 2. Neutralizing antibody titers in YF-17D vaccinated mice.
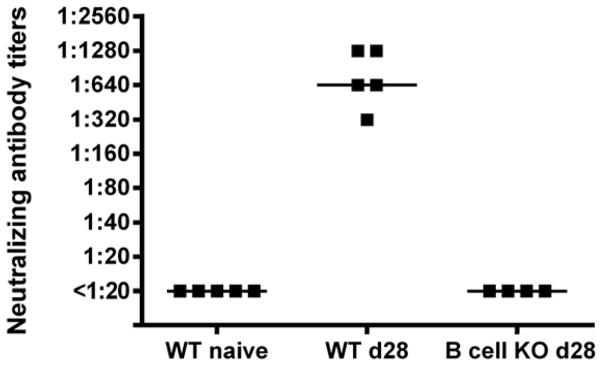
WT B6 mice were vaccinated with YF-17D virus and the serum neutralizing antibody titers were measured at day 28 post vaccination. No neutralization was observed with serum from naïve WT B6 mice or from YF-17D vaccinated B cell KO mice at day 28 post vaccination. Each dot represents an individual animal.
Figure 3. Transfer of YF-immune serum or YF-primed splenocytes to naïve mice confers protection towards viral challenge.

A) Percent survival of mice transfused with YF-immune serum (□), naive serum (○) or PBS (x) 3 days (0.5 ml i.v. and 0.5 ml i.p.) and one day (0.3 ml i.v.) prior to i.c. viral challenge; □: n=6; ○: n=5; x: n=5. Experiment was performed twice with similar results. B) Percent survival of mice adoptively transferred with 7×107 YF-17D-primed splenocytes (□), naïve splenocytes (○) or of mice receiving PBS (x) 2 days prior to i.c. challenge; □: n=10; ○: n=10; x: n=5. Experiment was performed twice with similar results.
The finding that transfer of antibodies sufficed for protection in the YF model does not necessarily exclude a potential contribution of cell mediated immunity. To address this issue, we performed adoptive transfer experiments in which YF-primed splenocytes were transferred to naïve recipients prior to i.c. challenge. Spleen cells were removed from vaccinated donor mice at day 28 p.i. and injected i.v. into naïve recipients (7 × 107/mouse); recipients were then challenged with 104 pfu of YF-17D virus i.c. (Fig. 3B). Interestingly, we observed that transfer of primed splenocytes resulted in 70% protection from i.c. challenge, while no protection was observed in the groups that received either a sham injection (PBS) or naïve splenocytes (Fig. 3B). Together with earlier observations in vaccinated B-cell deficient mice, which did not all succumb to i.c. challenge, these data could point to a role for cellular immunity in the protective immune response to YF-17D vaccine in mice.
Viral replication in the brains of non-immunized and immunized wt B6 mice following i.c. challenge
Since only i.c. infected animals die from YF infection, virus replication in the brain appear to be essential, and for that reason we wanted to study virus titers in the brains of wt B6 mice at different time points after i.c. inoculation. This was done to address two issues: i) whether the brain was a site of active viral replication following i.c. injection and if so, what was its time course; ii) whether prior vaccination would lead to virus control and clearance, as suggested from the lack of clinical disease in previously vaccinated animals. To this end, brains of naive mice and mice immunized 28 days prior to challenge were removed at day 3, 5 and 7 post i.c. challenge (Fig. 4); day 7 represents the latest time point for an unbiased analysis of acutely challenged mice that would invariably succumb between day 8 and 9 p.c. In the unvaccinated group, increasing virus titers were observed over time, with all the animals uniformly displaying high viral loads (106–107 pfu/g of organ) on day 7 p.c. (Fig. 4). In contrast, only transient replication was observed in vaccinated mice, and infectious virus was undetectable in the CNS by day 7 p.c. (Fig. 4). These results indicated that the brain is a site of active viral replication following i.c. challenge with YF-17D virus, and that vaccination leads to improved virus control in the brain. Notably, the presence of transient low-grade viral replication in the vaccinated group suggests that the i.c. viral inoculum is not completely neutralized by pre-existing YF-specific antibodies. This observation is suggestive of a more complex mechanism of protection than the mere presence of YF-neutralizing antibodies in the brain, requiring effectors capable of controlling virus in infected host cells.
Figure 4. YF-17D vaccinated mice clear virus infection from the CNS by day 7 post challenge.

WT B6 mice that were either naïve or had been immunized s.c. with 105 pfu of YF-17D virus 28 days earlier were challenged i.c. with 104 pfu of YF-17D virus. Viral loads in the brains at days 3, 5 and 7 post challenge are compared for unvaccinated (open squares) and YF-17D vaccinated (black squares) mice. Each dot represents an individual animal. The detection limit for virus in organs was 200 pfu/g organ.
Detection of T cells in virus-infected brains: vaccination induces an accelerated recruitment of T cells into the CNS following i.c. challenge
Infiltration of CD4+ and CD8+ T cells into the CNS has been observed in several models of flavivirus-mediated encephalitis in rodents (34–36). Furthermore, CD19+ B cells can also traffic to the virus infected CNS, where both IgM producing extrafollicular plasmablasts and IgG producing germinal center plasma cells can be found (37,38).
To test the hypothesis that i.c. infection of mice with YF virus would result in T cell influx into the brain, we first performed a comparative histological analysis of sham inoculated mice and mice infected i.c. with YF virus 7 days earlier (Fig. 5). As expected, cerebral injection of PBS did not result in migration of CD4+ or CD8+ cells into the brain (Fig. 5A & F), except at the injection site (not shown). In contrast, multiple CD4+ cells were observed in the meningeal membrane (Fig. 5B & C), the vascular and perivascular spaces (Fig. 5B & D) at day 7 post YF infection. CD4+ cells were also abundant in the brain parenchyma as shown for the hippocampal formation (Fig. 5B) and neocortex (Fig. 5B & E). Similarly, CD8+ cells had also infiltrated the meningeal membrane (Fig. 5G & H), corpus callosum (Fig. 5G & I), the external capsule (Fig. 5I), and fimbria (Fig. 5I & J) at day 7 post YF infection. The presence of both CD4+ and CD8+ T cells in the YF infected brain was confirmed by flow cytometric analysis of mononuclear cells isolated from the perfused brains of d.7 infected mice (Fig. 6).
Figure 5. Immunohistochemical analysis of CD4+ and CD8+ T cells into YF-17D infected brains.

A–E) Immunohistochemical analysis of CD4 showed absence of CD4+ cells in brains of PBS injected mice (A), while CD4+ cells had migrated into the brains of YF-17D i.c. infected mice day 7 post injection (B–E). F–J) Similarly, immunohistochemical analysis of CD8 showed absence of CD8+ cells in brains of PBS injected mice (F), while CD8+ cells had migrated into the brains of YF-17D i.c. infected mice day 7 post injection (G–J). vCA1-3: cornu ammonis 1 and 3; cc: corpus callosum; Ctx: cortex; DG: dentate gyrus; v: vessel; MM: meningeal membrane; fi: fimbria. Scale bar: (A, B, F, G) 400 μm, (C–E, H–J) 50 μm. Sections stained with control abs are depicted in a supplementary figure (Suppl. Fig. S1).
Figure 6. Infiltration of T cells into YF-17D infected brains detected by flow cytometry.

Lymphocytes were isolated from brains of naïve mice and of YF-17D i.c. infected WT mice at day 7 p.i.; FACS analysis revealed the presence of CD4+ and CD8+ cells within the infiltrated population. Dot plots, gated on FSC/SSC as indicated, are representative of cells pooled from 3 mice. All CD4+ and CD8+ cells were confirmed to be CD3+CD45highCD44high (not shown).
To address the kinetics of lymphocyte infiltration and the role of prior immunization in this context, we used Q-PCR-based analysis of the CD4, CD8 and CD19 mRNA expression levels in the YF-infected brains (Fig. 7). Wt B6 mice that were either naïve or had previously received YF immunization were challenged i.c. with YF-17D virus, and on days 3, 5, and 7 p.c., brains were collected for analysis; mice receiving an i.c. injection with PBS served as controls. We observed a significantly higher expression of CD8 mRNA at days 3 and 5 p.c. in the brains of previously immunized mice compared to unvaccinated animals, while expression levels became similar in the two groups by day 7 p.c. (Fig. 7). The expression of mRNA for CD4 also significantly increased as the infection progressed, but no statistically significant difference between vaccinated and unvaccinated mice was observed at any time point. In contrast, within the time frame studied here, we found no evidence of B cell infiltration into the YF-infected CNS, as CD19 mRNA expression levels in both vaccinated and unvaccinated animals was similar to baseline as observed in the control group (data not shown).
Figure 7. Expression levels of CD8 and CD4 mRNA in YF-17D infected brains.
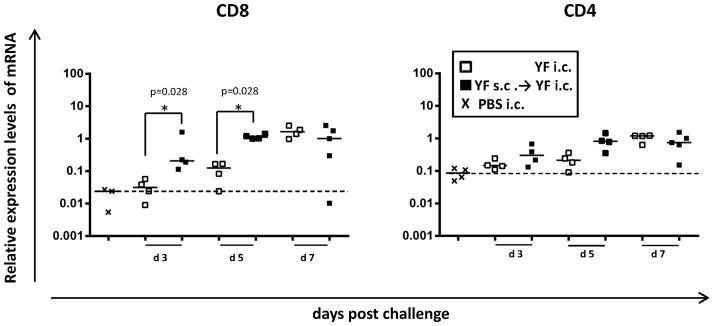
mRNA expression levels in the brains of vaccinated (black squares) and non-vaccinated (open squares) WT B6 mice were compared at days 3, 5 and 7 post i.c. challenge. The mRNA level in the brain of an immunized mouse at day 7 post challenge is set to R = 1. mRNA levels in the brains of mice that had received a sham (PBS) i.c. injection (x) was used to determine the baseline mRNA expression for each molecule (dashed lines). Each dot represents an individual animal.
Finally, to confirm that prior vaccination causes an accelerated recruitment of T cells to the CNS including antigen-specific CD8+ T cells, we studied the numbers and composition of mononuclear cells isolated from the perfused brains of day 5 infected mice (Fig. 8). As expected, our results revealed increased numbers of both CD4+ and CD8+ T cells in the CNS of vaccinated mice, and the infiltrating CD8+ T cells contained large numbers of YF-specific CD8+ T cells (Fig. 8).
Figure 8. Composition of mononuclear cells isolated from YF-17D infected brains.

WT B6 mice that were either naïve or had been immunized s.c. with 105 pfu of YF-17D virus 28 days earlier were challenged with 104 pfu of YF-17D virus i.c., and 5 days later cells were isolated from the brains; naïve mice served as controls. Absolute numbers of mononuclear cells, CD4+, CD8+ and YF specific CD8+ cells are depicted. Antigen-specific CD8+ T cells were detected by tetramer staining using NS3(268-275) or E(4-12) PE and APC-conjugated tetramers (tet); LCMV specific PE and APC-conjugated tetramers were used as controls (Ctrl tet). Each dot represents pooled cells from 2 mice.
YF-17D-induced protection in B cell KO mice is mediated by CD8+ T cells
The relative contribution of T cells to the YF-17D-induced protective response may be concealed by the presence of YF-specific antibodies in vaccinated wt B6 mice. Therefore, we decided first to compare the vaccine induced virus control in brains of immunized B cell deficient mice that were/were not in vivo depleted of CD8+ or of CD4+ T cells prior to i.c. viral challenge; non-immunized animals were included for control (Fig. 9). Mice were vaccinated s.c. with 105 pfu of YF-17D virus and challenged i.c. 28 days post priming; one day prior to challenge, the animals to be depleted were injected i.p. with anti-CD8 or anti-CD4 monoclonal antibodies, and this treatment was repeated on days 1 and 4 p.c. At day 7 p.c., mice were euthanized, and virus titers in the brains were determined. Intact vaccinated mice displayed a significantly lower viral load compared to unvaccinated mice (Fig. 9). Interestingly, we also noticed that the vaccinated group contained animals displaying both low and high viral loads; this is consistent with our previous finding that vaccination of B cell deficient mice resulted in survival of about 25% of the animals tested (Fig. 1C). Strikingly, depletion of CD8+ T cells from immunized B cell deficient mice uniformly abolished the vaccine induced virus control, as viral loads in the CNS of these animals were similar to those of unvaccinated mice (Fig. 9). On the other hand, when CD4+ T cells were depleted, vaccinated B cell deficient mice were still able to control the infection, as viral titers in the brains of these animals were significantly lower than those observed in unvaccinated animals and in vaccinated/CD8-depleted mice (Fig. 9). However, it should also be noted that none of the CD4-depleted mice matched the most efficient virus controllers found in the group of un-depleted animals, suggesting that in the absence of antibodies not only CD8+ T cells, but also CD4+ T cells – albeit to a lesser extent - might contribute in controlling the virus spread in the CNS.
Figure 9. CD8+ T cells mediate protection in YF-17D vaccinated B cell KO mice.

Viral titers in the brains of unvaccinated (open squares) and vaccinated (black squares) B cell KO mice at day 7 post challenge. Some vaccinated animals were in vivo depleted of CD8+ (α-CD8) or CD4+ (α-CD4) T cells 1 day prior to i.c. challenge, and again at days 1 and 4 post challenge. Each dot represents an individual animal. The detection limit for virus in organs was 200 pfu/g organ.
In vivo depletion of CD8+ T cells from vaccinated wt B6 mice results in impaired virus clearance from the CNS following challenge
The above results clearly showed that the YF vaccine-induced protection in B cell deficient mice relied almost exclusively on CD8+ T cells. On the other hand, in the presence of a humoral immune response CD8+ T cells appeared redundant for long term survival of immunized mice in vivo (cf. Table II). However, the faster recruitment of CD8+ T cells to the CNS of immunized wt mice following challenge suggested to us that this subset of lymphocytes might also carry out antiviral effector functions in the presence of YF-specific antibodies. To test this hypothesis, we analyzed the virus control in brains of immunized wt B6 mice there were/were not in vivo depleted of CD8+ and/or CD4+ T cells one day prior to YF i.c. challenge (Fig. 10A). Animals were vaccinated s.c. with 105 pfu of YF-17D virus and challenged i.c. 28 days later; unvaccinated wt mice were included as controls. The mice to be depleted were injected i.p. with monoclonal antibodies as described above. At day 7 p.c., brains were collected and viral titers were determined. As expected, unvaccinated animals all exhibited high viral loads, while vaccinated mice had completely cleared infectious virus from the CNS by day 7 p.c. (Fig. 10A). Interestingly, vaccinated mice that were depleted of CD8+ T cells showed defective viral clearance, as part of these animals still displayed detectable amounts of infectious virus in the CNS (Fig. 10A); however, the viral loads of CD8-depleted, vaccinated mice were lower than those of unvaccinated animals, suggesting that CD8+ T cells as well as other immunological mechanisms are required for optimal control of YF-17D virus infection in the CNS. Depletion of CD4+ T cells did not significantly impaired vaccine induced, virus control, while mice depleted of both CD4+ and CD8+ T cells resembled CD8-depleted mice. To address the antiviral effector mechanism(s) underlying cell-mediated immunity to YF, we analyzed the virus titers in brains of immunized wt, IFN-γ KO, prf KO and IFN-γ/prf double KO mice challenged i.c. 28 days after vaccination with 105 pfu of YF-17D; strain-matched unvaccinated mice were included as controls (Fig. 10B). Expanding on the results regarding survival of vaccinated, i.c. challenged immunodeficient mice (see Table II), we observed that in the absence of IFN-γ or perforin, vaccination still resulted in highly efficient control of virus replication in the CNS. On the other hand, in vaccinated mice lacking both effector molecules we observed that all but one mouse contained high amounts of virus in the CNS following i.c. challenge (Fig. 10B). Taken together, these findings demonstrate that T cells expressing IFN-γ and/or perforin contribute significantly to protection against infection with YF virus in vivo.
Figure 10. Impaired YF-17D virus clearance from the CNS of wild type B6 mice in the absence of CD8+ effector T cells.

A) Viral titers in the brains of unvaccinated (open squares) and vaccinated (black squares) WT B6 mice at day 7 post challenge. Some vaccinated animals were in vivo depleted of CD8+ (α-CD8) and/or CD4+ (α-CD8/CD4; α-CD4) T cells 1 day prior to i.c. challenge, and again at days 1 and 4 post challenge. B) WT B6 mice, IFN-γ KO, prf KO and IFN-γ/prf double KO mice that were either naïve (open squares) or had been immunized s.c. with 105 pfu of YF-17D virus 28 days earlier (black squares) were challenged i.c. with 104 pfu of YF-17D virus. Viral loads in the brains at day 7 post challenge are compared for unvaccinated and YF-17D vaccinated mice of each strain. Each dot represents an individual animal. The detection limit for virus in organs was 200 pfu/g organ.
Discussion
The YF-17D vaccine represents a good opportunity to study both the establishment of a protective response following vaccination and the host immune response to an acute viral infection, given the live attenuated nature of the YF-17D virus. The importance of neutralizing antibodies in protection from YF has been suggested by earlier studies (4). Similarly, numbers of circulating CD8+ T cells has also been found to increase as a result of YF vaccination, and for some time, this more or less represented what was known about the vaccine-induced immune response. In recent years, however, several studies have shown that the YF vaccine-induced immune response in humans is also characterized by the presence of a broad and polyfunctional CD8+ T cell response that can generate long-term immunological memory (5,13,14). However, until now the importance of this CD8+ T cell response in protection from YF infection has not been clear, and studies on this question have been hampered by the lack of a small animal model for YF infection.
In this report, we describe a murine model for YF-17D infection, which allows us to study in details the generation of a protective immune response following vaccination. This was possible because, as in humans, peripheral infection with YF-17D virus in wt B6 mice resulted in immunity towards a subsequent viral challenge. Interestingly, we observed that, even in the absence of B and T cell responses (Rag1 deficient mice), mice did not succumb to peripheral infection with YF-17D virus, suggesting that effectors of the innate immune system may suffice to contain the limited peripheral infection resulting from even i.v. inoculation of the attenuated virus. This is in agreement with the finding that the vaccine-associated serious adverse events that can be observed in human vaccinees typically do not reflect virus reversion to virulence, but rather seems to be caused by host genetic factors (39,40), possibly specific deficiencies in innate immune mechanisms.
In our model, we induce a uniformly lethal CNS disease by i.c. administration of YF-17D virus. The relevance of this challenge model compared to natural infection has been debated, and mice lacking both type I and type II IFN signaling pathways have been claimed to represent a better model for human YF infection due to their susceptibility to peripheral challenge with the 17D virus (23,24). This may hold true if one aims to use YF-17D as a paradigm for wt YF virus, but this model is obviously not suitable for analysis of the vaccine-induced immunity, since no protective response can be mounted in those animals. Moreover, in that model only a mild transient viral replication in visceral organs was observed, while the YF-17D viral burden in the brain increased steadily until the animals succumbed (24), suggesting that in fact mortality may be due to viral replication inside the CNS, similarly to what is the case in our challenge model.
Using a panel of gene targeted mice lacking selected genes relevant to the immune system, we unequivocally demonstrated the central role of humoral immunity in the vaccine-induced protection; moreover, we corroborated this finding by showing that immunity to YF can be passively transferred through transfusion with YF immune serum, as previously described (21,41,42). Notably, we also found that adoptively transferred YF-primed splenocytes could mediate protection, suggesting a functional role for cell-mediated immunity, although the possibility that activated B cells within the splenocyte pool may produce antibodies in the recipient mice cannot be ruled out (43). Also supporting a role for T-cell mediated immunity, we observed an accelerated recruitment of both CD8+ T cells and CD4+ T cells to the brain in previously vaccinated mice. The appearance of infiltrating T cells in the CNS of vaccinated mice nicely matched the observed kinetics regarding the reduction in virus content. Thus, the presence of detectable virus in the CNS of most vaccinated mice on days 3 and 5 post challenge followed by complete clearance of infectious virus by day 7 p.c., demonstrate quite clearly that the i.c. inoculum is not completely neutralized by pre-existing antibodies, and indicate that additional mechanisms are essential for optimal clearance of this virus.
More directly addressing the contribution of non-B cells to the in vivo protection, we observed that 23% of immunized B cell KO mice survived viral challenge. The presence of some resistance in the absence of B cells/antibodies, suggested to us that effector T cells could mediate protection in this settings, and this hypothesis was confirmed by the impaired virus control noted in immunized B cell KO mice that were depleted of CD8+ T cells in vivo. When testing CD40L KO mice in vaccination challenge experiments, we found that protection is completely impaired, first of all suggesting a rather stringent requirement for isotype switching. However, the higher mortality observed in the absence of CD40L, as compared to the lack of B cells, may point to an additional direct role for this molecule in establishing an effective antiviral CD8+ T cell response to YF-17D. This has previously been demonstrated for other murine viral infections (30,31,44), as well as in the context of infection with West Nile virus, a related flavivirus, where CD8+ T cell trafficking to the CNS is markedly impaired in the absence of CD40-CD40L interaction (45). However, as CD40L is also expressed by macrophages and NK cells, among other cell subsets, we cannot formally rule out the contribution of additional cell types in influencing the outcome of YF-17D vaccination in CD40L KO mice. Similar to CD40L deficient mice, no YF-17D vaccinated MHC class II deficient mice survived viral challenge. Again this could reflect a requirement for CD4+ T cells, either as antiviral effector cells (note that the virus infection causes a higher mortality in vaccinated perforin/IFN-γ virus double KO mice than in CD8+ T-cell deficient mice) or as helper cells required for the generation and/or maintenance of memory CD8 T cells. Regarding these possibilities it is of interest to note that depletion of CD4+ T cells immediately before challenge did not impact the vaccine-induced virus control, suggesting that these cells are not essential during the secondary response. In contrast, several studies have shown an accelerated decline of CD8+ T cell memory in the absence of CD4+ T cells (46–50).
Underscoring a role for CD8+ T cells as important mediators of virus clearance in the CNS, we observed that in vivo depletion of CD8+ T cells from vaccinated wt mice resulted in impaired and/or delayed viral clearance from the brain. Regarding the molecular mechanism(s) underlying the virus control exerted by CD8+ T cells, it is interesting to note that neither perforin nor IFN-γ deficient mice appear to be seriously impaired in their ability to control YF infection of the CNS, at least when antibodies are also produced. However, when both these effector mechanisms are deficient virus control is significantly impaired, suggesting that the effector T cell dependent virus control may be mediated by several internally redundant mechanisms. However, precisely how these molecular pathways interact and contribute in inhibiting YF virus replication remains to be determined. Indeed, it also remains to be determined precisely how antibodies combat YF infection in the CNS of immunized mice after viral challenge; thus an important question is whether these antibodies can perform antiviral functions beyond neutralization of extracellular virions, in order to eliminate YF-17D virus present within CNS cells?
Interestingly, our observation on the role of CD8+ T cells in the control of virus replication in the YF-infected CNS seemingly contradicts the only previous report investigating the T cell response in the YF-infected murine brain. In the latter study, the authors isolated CD4+ and CD8+ T cells from the brains of immunized, challenged mice and, upon ex vivo stimulation with viral antigen, they found a significant increase in IFN-γ production only in the CD4+ subset (51). However, by using whole antigen for stimulation ex vivo, their analysis was strongly biased for the detection of MHC class II restricted responses. Furthermore, that study was carried out using the neuroadapted Porterfield strain of YF-17D virus (PYF) to immunize young mice (4 to 5 weeks old), and that may also account for some discrepancies.
Importantly, our data reveal that the YF-induced CD8+ T cells not only function as a back-up defense system when antibodies are absent, but they also actively contribute to viral clearance from the CNS of wt mice with a normal humoral response. These findings are consistent with the existing knowledge on the contribution of CD8+ T cells to the recovery from related flaviviral infections in mice. For instance, CD8+ T cells have been demonstrated to have an important role in the murine host defense against dengue virus (DENV), and peptide immunization targeting DENV-specific CD8+ T cells was shown to accelerate viral clearance in infected mice (52). Furthermore, challenge of CD8+ T cell deficient mice with West Nile virus (WNV) resulted in increased mortality and virus persistence in the CNS of the surviving mice as compared to wt mice (53), and, matching this observation, a recent study on vaccination against this virus revealed a complementary role for CD8+ T cells in protection against CNS disease (54). Moreover, in murine models of Japanese Encephalitis virus (JEV) infection, where humoral immunity has been shown to be sufficient in preventing virus spread to the CNS (55–57), an auxiliary contribution of CD8+ T cells has also been disclosed, and adoptive transfer of virus-specific cytotoxic T lymphocytes was shown to confer protection against lethal i.c. viral challenge (58).
In conclusion, our results for the first time formally demonstrate that YF-17D-induced CD8+ T cells are functional in vivo and may complement antibodies in controlling viral infection. Our findings provide new insights regarding the mode of action of the YF vaccine, and open up the possibility that CD8+ T cell memory contributes to the strong and long-lasting YF-17D-induced immunity in humans. Moreover, they highlight the potential of using YF-17D as a recombinant vector for vaccination against human diseases in which the induction of a functional T cell response is crucial, such us HIV, TB and Malaria.
Supplementary Material
Acknowledgments
Grant support: This study was supported by a contract from the National Institutes of Health (NIH) (NIAID Ref No N01-AI-2008032). Additionally, we thank the Faculty of Health and Medical Sciences, University of Copenhagen, Leo Nielsen Foundation, Jacob and Olga Madsen Foundation, the Augustinus Foundation and Christian Larsen and Judge Ellen Larsen’s Foundation for financial support.
We are extremely grateful to P. Rasmussen, D. Bardenfleth and B. Jensen for expert technical assistance throughout the study. Sussanne Petersen is greatly acknowledged for technical assistance with regard to brain sectioning. The authors thank Jason M Millward for his help in setting up the procedure for isolation of mononuclear cells from the CNS.
Abbreviations used
- i.c
intracerebral
- IFA
immune focus assay
- LCMV
lymphocytic choriomeningitis virus
- p.c
post challenge
- YF
yellow fever
- YF-17D
attenuated YF vaccine strain
Reference List
- 1.Monath TP. Yellow fever vaccine. Expert Rev Vaccines. 2005;4:553–574. doi: 10.1586/14760584.4.4.553. [DOI] [PubMed] [Google Scholar]
- 2.Theiler M, Smith HH. The use of yellow fever virus modified by in vitro cultivation for human immunization. J Exp Med. 1937;65:787–800. doi: 10.1084/jem.65.6.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theiler M, Smith HH. The effect of prolonged cultivation in vitro upon the pathogenicity of yellow fever virus. J Exp Med. 1937;65:767–786. doi: 10.1084/jem.65.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinhardt B, Jaspert R, Niedrig M, Kostner C, L’age-Stehr J. Development of viremia and humoral and cellular parameters of immune activation after vaccination with yellow fever virus strain 17D: a model of human flavivirus infection. J Med Virol. 1998;56:159–167. doi: 10.1002/(sici)1096-9071(199810)56:2<159::aid-jmv10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 5.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, Germon S, Del RC, Mulligan MJ, Staprans SI, Altman JD, Feinberg MB, Ahmed R. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 6.Co MD, Terajima M, Cruz J, Ennis FA, Rothman AL. Human cytotoxic T lymphocyte responses to live attenuated 17D yellow fever vaccine: identification of HLA-B35-restricted CTL epitopes on nonstructural proteins NS1, NS2b, NS3, and the structural protein E. Virology. 2002;293:151–163. doi: 10.1006/viro.2001.1255. [DOI] [PubMed] [Google Scholar]
- 7.Bonaldo MC, Martins MA, Rudersdorf R, Mudd PA, Sacha JB, Piaskowski SM, Costa Neves PC, Veloso de Santana MG, Vojnov L, Capuano S, III, Rakasz EG, Wilson NA, Fulkerson J, Sadoff JC, Watkins DI, Galler R. Recombinant yellow fever vaccine virus 17D expressing simian immunodeficiency virus SIVmac239 gag induces SIV-specific CD8+ T-cell responses in rhesus macaques. J Virol. 2010;84:3699–3706. doi: 10.1128/JVI.02255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guirakhoo F, Weltzin R, Chambers TJ, Zhang ZX, Soike K, Ratterree M, Arroyo J, Georgakopoulos K, Catalan J, Monath TP. Recombinant chimeric yellow fever-dengue type 2 virus is immunogenic and protective in nonhuman primates. J Virol. 2000;74:5477–5485. doi: 10.1128/jvi.74.12.5477-5485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAllister A, Arbetman AE, Mandl S, Pena-Rossi C, Andino R. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J Virol. 2000;74:9197–9205. doi: 10.1128/jvi.74.19.9197-9205.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nogueira RT, Nogueira AR, Pereira MC, Rodrigues MM, Neves PC, Galler R, Bonaldo MC. Recombinant yellow fever viruses elicit CD8+ T cell responses and protective immunity against Trypanosoma cruzi. PLoS One. 2013;8:e59347. doi: 10.1371/journal.pone.0059347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barba-Spaeth G, Longman RS, Albert ML, Rice CM. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J Exp Med. 2005;202:1179–1184. doi: 10.1084/jem.20051352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, Quyyumi F, Garg S, Altman JD, Del RC, Keyserling HL, Ploss A, Rice CM, Orenstein WA, Mulligan MJ, Ahmed R. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183:7919–7930. doi: 10.4049/jimmunol.0803903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blom K, Braun M, Ivarsson MA, Gonzalez VD, Falconer K, Moll M, Ljunggren HG, Michaelsson J, Sandberg JK. Temporal dynamics of the primary human T cell response to yellow fever virus 17D as it matures from an effector- to a memory-type response. J Immunol. 2013;190:2150–2158. doi: 10.4049/jimmunol.1202234. [DOI] [PubMed] [Google Scholar]
- 15.Nansen A, Jensen T, Christensen JP, Andreasen SO, Ropke C, Marker O, Thomsen AR. Compromised virus control and augmented perforin-mediated immunopathology in IFN-gamma-deficient mice infected with lymphocytic choriomeningitis virus. J Immunol. 1999;163:6114–6122. [PubMed] [Google Scholar]
- 16.van der Most RG, Harrington LE, Giuggio V, Mahar PL, Ahmed R. Yellow fever virus 17D envelope and NS3 proteins are major targets of the antiviral T cell response in mice. Virology. 2002;296:117–124. doi: 10.1006/viro.2002.1432. [DOI] [PubMed] [Google Scholar]
- 17.Toft-Hansen H, Nuttall RK, Edwards DR, Owens T. Key metalloproteinases are expressed by specific cell types in experimental autoimmune encephalomyelitis. J Immunol. 2004;173:5209–5218. doi: 10.4049/jimmunol.173.8.5209. [DOI] [PubMed] [Google Scholar]
- 18.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cobbold S, Martin G, Waldmann H. Monoclonal antibodies for the prevention of graft-versus-host disease and marrow graft rejection. The depletion of T cell subsets in vitro and in vivo. Transplantation. 1986;42:239–247. doi: 10.1097/00007890-198609000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Brandriss MW, Schlesinger JJ, Walsh EE, Briselli M. Lethal 17D yellow fever encephalitis in mice. I. Passive protection by monoclonal antibodies to the envelope proteins of 17D yellow fever and dengue 2 viruses. J Gen Virol. 1986;67(Pt 2):229–234. doi: 10.1099/0022-1317-67-2-229. [DOI] [PubMed] [Google Scholar]
- 21.Schlesinger JJ, Brandriss MW, Walsh EE. Protection against 17D yellow fever encephalitis in mice by passive transfer of monoclonal antibodies to the nonstructural glycoprotein gp48 and by active immunization with gp48. J Immunol. 1985;135:2805–2809. [PubMed] [Google Scholar]
- 22.Putnak JR, Schlesinger JJ. Protection of mice against yellow fever virus encephalitis by immunization with a vaccinia virus recombinant encoding the yellow fever virus non-structural proteins, NS1, NS2a and NS2b. J Gen Virol. 1990;71(Pt 8):1697–1702. doi: 10.1099/0022-1317-71-8-1697. [DOI] [PubMed] [Google Scholar]
- 23.Meier KC, Gardner CL, Khoretonenko MV, Klimstra WB, Ryman KD. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009;5:e1000614. doi: 10.1371/journal.ppat.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thibodeaux BA, Garbino NC, Liss NM, Piper J, Blair CD, Roehrig JT. A small animal peripheral challenge model of yellow fever using interferon-receptor deficient mice and the 17D-204 vaccine strain. Vaccine. 2012;30:3180–3187. doi: 10.1016/j.vaccine.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christensen JP, Kauffmann SO, Thomsen AR. Deficient CD4+ T cell priming and regression of CD8+ T cell functionality in virus-infected mice lacking a normal B cell compartment. J Immunol. 2003;171:4733–4741. doi: 10.4049/jimmunol.171.9.4733. [DOI] [PubMed] [Google Scholar]
- 26.Thomsen AR, Johansen J, Marker O, Christensen JP. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II-deficient mice and B cell-deficient mice. J Immunol. 1996;157:3074–3080. [PubMed] [Google Scholar]
- 27.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 28.Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voigt I, Camacho SA, de Boer BA, Lipp M, Forster R, Berek C. CXCR5-deficient mice develop functional germinal centers in the splenic T cell zone. Eur J Immunol. 2000;30:560–567. doi: 10.1002/1521-4141(200002)30:2<560::AID-IMMU560>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 30.Andreasen SO, Christensen JE, Marker O, Thomsen AR. Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T cell responses. J Immunol. 2000;164:3689–3697. doi: 10.4049/jimmunol.164.7.3689. [DOI] [PubMed] [Google Scholar]
- 31.Wiesel M, Joller N, Ehlert AK, Crouse J, Sporri R, Bachmann MF, Oxenius A. Th cells act via two synergistic pathways to promote antiviral CD8+ T cell responses. J Immunol. 2010;185:5188–5197. doi: 10.4049/jimmunol.1001990. [DOI] [PubMed] [Google Scholar]
- 32.Clarke SR. The critical role of CD40/CD40L in the CD4-dependent generation of CD8+ T cell immunity. J Leukoc Biol. 2000;67:607–614. doi: 10.1002/jlb.67.5.607. [DOI] [PubMed] [Google Scholar]
- 33.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh MF, Lai SL, Chen JP, Sung JM, Lin YL, Wu-Hsieh BA, Gerard C, Luster A, Liao F. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J Immunol. 2006;177:1855–1863. doi: 10.4049/jimmunol.177.3.1855. [DOI] [PubMed] [Google Scholar]
- 35.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larena M, Regner M, Lee E, Lobigs M. Pivotal role of antibody and subsidiary contribution of CD8+ T cells to recovery from infection in a murine model of Japanese encephalitis. J Virol. 2011;85:5446–5455. doi: 10.1128/JVI.02611-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metcalf TU, Griffin DE. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J Virol. 2011;85:11490–11501. doi: 10.1128/JVI.05379-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tschen SI, Stohlman SA, Ramakrishna C, Hinton DR, Atkinson RD, Bergmann CC. CNS viral infection diverts homing of antibody-secreting cells from lymphoid organs to the CNS. Eur J Immunol. 2006;36:603–612. doi: 10.1002/eji.200535123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galler R, Pugachev KV, Santos CL, Ocran SW, Jabor AV, Rodrigues SG, Marchevsky RS, Freire MS, Almeida LF, Cruz AC, Yamamura AM, Rocco IM, da Rosa ES, Souza LT, Vasconcelos PF, Guirakhoo F, Monath TP. Phenotypic and molecular analyses of yellow fever 17DD vaccine viruses associated with serious adverse events in Brazil. Virology. 2001;290:309–319. doi: 10.1006/viro.2001.1168. [DOI] [PubMed] [Google Scholar]
- 40.Pulendran B. Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat Rev Immunol. 2009;9:741–747. doi: 10.1038/nri2629. [DOI] [PubMed] [Google Scholar]
- 41.Schlesinger JJ, Foltzer M, Chapman S. The Fc portion of antibody to yellow fever virus NS1 is a determinant of protection against YF encephalitis in mice. Virology. 1993;192:132–141. doi: 10.1006/viro.1993.1015. [DOI] [PubMed] [Google Scholar]
- 42.Gould EA, Buckley A, Barrett AD, Cammack N. Neutralizing (54K) and non-neutralizing (54K and 48K) monoclonal antibodies against structural and non-structural yellow fever virus proteins confer immunity in mice. J Gen Virol. 1986;67(Pt 3):591–595. doi: 10.1099/0022-1317-67-3-591. [DOI] [PubMed] [Google Scholar]
- 43.Larena M, Prow NA, Hall RA, Petrovsky N, Lobigs M. JE-ADVAX vaccine protection against Japanese encephalitis virus mediated by memory B cells in the absence of CD8(+) T cells and pre-exposure neutralizing antibody. J Virol. 2013;87:4395–4402. doi: 10.1128/JVI.03144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bedenikovic G, Crouse J, Oxenius A. T-cell help dependence of memory CD8(+) T-cell expansion upon vaccinia virus challenge relies on CD40 signaling. Eur J Immunol. 2014;44:115–126. doi: 10.1002/eji.201343805. [DOI] [PubMed] [Google Scholar]
- 45.Sitati E, McCandless EE, Klein RS, Diamond MS. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J Virol. 2007;81:9801–9811. doi: 10.1128/JVI.00941-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC. Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J Virol. 2002;76:12388–12393. doi: 10.1128/JVI.76.23.12388-12393.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.von Herrath MG, Yokoyama M, Dockter J, Oldstone MB, Whitton JL. CD4-deficient mice have reduced levels of memory cytotoxic T lymphocytes after immunization and show diminished resistance to subsequent virus challenge. J Virol. 1996;70:1072–1079. doi: 10.1128/jvi.70.2.1072-1079.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalams SA, Walker BD. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med. 1998;188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christensen JP, Marker O, Thomsen AR. The role of CD4+ T cells in cell-mediated immunity to LCMV: studies in MHC class I and class II deficient mice. Scand J Immunol. 1994;40:373–382. doi: 10.1111/j.1365-3083.1994.tb03477.x. [DOI] [PubMed] [Google Scholar]
- 50.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu T, Chambers TJ. Yellow fever virus encephalitis: properties of the brain-associated T-cell response during virus clearance in normal and gamma interferon-deficient mice and requirement for CD4+ lymphocytes. J Virol. 2001;75:2107–2118. doi: 10.1128/JVI.75.5.2107-2118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yauch LE, Zellweger RM, Kotturi MF, Qutubuddin A, Sidney J, Peters B, Prestwood TR, Sette A, Shresta S. A protective role for dengue virus-specific CD8+ T cells. J Immunol. 2009;182:4865–4873. doi: 10.4049/jimmunol.0801974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shrestha B, Ng T, Chu HJ, Noll M, Diamond MS. The relative contribution of antibody and CD8+ T cells to vaccine immunity against West Nile encephalitis virus. Vaccine. 2008;26:2020–2033. doi: 10.1016/j.vaccine.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goncalvez AP, Chien CH, Tubthong K, Gorshkova I, Roll C, Donau O, Schuck P, Yoksan S, Wang SD, Purcell RH, Lai CJ. Humanized monoclonal antibodies derived from chimpanzee Fabs protect against Japanese encephalitis virus in vitro and in vivo. J Virol. 2008;82:7009–7021. doi: 10.1128/JVI.00291-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gupta AK, Lad VJ, Koshy AA. Protection of mice against experimental Japanese encephalitis virus infections by neutralizing anti-glycoprotein E monoclonal antibodies. Acta Virol. 2003;47:141–145. [PubMed] [Google Scholar]
- 57.Kimura-Kuroda J, Yasui K. Protection of mice against Japanese encephalitis virus by passive administration with monoclonal antibodies. J Immunol. 1988;141:3606–3610. [PubMed] [Google Scholar]
- 58.Murali-Krishna K, Ravi V, Manjunath R. Protection of adult but not newborn mice against lethal intracerebral challenge with Japanese encephalitis virus by adoptively transferred virus-specific cytotoxic T lymphocytes: requirement for L3T4+ T cells. J Gen Virol. 1996;77(Pt 4):705–714. doi: 10.1099/0022-1317-77-4-705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.