

Abstract
Autotaxin (ENPP2/ATX) and lysophosphatidic acid (LPA) receptors represent two key players in regulating cancer progression. The present study sought to understand the mechanistic role of LPA G protein-coupled receptors (GPCR), not only in the tumor cells but also in stromal cells of the tumor microenvironment. B16F10 melanoma cells predominantly express LPA5 and LPA2 receptors but lack LPA1. LPA dose-dependently inhibited invasion of cells across a Matrigel layer. RNAi-mediated knockdown of LPA5 relieved the inhibitory effect of LPA on invasion without affecting basal invasion. This suggests that LPA5 exerts an anti-invasive action in melanoma cells in response to LPA. In addition, both siRNA-mediated knockdown and pharmacological inhibition of LPA2 reduced the basal rate invasion. Unexpectedly, when probing the role of this GPCR in host tissues, it was found that the incidence of melanoma-derived lung metastasis was greatly reduced in LPA5 knockout (KO) mice compared to wild-type (WT) mice. LPA1- but not LPA2-KO mice also showed diminished melanoma-derived lung metastasis, suggesting that host LPA1 and LPA5 receptors play critical roles in the seeding of metastasis. The decrease in tumor cell residence in the lungs of LPA1- and LPA5-KO animals was apparent 24 h after injection. However, KO of LPA1, LPA2 or LPA5 did not affect the subcutaneous growth of melanoma tumors.
Implications
These findings suggest that tumor- and stromal-LPA receptors, in particular LPA1 and LPA5, play different roles in invasion and the seeding of metastasis.
Keywords: Autotaxin, Lysophosphatidic acid receptor, Invasion, Metastasis, Tumor microenvironment
Introduction
One of the first indications for the involvement of autotaxin (ATX) in cancer was the identification of this secretory protein in human melanoma cells capable of stimulating cancer cell motility (1). The mechanism by which ATX acts as a motogen remained elusive until it was established that ATX has lysophospholipase D activity and is the primary enzyme responsible for lysophosphatidic acid (LPA) production in biological fluids (2, 3). Notably, plasma LPA levels in heterozygous knockout (KO) mice for the ATX gene (atx+/-) were reduced by half compared to levels in wild-type (WT) littermates (4, 5). Aberrantly high expression of ATX has been detected in breast cancer (6), glioblastoma multiforme (7), prostate cancer (8), hepatocellular carcinoma (9), and melanoma (10). Overexpression of ATX in these malignancies promotes tumor motility and invasiveness, enhances metastatic potential, and is commonly associated with poor clinical outcomes (11).
LPA appears to be responsible for many of ATX's biological activities. This is not surprising, as early studies have demonstrated that LPA has pro-tumorigenic effects on ovarian, breast, and prostate cancer cells (12, 13). High levels of LPA were also reportedly present in ascites of patients with ovarian cancer (14). In cases of follicular lymphoma and of pancreatic cancer, elevated levels of ATX were detected in sera of patients with a concomitant increase in plasma LPA levels (15, 16).
LPA exerts its cellular functions by acting on specific G protein-coupled receptors (GPCR). There are at least nine GPCR that were reported to be activated by LPA, amongst which LPA1, LPA2, and LPA3 receptors have been extensively studied. LPA1-3 receptors belong to the same endothelial differentiation gene (edg) GPCR subfamily as the sphingosine-1-phosphate receptors (17). Consistent with the reported roles of ATX and LPA in cancer, it seems apparent that LPA receptors are also involved in tumorigenesis. Indeed, compelling evidence shows that LPA receptors are overexpressed in most cancers (18, 19) and function to enhance cancer cell survival, motility, invasion, and metastasis (20-22). In particular, both LPA1 and LPA2 receptors are implicated in breast cancer progression; the LPA1 receptor was found to promote the metastasis of human breast cancer xenografts to the bone (23), whereas LPA2 receptor expression was elevated in the majority of postmenopausal breast cancers (24). In cases of ovarian cancer, abundant expression of LPA2 receptors was associated with an increase in invasiveness (25) and lipogenesis of ovarian cancer cells (26). In addition, expression levels of LPA2 receptors in colorectal cancer were reportedly high and further increased during malignant transformation (27). A role for the LPA3 receptor in regulating growth and survival of metastatic melanoma has also been documented. However, it is important to note that not all metastatic melanomas express the LPA3 receptor, as data from gene expression analyses of human melanoma specimens revealed high variability in expression profiles amongst samples (10).
In contrast to LPA1-3 receptors, little is known about the roles of LPA4 (GPR23/P2Y9), LPA5 (GPR92), and LPA6 (P2Y5) receptors in cancer (11); and characterization of the putative LPA GPCR GPR87, P2Y10, and GPR35 awaits further research. Only a few studies have been carried out to elucidate the function of LPA4 in tumorigenesis. Specifically, work by Harper and colleagues demonstrated that the LPA4 receptor was responsible for mediating ATX-induced invadopodia formation in fibrosarcoma cells, a critical step in the mesenchymal form of invasion (28). This report however, contradicts the findings from an earlier study, in which LPA4 was found to inhibit cell motility and invasion of mouse embryonic fibroblasts (29). It has recently been shown that the LPA5 receptor may potentially inhibit migration of B16 melanoma cells by activating the cAMP-PKA pathway and diminishing PIP3 signaling (30).
Although much interest has been shown in elucidating the functions of ATX and LPA receptors in cancer cells, the effect of stromal or host LPA receptors on tumor-microenvironment (TME) interactions has not been addressed. It is becoming clear that the TME plays a fundamental role not only in supporting tumor growth but also in its progression toward a metastatic disease. In fact, tumor cells are capable of establishing a reciprocal communication with their host microenvironment to create the most favorable environment for them to proliferate, migrate, invade, and metastasize (31). In the present study, we evaluated the function of the ATX-LPA receptor signaling axis in TME interaction, particularly in the homing of metastasizing cancer cells. The syngeneic B16 murine melanoma model was chosen as our platform for assessing tumor-stroma interactions, with good reasons. We have previously shown that the metastasizing capacity of B16F10 melanoma cells in vivo is in part attributable to ATX (32, 33). In addition, the availability of LPA receptor KO mice generated in a mixed C57BL/6 and 129/Sv genetic background allows the use of this model to study the role of host LPA receptors in metastasis. Our data demonstrate that the homing of metastasizing B16F10 melanoma cells to the lungs is substantially reduced by the absence of host LPA1 and almost completely reduced by the absence of LPA5, whereas LPA2 and LPA5 expressed in the tumor cell promote and inhibit invasion, respectively.
Materials and Methods
Materials
Lysophosphatidic acid (18:1) and lysophosphatidylcholine (18:1) were purchased from Avanti Polar Lipids (Alabaster, AL). ATX inhibitor BMP22 and specific LPA2 antagonist compound 35 were synthesized as described in (32) and (34). Ki16425 was purchased from Cayman Chemical (Ann Arbor, MI). Stock solutions (10 mM) of BMP22, compound 35, and Ki16425 were prepared in dimethyl sulfoxide (DMSO) for in vitro studies. A stock solution of LPA (1 mM) was prepared as an equimolar complex with charcoal-stripped, fatty acid-free bovine serum albumin (BSA) (Sigma-Aldrich; St. Louis, MO) in phosphate-buffered saline (PBS). Polyethylene glycol 400 (PEG 400) was purchased from Sigma-Aldrich. All cell culture media were purchased from Corning Cellgro (Manassas, VA), and cell culture reagents were from Life Technologies (Grand Island, NY) unless otherwise stated. The fluorescent ATX substrate FS-3 was purchased from Echelon Biosciences (Salt Lake City, UT).
Cell culture
B16F10 melanoma cells (gift from Dr. Gordon Mills, University of Texas, MD Anderson Cancer Center) were cultured in MEM supplemented with 5% heat-inactivated HyClone® FBS (Thermo Scientific; Pittsburgh, PA), 2 mM L-glutamine, 1X MEM vitamin, 1X MEM non-essential amino acid (NEAA), 1 mM sodium pyruvate, and 1X antibiotic-antimycotic. The highly invasive MM1 rat hepatoma suspension cells (gift from Dr. Michiko Mukai, Osaka University, Japan) were grown in DMEM supplemented with 10% (V/V) FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human umbilical vein endothelial cells (HUVEC) and rat lung microvascular endothelial cells (RLMVEC) were purchased from VEC Technologies Inc. (Rensselaer, NY) and cultured in MCDB-131 complete medium supplemented with 10% (V/V) FBS, 90 μg/ml heparin, 10 ng/m EGF, 1 μg/ml hydrocortisone, 0.2 mg/ml EndoGrowth, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 25 μg/ml amphotericin B. The isolation and culture of mesothelial cells from mice has been previously described elsewhere (35). Primary lung microvascular endothelial cells (MLVEC) from C57BL/6 mice were purchased from Cell Biologics, Inc. (Chicago, IL) and cultured in M1168 complete medium as described by the manufacturer's protocol.
Primary rat ATII cells were isolated according to the methods described previously (36, 37). Briefly, ATII cells were isolated from male Sprague-Dawley rats by elastase digestion and differential adherence on IgG-coated dishes. ATII cells were identified using Nile Red (Sigma-Aldrich) staining of lamellar bodies, and >95% of the cells were Nile Red-positive on day 2. Six-well plates that were coated with matrix deposited by rat lung fibroblasts (American Type Culture Collection; Manassas, VA) were used for culture of ATII cells. Freshly isolated cells were seeded to confluence at 3.5 × 106 cells/well in ATII culture medium (DMEM with 10% FBS, 4 mM glutamine, 1% penicillin/streptomycin, and 0.25 μM amphotericin B), and experiments were performed on day 2 after isolation. To obtain ATI-like cells, ATII cells were cultured until day 6 from the day of isolation, changing the media every day. On day 2 or day 6, cells were harvested and total RNA was isolated using the RNA-isolation kit, RNeasy (Qiagen; Valencia, CA).
Short hairpin RNA (shRNA) knockdown in B16F10 melanoma cells
Five clones of lentiviral shRNA LPA5 constructs (SHCLNG-XM_355812) were purchased from Sigma-Aldrich. Viral particles were produced by co-transfection of 293FT cells with an shRNA-expressing plasmid (CCGGCGTGCTGGATCCACTGGTTTACTCGAGTAAACCAGTGGATCCAGCACGTTTTTG), or pLK01 scramble, and a lentiviral packaging mix (Virapower) as described previously (38). B16F10 cells were plated onto a 35-mm dish at a density of 2 × 105 in growth medium, and incubated overnight. The following day, the cells were transduced with the lentiviral shRNA with 6 μg/ml polybrene in 1 ml of growth medium. One day after the transduction, the medium was replaced with fresh growth medium containing 1μg/ml puromycin (Sigma-Aldrich), and the cells were cultured for at least an additional week before performing functional studies.
Small interfering RNA (siRNA) knockdown in B16F10 melanoma cells
siRNA pools specific for LPA2 and non-targeting scrambled siRNA were purchased from Dharmacon (GE Healthcare; Wauwatosa, WI). B16F10 cells were plated onto a 35-mm dish at a density of 2 × 105 in growth medium, and incubated overnight. The following day, the cells were transfected with 30pmol of siRNA using Lipofectamine® RNAiMAX reagent (Life Technologies; Grand Island, NY) according to the manufacturer's protocol. 24 hours after siRNA transfection, cells were harvested and used in the invasion assay.
Lentiviral overexpression of LPA1 in B16F10 melanoma cells
B16F10 cells were transduced with enhanced green fluorescent protein (GFP), empty vector, or LPA1-containing lentiviruses, respectively, in the presence of 6 μg/ml polybrene in 1 ml of growth medium. One day after transduction, the medium was replaced with fresh growth medium, and cells were selected with 1 μg/ml puromycin for one week.
RNA isolation and real-time quantitative polymerase chain reaction (PCR)
Total RNA was extracted using Trizol® reagent (Life Technologies; Grand Island, NY) followed by treatment with DNase I (Sigma-Aldrich) to remove any trace of genomic DNA contamination. Single-stranded cDNA was synthesized using the ThermoScript™ RT System (Life Technologies). Quantitative real-time PCR was performed using StepOnePlus™ instrument (Applied Biosystem) using the RT2 RealTime™ SYBR Green/ROX PCR Master Mix Kit (Qiagen). The mRNA amplification cycle was as follows: 40 cycles of 95°C for 15 s and 60°C for 60 s. Each sample was normalized to the relative expression levels of GAPDH. Primer sequences are listed in Supplementary Table 1.
Measurement of ATX activity
Conditioned media (CM) from B16F10, MLVEC, MM1, or mesothelial cells (HUVEC or RLMVEC) were respectively prepared by incubating 5 × 106 cells for 20 h in serum-free MEM, M1168, DMEM, or MCDB-131. Control media were prepared from MEM, M1168, DMEM, or MCDB-131 without exposure to cells. Collected CM were centrifuged, filtered through a 0.22-μm filter, and concentrated (∼25-fold) using Amicon Ultra 30 000 Centrifugal Filter Units (Millipore; Billerica, MA). ATX/lyso-PLD activity was measured by incubating 20μl of concentrated CM or mouse plasma with 2μM of the fluorogenic substrate FS-3 and 10μM of BSA. The change in fluorescence intensity was monitored for 3.5 h at 37°C using the FLEXStation II, with excitation and emission wavelengths of 485 nm and 538 nm, respectively. The differences in fluorescence intensity for each time point were normalized to that of time zero.
MM1 hepatoma cell invasion of endothelial monolayers
For in vitro tumor cell invasion of the endothelial barrier, 1.3 × 105 HUVEC (passages 4-6) were seeded into each well of a 12-well plate pre-coated with 0.2% gelatin (Sigma-Aldrich) and incubated for 2 d to form a confluent monolayer. Mesothelial cells harvested from three mice were initially plated in 6-well plates pre-coated with poly-L-lysine (Sigma-Aldrich) and grown to confluence in DMEM supplemented with 10% (V/V) FBS in the presence of 2 mM L-glutamine, 100 U/ml penicillin G, and 10 μg/ml streptomycin. When confluent, 1.8 × 105 mesothelial cells were plated into each well of a 12-well plate and grown to confluence. For all invasion assays, MM1 cells were pre-stained with 2 μg/ml calcein AM (Invitrogen; Grand Island, NY) for 2 h, rinsed once, and seeded at a density of 5 × 104 cells per well over the monolayers. Tumor cells were left to invade the monolayers for 24 h either in serum-free MCDB-131 media (for HUVEC monolayer) or DMEM (for mesothelial monolayer) supplemented with 0.1% fatty acid free BSA. The day after MM1 cell seeding, non-invaded tumor cells were removed by repeated rinses of the monolayer with PBS (containing Ca2+ and Mg2+), followed by fixation with 10% buffered formalin. The number of tumor cells that penetrated the monolayer was photographed under a NIKON TiU inverted microscope using phase-contrast and fluorescence illumination in a minimum of five non-overlapping fields at 100x magnification. The fluorescent images were overlaid on top of the phase contrast images using Elements BR software (version 3.1x), and the invaded MM1 cells showing the characteristic flattened morphology underneath the monolayer were counted.
Boyden chamber invasion of B16 melanoma cells
Cell invasion across a Matrigel-coated membrane was performed using the 8-μm pore size, 24-well BD Biocoat™ tumor invasion system (BD Biosciences; San Jose, CA). The Matrigel coating of the plates was rehydrated with PBS for 2 h at 37°C according to the manufacturer's protocol. The PBS was removed, and 1 × 105 cells in serum-free MEM supplemented with 0.1% BSA were added to each upper chamber; 0.75 ml of serum-free MEM/0.1% BSA containing LPA was added to the bottom chamber as a chemoattractant. In experiments with compound 35 or BMP22 respectively, the compounds were added to both the top and bottom chambers. Cells were left to invade the Matrigel for 20 h at 37°C. After incubation, the medium in the upper chamber was removed, and the insert was transferred into a new 24-well plate containing 4μg/ml of calcein AM in Hank's balanced salt solution (HBSS). The plates were incubated for 1 h at 37°C. The fluorescence of invaded cells was then measured with a FLEXStation II plate reader at excitation and emission wavelengths of 485 and 530 nm, respectively.
B16F10 lung experimental metastasis model
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Tennessee and were consistent with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication 85-23, revised 1985). B16F10 cells (1 × 105) were injected into the various LPA receptor KO female mice and their respective female WT littermates via tail vein. All mice were sacrificed at day 21. The lungs were harvested and inflated, and the number of metastatic tumor nodules was counted. Necropsy was also conducted to detect extrapulmonary metastases.
Where indicated, 1 × 105 B16F10 cells transduced with either lentivirus containing scrambled shRNA or shRNA for LPA5 were injected into the tail vein of 8- to 12-wk-old female C57BL/6 mice (purchased from The Jackson Laboratory; Bar Harbor, Maine). In experiments involving treatment with BMP22 and/or Ki16425, 1 × 105 B16F10 cells were injected into the tail vein of 8- to 12-wk-old female C57BL/6 mice. Thirty minutes later, each group of mice received treatment with vehicle (PBS with 10% PEG), or Ki16425 (1.5 mg/kg-1), or BMP22 (0.1 mg/kg-1), or a combination of Ki16425 and BMP22 via intraperitoneal injection daily for 20 days.
Histopathological examination
Lungs were excised from mice on day 21 and fixed in 10% buffered formalin. Multiple paraffin-embedded 5-μm sections of the lungs were stained with H&E.
Lung extravasation assay
In some experiments, 1 × 106 B16F10 cells stably expressing GFP were injected via tail vein into the various LPA receptor KO female mice and their respective WT littermates. Mice were sacrificed 6 h and 24 h after injection. Lungs were harvested and filled with PBS, and the GFP-expressing B16F10 cells on the lung surface were imaged using a Nikon TiU inverted fluorescence microscope (using a low-magnitude 4X objective lens). At least 12 images were taken for each mouse, and tumor burden was measured as total area of GFP fluorescence (in pixel units) using the Nikon NIS element software.
Subcutaneous inoculation of B16F10 cells
For subcutaneous tumor growth, 4 × 105 B16F10 cells were injected subcutaneously into the left flank of each mouse in a 100-μl volume. The cells were prepared by reconstituting with Extracel-X® Hydrogel (Glycosan BioTime, Inc.; Alameda, CA) in a 1:1 dilution with culture medium as recommended by the manufacturer's protocol. All mice were sacrificed on day 10, and tumors were measured using a caliper, harvested, and weighed. Tumor volume was calculated using the modified ellipsoid formula: Tumor volume = 1/2 (length × width2), where length is the greatest longitudinal diameter.
Statistical analysis
All statistical analysis were performed with GraphPad Prism software (version 5.0) using the two-tailed unpaired Student's t-test or the one-way ANOVA, followed by either a Bonferroni post-test or Newman-Keuls multiple comparison test.
Results
Differential expression of ATX and LPA receptors in tumor and stromal cells
To understand the roles of ATX and LPA receptors in TME interaction, we first sought to determine their expression profiles in tumor and stromal cells. Quantitative real-time PCR was performed on mRNA isolated from B16F10 melanoma cells and C57BL/6 lung microvascular endothelial cells (MLVEC). We found that B16F10 cells showed robust expression of transcripts encoding ATX, LPA5, LPA2, and LPA6 receptors (Fig. 1A). In contrast, MLVEC had very low expression of ATX and LPA5 transcripts and predominantly expressed LPA6, LPA1, and LPA4 (Fig. 1B). We observed a similar trend in our previous studies, in which expression of ATX was higher in MM1 rat hepatocarcinoma cells compared to expression in stromal cells such as HUVEC and isolated murine mesothelial cells (32). Because ATX is primarily a secreted enzyme that hydrolyzes lysophosphatidylcholine (LPC) to LPA, we decided to measure ATX activity in concentrated CM of B16F10 cells and MLVEC. To do this, we utilized the synthetic fluorogenic substrate FS-3, which was cleaved by ATX to release the fluorescent moiety from the quencher (39). Consistent with the ATX expression profiles in these cells, a higher level of ATX activity was detected in CM from B16F10 cells but not in CM from MLVEC (Fig. 1C and D). We extended this assessment to include concentrated CM from MM1 hepatocarcinoma, HUVEC, rat lung microvascular endothelial cells (RLMVEC), and mesothelial cells. Except in MM1 cells, we were unable to detect ATX activity in the CM from any of these stromal cells (Supplementary Fig. 1). On the basis of these observations, we concluded that B16F10 cells may provide a rich source of ATX within the tumor microenvironment.
Figure 1.

ATX and LPA receptor profiling in (A) B16F10 and (B) mouse lung vascular endothelial cells (MLVEC) using quantitative real-time PCR. Time course of ATX activity measured in the conditioned media (CM) of (C) B16F10 and (D) MLVEC. Control represents media that were not exposed to cells. Data are expressed as mean ± SD of an experiment performed twice in quadruplicates (for quantitative real-time PCR) and three times in triplicates for the ATX activity assay.
LPA5 receptor in B16F10 cells inhibits cell invasion in vitro
We have previously determined that ATX is involved in the formation of B16F10 lung metastases in C57BL/6 mice (32, 33). To determine whether LPA receptors contribute to the invasive behavior of these cells, Boyden chamber assays were conducted. We observed that B16F10 cells had a high basal invasion rate across the Matrigel layer; but when LPA was added to the bottom chamber as a chemoattractant, cell invasion was reduced (data not shown). To determine which LPA receptor was responsible for this effect, we used a lentiviral system to deliver an shRNA construct to knock down the LPA5 receptor, the predominant LPA receptor expressed in B16F10 cells. This system effectively reduced the levels of LPA5 mRNA by 80% compared to the levels in B16F10 cells transduced with scrambled shRNA (Fig. 2A). We found that knockdown of LPA5 in B16F10 cells moderately attenuated the inhibitory effect of LPA on invasion. This effect was observed only when concentrations higher than 30 nM LPA were used (Fig. 2B). In addition, the basal invasion rate of B16F10 cells was unaffected by the knockdown of LPA5.
Figure 2.

Inhibition of B16F10 cell invasion by LPA is mediated through LPA5. (A) Validation of shRNA knockdown of LPA5 in B16F10 using quantitative real-time PCR. (B) Invasion of LPA5 knockdown-B16F10 across a Matrigel layer in the presence or absence of increasing concentrations of LPA. (C) Validation of siRNA knockdown of LPA2 in B16F10 using quantitative real-time PCR. (D) Invasion of LPA2 knockdown-B16F10 or non-targeting scrambled siRNA-B16F10 (control) across a Matrigel layer. (E) Invasion of B16F10 in the presence of the LPA2 antagonist, compound 35. (F) Validation of LPA1 overexpression in B16F10 using quantitative real-time PCR. (G) Invasion of B16F10 cells overexpressing LPA1 across a Matrigel layer in the presence or absence of increasing concentrations of LPA. Data are representative of an experiment performed twice in quadruplicates and are expressed as mean ± SD. *Denotes p value < 0.05 using one-way ANOVA followed by a Bonferroni post-test. (H) Effect of LPA5 knockdown-B16F10 cells on lung metastasis in C57BL/6 mice. n = 22 mice injected with scrambled shRNA B16F10 cells, and n = 21 mice injected with shLPA5 B16F10 cells.
We next examined the role of the LPA2 receptor in B16F10 cell invasion. To do this, we utilized a siRNA targeting system to knockdown LPA2 in B16F10 by 75% (Fig. 2C) and assessed the effect on invasion. We found that knockdown of LPA2 significantly reduced the basal invasion rate of B16F10 compared to the scrambled siRNA control (Fig. 2D). To complement this finding, we used a potent and selective small-molecule antagonist for LPA2, designated as compound 35 by Beck et al. (34). Consistent with the siRNA knockdown results, treatment with compound 35 alone dose-dependently inhibited the basal invasion rate of B16F10 across the Matrigel layer. These results suggest that LPA2 may be, in part, responsible for the high basal invasion rate observed in B16F10 cells (Fig. 2E).
To further validate these results, we transduced B16F10 cells, which do not express LPA1 mRNA (Fig. 1A), with an LPA1 construct using the lentiviral system (Fig. 2F). We hypothesized that introducing the pro-invasive LPA1 receptor might affect the outcome of invasion in these cells. Indeed, we noted that the invasion rate of LPA1-overexpressing B16F10 cells in response to LPA increased significantly compared to the rate of vector-transduced B16F10 cells (Fig. 2G). In fact, cell invasion was greatest at 10 nM of LPA, and it decreased in magnitude at higher concentrations of LPA. We reasoned that this might be attributed to the preference of LPA1 receptors for the acyl form of LPA (18:1) used in the assay, whereas the more preferred ligand for the LPA5 receptors is alkyl-glycerophosphate (AGP, also known as alkyl-LPA) (40).
Although these findings seemingly suggest that LPA5 acts as an anti-invasive receptor in B16F10 cells in vitro, we found it difficult to reconcile them with the high metastatic capacity of these cells when injected into mice. Hence, we assessed the effect of shLPA5 knockdown in B16F10 cells on the formation of lung metastases in C57BL/6 mice. Interestingly, we observed no significant difference in the number of lung metastases formed in mice inoculated with either shLPA5 or scrambled shRNA B16F10 cells (Fig. 2H).
Host LPA1 and LPA5 receptors affect the homing of metastasizing B16F10 cells in vitro
Next, we investigated whether host LPA receptors play a role in influencing the homing of metastasizing tumor cells. For this purpose, we utilized a transcellular invasion model described previously by Mukai and colleagues (41). This model measures the invasion of MM1 rat hepatocarcinoma cells across a mesothelial monolayer that commonly lines the body's serous cavities. We isolated mesothelial cells from LPA1-, LPA2-, and LPA5-KO mice and subjected the cultured monolayers to invasion by tumor cells. We found that the rate of MM1 invasion was significantly reduced in mesothelial monolayers generated from LPA1- but not from LPA2- and LPA5-KO mice (Fig. 3A). This was the first indication of a possible involvement of stromal LPA1 receptors in tumor invasion. To confirm this finding, we applied the LPA1/3 receptor antagonist Ki16425 to co-cultures of MM1 cells and HUVEC monolayers, and quantified invasion after 24 h. Ki16425 dose-dependently inhibited the LPA-induced MM1 invasion across the HUVEC monolayer, thus reinforcing our earlier observations in the LPA1 KO mesothelium (Fig. 3B).
Figure 3.

Role of host LPA receptors in cell invasion and the seeding of metastasis. (A) MM1 rat hepatoma cell invasion across a mesothelial monolayer isolated from LPA1-, LPA2-, and LPA5-KO mice, respectively. (B) Ki16425 inhibits LPA-induced MM1 cell invasion of HUVEC monolayer. Data are expressed as mean ± SD of an experiment performed three times in duplicates. *Denotes p value <0.05 using one-way ANOVA followed by a Bonferroni post-test. (C) Effect of Ki16425, BMP22, and combination therapy on the lung metastasis of B16F10 cells in C57BL/6 mice. Vehicle treatment: n = 9 mice, Ki16425: n = 8 mice, BMP22: n = 8 mice; and combination treatment of Ki16425 and BMP22: n = 10 mice. P values were obtained using one-way ANOVA followed by Newman-Keuls multiple comparison test.
To complement our in vitro observations, we tested the effect of Ki16425 in the B16 experimental metastasis model. Briefly, 1 × 105 B16F10 cells were injected into the tail vein of female C57BL/6 mice. The mice were treated daily with Ki16425, and lungs were harvested on day 21 for quantification of metastases. As a positive control, a parallel group of mice was treated with BMP22, an ATX inhibitor that was characterized recently by our group as being effective in reducing the metastasis of B16F10 cells to the lungs (32). In this model, administering Ki16425 significantly reduced the number of metastatic nodules formed in mice. In this regard, we found Ki16425 to be as effective as BMP22 in reducing the number of metastatic foci (Fig. 3C). Next, we questioned whether metastasis could be further reduced by concurrent administration of Ki16425 and BMP22. Our data showed that combination therapy was more effective in reducing the number of lung tumor nodules than single therapy with either compound (Fig. 3C).
Because B16F10 cells do not express LPA1 receptors, we postulated that the effect of Ki16425 in reducing lung metastasis might be due to the inhibition of host LPA1 receptors. To further validate this hypothesis, we injected B16F10 cells into LPA1 KO mice and examined lung metastasis on day 21. Consistent with the data obtained using Ki16425, LPA1 KO mice had significantly fewer metastatic nodules compared to WT mice (Fig. 4A and B). We extended these studies to include LPA2- and LPA5-KO mice. Interestingly, we found that deletion of the host LPA2 receptor did not influence the seeding of lung metastasis (Fig. 4C and D). On the contrary, compared to WT mice, LPA5 KO mice showed substantially fewer metastases, with most of the mice having no detectable lung metastases (Fig. 4E and F). Although the lack of available antagonists for LPA5 receptor impeded further validation of these studies, these data from KO mice nonetheless demonstrate that host LPA5 receptors may potentially be involved in the homing of metastasizing cells. Analysis of LPA1-5 receptor expression profiles in whole lung tissue isolated from the respective knockout mice revealed no changes in the expression of LPA2, LPA4 and LPA5 across genotype. Only LPA1 expression was moderately elevated in LPA5 KO mice, whereas LPA3 expression was increased in LPA2 KO mice (Supplementary Fig. 2).
Figure 4.
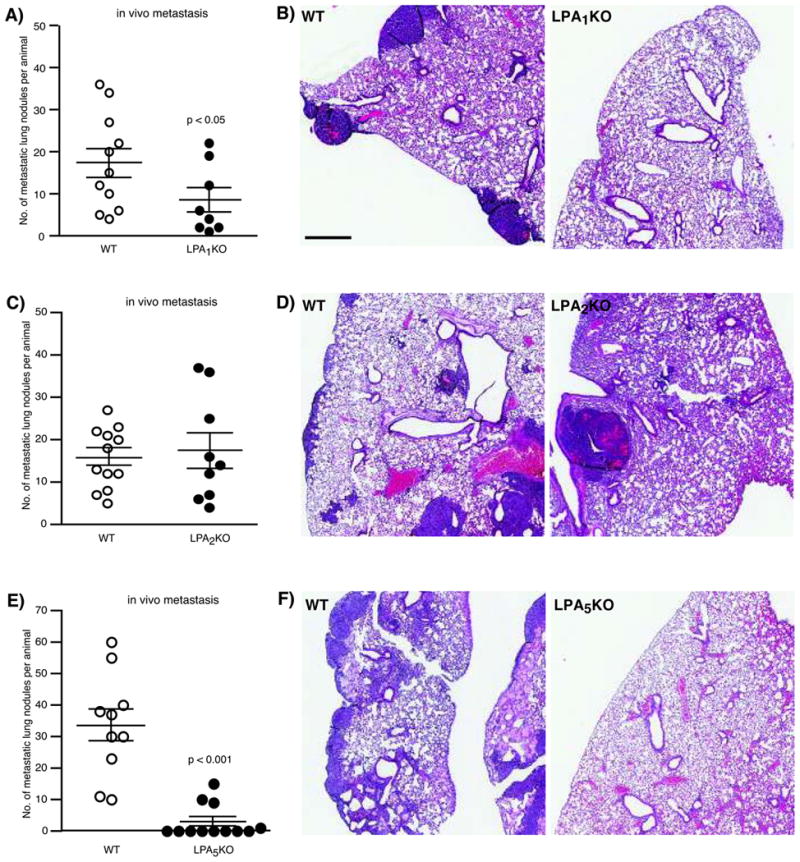
Effect of host LPA receptor on the homing of B16F10 lung metastasis. (A) LPA1 KO mice had a reduced number of lung nodules. n = 11 WT mice, and n = 8 LPA1 KO mice. (B) H&E staining of lungs from WT (left panel) and LPA1 KO mice (right panel). (C) No differences in the number of lung nodules were observed between WT and LPA2 KO mice. n = 12 WT mice, and n = 9 LPA2 KO mice. (D) H&E staining of lungs from WT (left panel) and LPA2 KO mice (right panel). (E) LPA5 KO mice had a reduced number of lung nodules. n = 10 WT mice, and n = 12 LPA5 KO mice. (F) H&E staining of lungs from WT (left panel) and LPA5 KO mice (right panel). P values are relative to control WT mice. Scale bar = 500 microns.
We also examined the effect of host LPA1, LPA2, and LPA5 receptors on tumor growth. However, no differences in the subcutaneous growth of B16F10 cells were observed between WT and the respective LPA receptor KO mice (Fig. 5A-F).
Figure 5.

Effect of host LPA receptor on the subcutaneous growth of B16F10. No significant differences in subcutaneous tumor growth were observed between WT and LPA1 KO (A & B), LPA2 KO (C & D), or LPA5 KO (E & F) mice.
Differential lung distribution of B16F10 cells at early time points in LPA1- and LPA5-KO mice
To gain insight into why lung metastases were reduced in LPA1- and LPA5-KO mice, we examined the distribution of B16F10 cells in mice at 24 h post-inoculation. To visualize cells at early time points, we transduced B16F10 cells with a GFP construct. The expression profiles of ATX and LPA receptors in GFP-B16F10 cells were unaltered, and the cells retained their ability to form lung metastasis when injected into mice (Supplementary Fig. 3). At 24 h post-injection, lungs were isolated, and GFP-B16F10 cells on the lung surface were visualized and quantified using an inverted fluorescent microscope. As shown in Fig. 6A and B, fewer GFP-B16F10 cells were seen on the lung surfaces of LPA1- and LPA5-KO mice, respectively. On the contrary, GFP-B16F10 cells appeared to be distributed to the same degree in both WT and LPA2 KO mice (Fig. 6A). No fluorescent cells were detected in control mice injected with unlabeled B16F10 cells (Fig. 6B). We demonstrated that the differences seen in the distribution of GFP-B16F10 cells in LPA1- and LPA5-KO mice were significant (Fig. 6C and D).
Figure 6.
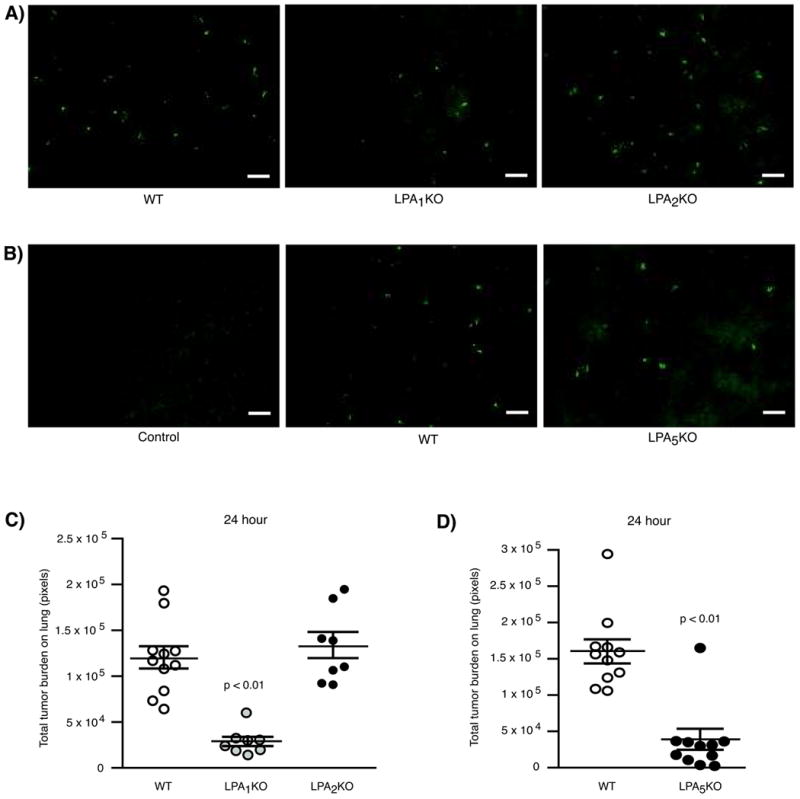
Distribution of GFP-B16F10 cells in mice at early time points. (A & B) Representative images of GFP-B16F10 cells on the lung surfaces of WT, LPA1-, LPA2-, and LPA5-KO mice, respectively. Control = injection with unlabeled B16F10 cells. Scale bar = 200 microns (C). Distribution of GFP-B16F10 cells in LPA1-, LPA2-, and (D) LPA5-KO mice at 24 h after injection via tail vein. n = 11 WT mice, n = 8 LPA1 KO mice, n = 8 LPA2 KO mice, and n = 10 LPA5 KO mice. P values are relative to control WT mice.
Discussion
In the present study, we showed that tumor- and stromal-LPA receptors play different roles in the invasion and metastasis of malignant melanoma. Our finding that LPA inhibits B16F10 invasion via the LPA5 receptor clearly supports recent data from Jongsma and colleagues, who demonstrated a similar anti-migratory effect of LPA5 in these cells in vitro (30). However, the anti-invasive attribute of LPA5 seen in these in vitro studies did not translate to in vivo observations. Even though knockdown of LPA5 in B16F10 cells did relieve the inhibitory effect of LPA on invasion, the magnitude of effect we observed in these in vitro assays was moderate (as shown in Fig. 2B), which could possibly explain the lack of differences seen in the in vivo studies. Another possible explanation for the discrepancy between the in vitro and in vivo findings is the involvement of additional pro-invasive mechanism(s) in vivo that are independent of LPA, which may override the anti-invasive attribute of LPA5 in B16F10 cells.
We also questioned the role of LPA6 and LPA2 on B16F10 invasion. Jongsma and colleagues ruled out the involvement of LPA6 in invasion, as knockdown of the receptor affected neither the basal invasion rate nor the inhibitory actions of LPA on B16F10 invasion (30). On the contrary, our data with the siRNA-mediated knockdown of LPA2 or the LPA2 antagonist compound 35 showed that the basal invasion rate of B16F10 was clearly reduced by blocking LPA2. Since these experiments were performed under serum-free conditions, one might question where the source of LPA is from and if ATX activity accounted for the basal invasion of B16F10 cells. We found that BMP22 dose-dependently reduced the rate of basal invasion in these cells (Supplementary Fig. 4). This suggests that B16F10 cells are capable of providing a source of substrate for ATX thereby generating its own pool of LPA for the activation of LPA2. Indeed, early studies have shown that cancer cell lines such as human A2058 melanoma cells and MDA-MB-231 breast cancer cells released detectable amounts of LPC into the culture medium (3), which could arguably be the case for B16F10 cells. The role of LPA in the regulation of matrix metalloproteinase (MMP) is well documented. In particular, the ATX-LPA-LPA1 signaling axis has been shown to induce MMP-9 expression in hepatocellular carcinoma (HCC) subsequently enhancing the invasive capacity of these cells (42). Similarly, studies by Do et al., reported a role for LPA in regulating MMP-2 activity in epithelial ovarian cancer (EOC) (43). Because MMPs play a vital role in invasion by degrading the extracellular matrix, it would be interesting to assess if LPA receptors differentially regulate MMP expression or activity in B16F10 cells.
Although we have unraveled a distinct role for host LPA1, LPA2, and LPA5 receptors in supporting the seeding of lung metastasis by B16F10 cells, the real challenge ahead lies in deciphering the mechanism(s) of action. The experimental metastasis model we used herein studies the late stages of the metastatic cascade. Nonetheless, it is still a complex process in which tumor cells must survive the harsh conditions in the systemic circulation, evade the host immune system, arrest or adhere at a distal vessel wall, and extravasate into the surrounding tissue to establish secondary metastasis (31). Hence, there is a possibility that host LPA1 and LPA5 receptors could be involved in any of these steps. Nevertheless, our observation that LPA1- and LPA5-KO mice had diminished GFP-B16F10 residence in the lungs at an early time point (24 h) suggests that an impaired tumor-platelet interaction, tumor-endothelial cell interaction or adhesion may be involved.
We found that mesothelial cells isolated from LPA1 KO mice were more resistant to invasion by MM1 hepatocarcinoma cells. Moreover, pharmacological inhibition of the LPA1 receptor with Ki16425 significantly reduced the invasion of MM1 cells across the HUVEC monolayer. It is important to note that MM1 cells predominantly expressed LPA2 and LPA6 receptors, whereas the expression level of LPA1 was low (32). Therefore, we postulate that Ki16425 most likely acts by antagonizing the LPA1 receptor in HUVEC to reduce the invasion by MM1 cells; HUVEC express LPA1 but not LPA3 receptors, thus ruling out the effect of Ki16425 on LPA3 receptors (44). In fact, earlier studies have shown that LPA is capable of causing endothelial barrier dysfunction and vascular permeability (45). A more recent study by Tager and colleagues described the involvement of LPA1 receptors in increasing vascular leakage following tissue injury in a bleomycin mouse model of pulmonary fibrosis (46). Although neither of these studies was conducted in the context of tumor-endothelial cell interaction, it certainly supports a positive role for the LPA-LPA1 receptor axis in regulating endothelial barrier function. Nonetheless, more studies are needed to further validate the involvement of endothelial LPA1 receptors in mediating tumor adhesion or invasion.
We determined that primary mouse lung microvascular endothelial cells (MLVEC) isolated from C57BL/6 mice predominantly express LPA6, LPA1, and LPA4 receptors. Since these cells do not express LPA5 receptors, we speculate that the diminished residence of GFP-B16F10 cells on the lung surface of LPA5 KO mice at an early time point may not be caused by an impaired tumor-endothelial cell interaction. Moreover, we demonstrated that the rate of MM1 invasion across mesothelial cells isolated from LPA5 KO mice was similar to that in WT mesothelium. Hence, what accounts for the reduced lung metastasis observed in LPA5 KO mice remains unknown. We considered one possible mechanism of action based on recent work by Oda et al., who found that activation of LPA5 receptors on cytotoxic CD8+ T cells inhibited T cell activation and proliferation. CD8+ T cells are a subset of immune cells that participate in tumor immunosurveillance; thus, inhibition of T cell activation may be advantageous for tumor cells in evading host immunity. Subsequently, the authors showed that the growth rate of established melanoma tumors in WT mice was reduced following the transfer of naïve LPA5-/- tumor-specific T cells (47). These findings suggest a potential role for LPA5 in regulating host immunity toward cancer progression. Therefore, additional studies are needed to assess the response of the host immune system in LPA5 KO mice toward metastasizing B16F10 cells. These studies should be focused on the early hours post tumor inoculation because by 24 h only few B16F10 cells can be detected in the lungs. Platelets interact with tumor cells and this interaction has been shown to increase LPA production and LPA-mediated bone metastasis formation by breast cancer cells (48). LPA5 has been shown to mediate platelet activation (40) and the lack of this receptor subtype in platelets might also impact LPA production in the tumor microenvironment. This hypothesis should be tested in subsequent studies.
To understand how host LPA receptors may potentially be educated by tumor cells to form a permissive microenvironment or “premetastatic niche,” we reviewed the LPA receptor expression profiles in other stromal elements of the lung microenvironment. Tager and colleagues have extensively profiled LPA receptor expression in fibroblast and leukocyte subsets of murine lungs. LPA1 was the predominant receptor expressed in lung fibroblasts, whereas alveolar macrophages predominantly expressed LPA5, followed by LPA4 and, to a lesser extent, LPA2 receptors. LPA2 and LPA5 were the most abundantly expressed receptors in CD4+ and CD8+ T lymphocytes (46). The majority of these cells play vital roles in shaping the microenvironment for cancer progression and metastasis, as reviewed in (49). It would be of interest to study whether host LPA receptors in these stromal elements affect TME interaction. We have also determined the ATX and LPA receptor expression in primary rat alveolar type II epithelial cells and differentiated type I-like epithelial cells (derived from type II cells), which line the pulmonary alveoli. We found that these cells express high levels of LPA2, P2Y10, and ATX and moderate levels of LPA6, LPA3, and LPA5 (Supplementary Fig. 5).
In summary, we show for the first time that LPA1- and LPA5-KO mice are protected from lung metastasis in this model. Although presently there is a lack of experimental evidence to elucidate the mechanism(s) involved in the homing of metastasizing tumor cells by host LPA receptors, future studies in which expression of selective LPA receptors can be regulated in a cell-type manner should help to clarify this issue.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Jerold Chun from the Scripps Research Institute (La Jolla, CA) for the generous gift of the LPA1- and LPA2-KO mice and Jin Emerson-Cobb for editorial assistance. This study was supported by the National Cancer Institute grant CA 092160 and the Harriet Van Vleet endowment.
Funding: National Cancer Institute grant CA092160 and the Harriet Van Vleet endowment (UTHSC).
Footnotes
Conflict of interest disclosure: The authors declare no competing financial interests.
References
- 1.Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, et al. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. The Journal of biological chemistry. 1992;267:2524–9. [PubMed] [Google Scholar]
- 2.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, et al. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. The Journal of biological chemistry. 2002;277:39436–42. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 3.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–33. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26:5015–22. doi: 10.1128/MCB.02419-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. The Journal of biological chemistry. 2006;281:25822–30. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- 6.Euer N, Schwirzke M, Evtimova V, Burtscher H, Jarsch M, Tarin D, et al. Identification of genes associated with metastasis of mammary carcinoma in metastatic versus non-metastatic cell lines. Anticancer research. 2002;22:733–40. [PubMed] [Google Scholar]
- 7.Kishi Y, Okudaira S, Tanaka M, Hama K, Shida D, Kitayama J, et al. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. The Journal of biological chemistry. 2006;281:17492–500. doi: 10.1074/jbc.M601803200. [DOI] [PubMed] [Google Scholar]
- 8.Nouh MA, Wu XX, Okazoe H, Tsunemori H, Haba R, Abou-Zeid AM, et al. Expression of autotaxin and acylglycerol kinase in prostate cancer: association with cancer development and progression. Cancer science. 2009;100:1631–8. doi: 10.1111/j.1349-7006.2009.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu JM, Xu Y, Skill NJ, Sheng H, Zhao Z, Yu M, et al. Autotaxin expression and its connection with the TNF-alpha-NF-kappaB axis in human hepatocellular carcinoma. Molecular cancer. 2010;9:71. doi: 10.1186/1476-4598-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altman MK, Gopal V, Jia W, Yu S, Hall H, Mills GB, et al. Targeting melanoma growth and viability reveals dualistic functionality of the phosphonothionate analogue of carba cyclic phosphatidic acid. Molecular cancer. 2010;9:140. doi: 10.1186/1476-4598-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brindley DN, Lin FT, Tigyi GJ. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochimica et biophysica acta. 2013;1831:74–85. doi: 10.1016/j.bbalip.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Fang XJ, Casey G, Mills GB. Lysophospholipids activate ovarian and breast cancer cells. The Biochemical journal. 1995;309(Pt 3):933–40. doi: 10.1042/bj3090933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie Y, Gibbs TC, Mukhin YV, Meier KE. Role for 18:1 lysophosphatidic acid as an autocrine mediator in prostate cancer cells. The Journal of biological chemistry. 2002;277:32516–26. doi: 10.1074/jbc.M203864200. [DOI] [PubMed] [Google Scholar]
- 14.Baker DL, Morrison P, Miller B, Riely CA, Tolley B, Westermann AM, et al. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA. 2002;287:3081–2. doi: 10.1001/jama.287.23.3081. [DOI] [PubMed] [Google Scholar]
- 15.Masuda A, Nakamura K, Izutsu K, Igarashi K, Ohkawa R, Jona M, et al. Serum autotaxin measurement in haematological malignancies: a promising marker for follicular lymphoma. British journal of haematology. 2008;143:60–70. doi: 10.1111/j.1365-2141.2008.07325.x. [DOI] [PubMed] [Google Scholar]
- 16.Nakai Y, Ikeda H, Nakamura K, Kume Y, Fujishiro M, Sasahira N, et al. Specific increase in serum autotaxin activity in patients with pancreatic cancer. Clinical biochemistry. 2011;44:576–81. doi: 10.1016/j.clinbiochem.2011.03.128. [DOI] [PubMed] [Google Scholar]
- 17.Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, et al. LPA Receptors: Subtypes and Biological Actions. Annual Review of Pharmacology and Toxicology. 2010;50:157–86. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 18.Muller R, Berliner C, Leptin J, Portner D, Bialecki W, Kleuser B, et al. Expression of sphingosine-1-phosphate receptors and lysophosphatidic acid receptors on cultured and xenografted human colon, breast, melanoma, and lung tumor cells. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2010;31:341–9. doi: 10.1007/s13277-010-0043-7. [DOI] [PubMed] [Google Scholar]
- 19.Willier S, Butt E, Grunewald TG. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: a focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biology of the cell / under the auspices of the European Cell Biology Organization. 2013;105:317–33. doi: 10.1111/boc.201300011. [DOI] [PubMed] [Google Scholar]
- 20.Ishdorj G, Graham BA, Hu X, Chen J, Johnston JB, Fang X, et al. Lysophosphatidic acid protects cancer cells from histone deacetylase (HDAC) inhibitor-induced apoptosis through activation of HDAC. The Journal of biological chemistry. 2008;283:16818–29. doi: 10.1074/jbc.M710177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M, Towers LN, O'Connor KL. LPA2 (EDG4) mediates Rho-dependent chemotaxis with lower efficacy than LPA1 (EDG2) in breast carcinoma cells. American journal of physiology Cell physiology. 2007;292:C1927–33. doi: 10.1152/ajpcell.00400.2006. [DOI] [PubMed] [Google Scholar]
- 22.Shida D, Fang X, Kordula T, Takabe K, Lepine S, Alvarez SE, et al. Cross-talk between LPA1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer research. 2008;68:6569–77. doi: 10.1158/0008-5472.CAN-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boucharaba A, Serre CM, Guglielmi J, Bordet JC, Clezardin P, Peyruchaud O. The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9643–8. doi: 10.1073/pnas.0600979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitayama J, Shida D, Sako A, Ishikawa M, Hama K, Aoki J, et al. Over-expression of lysophosphatidic acid receptor-2 in human invasive ductal carcinoma. Breast cancer research : BCR. 2004;6:R640–6. doi: 10.1186/bcr935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.So J, Wang FQ, Navari J, Schreher J, Fishman DA. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2) Gynecologic oncology. 2005;97:870–8. doi: 10.1016/j.ygyno.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee A, Wu J, Barbour S, Fang X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. The Journal of biological chemistry. 2012;287:24990–5000. doi: 10.1074/jbc.M112.340083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shida D, Watanabe T, Aoki J, Hama K, Kitayama J, Sonoda H, et al. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Laboratory investigation; a journal of technical methods and pathology. 2004;84:1352–62. doi: 10.1038/labinvest.3700146. [DOI] [PubMed] [Google Scholar]
- 28.Harper K, Arsenault D, Boulay-Jean S, Lauzier A, Lucien F, Dubois CM. Autotaxin promotes cancer invasion via the lysophosphatidic acid receptor 4: participation of the cyclic AMP/EPAC/Rac1 signaling pathway in invadopodia formation. Cancer research. 2010;70:4634–43. doi: 10.1158/0008-5472.CAN-09-3813. [DOI] [PubMed] [Google Scholar]
- 29.Lee Z, Cheng CT, Zhang H, Subler MA, Wu J, Mukherjee A, et al. Role of LPA4/p2y9/GPR23 in negative regulation of cell motility. Molecular biology of the cell. 2008;19:5435–45. doi: 10.1091/mbc.E08-03-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jongsma M, Matas-Rico E, Rzadkowski A, Jalink K, Moolenaar WH. LPA is a chemorepellent for B16 melanoma cells: action through the cAMP-elevating LPA5 receptor. PloS one. 2011;6:e29260. doi: 10.1371/journal.pone.0029260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, et al. Tumor Microenvironment Complexity: Emerging Roles in Cancer Therapy. Cancer research. 2012;72:2473–80. doi: 10.1158/0008-5472.CAN-12-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupte R, Patil R, Liu J, Wang Y, Lee SC, Fujiwara Y, et al. Benzyl and naphthalene methylphosphonic acid inhibitors of autotaxin with anti-invasive and anti-metastatic activity. ChemMedChem. 2011;6:922–35. doi: 10.1002/cmdc.201000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gotoh M, Fujiwara Y, Yue J, Liu J, Lee S, Fells J, et al. Controlling cancer through the autotaxin-lysophosphatidic acid receptor axis. Biochemical Society transactions. 2012;40:31–6. doi: 10.1042/BST20110608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beck HP, Kohn T, Rubenstein S, Hedberg C, Schwandner R, Hasslinger K, et al. Discovery of potent LPA2 (EDG4) antagonists as potential anticancer agents. Bioorganic & medicinal chemistry letters. 2008;18:1037–41. doi: 10.1016/j.bmcl.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 35.Uchiyama A, Mukai M, Fujiwara Y, Kobayashi S, Kawai N, Murofushi H, et al. Inhibition of transcellular tumor cell migration and metastasis by novel carba-derivatives of cyclic phosphatidic acid. Biochimica et biophysica acta. 2007;1771:103–12. doi: 10.1016/j.bbalip.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desai LP, Chapman KE, Waters CM. Mechanical stretch decreases migration of alveolar epithelial cells through mechanisms involving Rac1 and Tiam1. Am J Physiol Lung Cell Mol Physiol. 2008;295:L958–65. doi: 10.1152/ajplung.90218.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobbs LGI. solation and culture of alveolar type II cells. American journal of physiology Cell physiology. 1990;258:L134–47. doi: 10.1152/ajplung.1990.258.4.L134. [DOI] [PubMed] [Google Scholar]
- 38.Yue J, Sheng Y, Ren A, Penmatsa S. A miR-21 hairpin structure-based gene knockdown vector. Biochemical and biophysical research communications. 2010;394:667–72. doi: 10.1016/j.bbrc.2010.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferguson CG, Bigman CS, Richardson RD, van Meeteren LA, Moolenaar WH, Prestwich GD. Fluorogenic phospholipid substrate to detect lysophospholipase D/autotaxin activity. Organic letters. 2006;8:2023–6. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams JR, Khandoga AL, Goyal P, Fells JI, Perygin DH, Siess W, et al. Unique Ligand Selectivity of the GPR92/LPA5 Lysophosphatidate Receptor Indicates Role in Human Platelet Activation. Journal of Biological Chemistry. 2009;284:17304–19. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukai M, Nakamura H, Tatsuta M, Iwasaki T, Togawa A, Imamura F, et al. Hepatoma cell migration through a mesothelial cell monolayer is inhibited by cyclic AMP-elevating agents via a Rho-dependent pathway. FEBS letters. 2000;484:69–73. doi: 10.1016/s0014-5793(00)02129-3. [DOI] [PubMed] [Google Scholar]
- 42.Park SY, Jeong KJ, Panupinthu N, Yu S, Lee J, Han JW, et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene. 2010;30:1351–9. doi: 10.1038/onc.2010.517. [DOI] [PubMed] [Google Scholar]
- 43.Do TV, Symowicz JC, Berman DM, Liotta LA, Petricoin EF, Stack MS, et al. Lysophosphatidic Acid Down-Regulates Stress Fibers and Up-Regulates Pro-Matrix Metalloproteinase-2 Activation in Ovarian Cancer Cells. Molecular Cancer Research. 2007;5:121–31. doi: 10.1158/1541-7786.MCR-06-0319. [DOI] [PubMed] [Google Scholar]
- 44.Ptaszynska MM, Pendrak ML, Stracke ML, Roberts DD. Autotaxin signaling via lysophosphatidic acid receptors contributes to vascular endothelial growth factor-induced endothelial cell migration. Molecular cancer research : MCR. 2010;8:309–21. doi: 10.1158/1541-7786.MCR-09-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amerongen GPvN, Vermeer MA, van Hinsbergh VWM. Role of RhoA and Rho Kinase in Lysophosphatidic Acid-Induced Endothelial Barrier Dysfunction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:e127–e33. doi: 10.1161/01.atv.20.12.e127. [DOI] [PubMed] [Google Scholar]
- 46.Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nature Medicine. 2007;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 47.Oda SK, Strauch P, Fujiwara Y, Al-Shami A, Oravecz T, Tigyi G, et al. Lysophosphatidic Acid Inhibits CD8 T-cell Activation and Control of Tumor Progression. Cancer Immunology Research. 2013;1:245–55. doi: 10.1158/2326-6066.CIR-13-0043-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boucharaba A, Serre CM, Grès S, Saulnier-Blache JS, Bordet JC, Guglielmi J, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. Journal of Clinical Investigation. 2004;114:1714–25. doi: 10.1172/JCI22123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.