

Abstract
Exocyclic etheno-DNA adducts are mutagenic and carcinogenic and are formed by the reaction of lipidperoxidation (LPO) products such as 4-hydoxynonenal or malondialdehyde with DNA bases. LPO products are generated either via inflammation driven oxidative stress or via the induction of cytochrome P-450 2E1 (CYP2E1). In the liver CYP2E1 is induced by various compounds including free fatty acids, acetone and ethanol. Increased levels of CYP2E1 and thus, oxidative stress are observed in the liver of patients with non-alcoholic steatohepatitis (NASH) as well as in the chronic alcoholic. In addition, chronic ethanol ingestion also increases CYP2E1 in the mucosa of the oesophagus and colon. In all these tissues CYP2E1 correlates significantly with the levels of carcinogenic etheno-DNA adducts. In contrast, in patients with non-alcoholic steatohepatitis (NASH) hepatic etheno-DNA adducts do not correlate with CYP2E1 indicating that in NASH etheno-DNA adducts formation is predominately driven by inflammation rather than by CYP2E1 induction. Since etheno-DNA adducts are strong mutagens producing various types of base pair substitution mutations as well as other types of genetic damage, it is strongly believed that they are involved in ethanol mediated carcinogenesis primarily driven by the induction of CYP2E1.
Keywords: Cytochrome P450-2E1, Etheno-DNA adducts, Lipidperoxidation products, Ethanol, Carcinogenesis, Non-alcoholic fatty liver disease
Graphical abstract
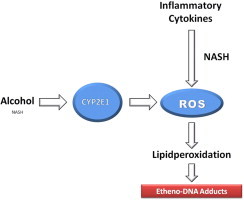
Generation of reactive oxygen species (ROS) either via inflammatory cytokines (predominately in NASH) or via cytochrome P-450 2E1 induction (predominately by ethanol, but also in NASH). ROS lead to lipidperoxidation and lipidperoxidation products result in the formation of carcinogenic etheno- or propano-DNA adducts.
Highlights
-
•
Cytochrome P-450 2E1 is induced following chronic ethanol ingestion.
-
•
CYP2E1 correlates with carcinogenic etheno-DNA formation.
-
•
CYP2E1 and oxidative stress are important mechanisms in alcohol mediated carcinogenesis in the liver, undefined and colon.
-
•
In NASH hepatic etheno-DNA adducts occur but possible due to inflammation.
Introduction
Oxidative stress is an important mechanism in the pathogenesis of many diseases including cancer. The generation of reactive oxygen species (ROS) with consecutive DNA damage is an initial step in carcinogenesis induced by inflammatory processes. During inflammation ROS is generated among others through various cytokines, but also through other mechanisms such as the induction of cytochrome P4502E1 (CYP2E1) as demonstrated following chronic alcohol consumption [1,2]. This review will focus on the effect of alcohol on CYP2E1 and its role in ROS formation, but major emphasis will be led on the generation of carcinogenic exocyclic etheno-DNA adducts as a consequence of the reaction between lipidperoxidation products generated by ROS and DNA bases following ethanol administration in vitro and in vivo. It will be shown that these etheno-DNA adducts following chronic ethanol consumption are of major importance with respect to ethanol mediated carcinogenesis in the liver and in other tissues.
Inflammation, oxidative stress and DNA damage
Chronic inflammation induced by various agents including viruses and bacteria is associated with an increased cancer risk due to tissue damage and genetic instability [3–7]. Oxidative stress with the generation of ROS may occur in chronic infection and inflammation primarily due to the generation of nitric oxide (NO), superoxide anion (O2.−) and other ROSs by macrophages and neutrophils that infiltrate the inflamed tissue [8,9].
Activated inflammatory cells in various tissues including the liver in turn induce oxidant generating enzymes such as NADPH oxidase, inducible nitric oxide synthetase (iNOS), xanthine oxidase (XO) and myeloperoxidase (MPO) [10,11]. In such conditions ROS and reactive nitrogen species (RNS) are generated. As a consequence ROS and RNS can damage DNA, RNA, lipids and proteins through nitration and oxidation resulting in an increased mutation load [10,11].
Furthermore, cytokines are released in inflammatory tissues which not only activate the above mentioned enzymes to create ROS and RNS, which also activate NFκB a nuclear transcription factor which among others stimulates cyclooxygenase 2(COX2), lipoxygenase (LOX), and iNOS [4,10–12]. Upregulation of iNOS, COX2, and LOX results also in an overproduction of ROS and RNS [10].
iNOS catalyses nitric oxide (NO) generation which reacts with oxygen to produce N2O3 a strong nitrosating compound which deaminates DNA bases and react with secondary amines to form N-nitrosoamines which are highly carcinogenic [10].
Another reaction with O2 leads to peroxynitrite with the formation of 8-nitroguanine. Peroxynitrite also results in single strand breakage of DNA [7,10].
COX2 catalyse the conversion of arachidonic acid (AA) to prostaglandins is inducible by various factors including NFκB, cytokines and tumour promoters and may influence apoptosis, angiogenesis, tumour invasion, but also the generation of oxidative stress [12,13]. An upregulation of COX2 has been shown in familial adenomatous polyposis (FAP) and in the Apc Min mouse model which resembles FAP and this was associated with a highly significant increase in various etheno-DNA lesions [13–16].
LOX metabolizes AA to hydroxyeicosatetraenoic acids (HETEs) or leukotrienes. It has been shown in mouse skin carcinogenesis that LOX isoenzymes are overactivated and some of the metabolites cause chromosomal damage which was found to be inhibited by LOX inhibitors [17,18].
Thus, all the factors mentioned above lead to the generation of ROS and RNS with consequent lipid peroxidation and the production of lipidperoxidation products such as 4-hydoxynonenal (4HNE), 4-hydoxyhydroperoxy-2-nonenal (HPNE) and malondialdehyde (MDA) (Fig. 1). These lipidperoxidation products react with DNA either directly or through bifunctional intermediates to form various promutagenic exocyclic etheno-DNA adducts. Some major types are depicted in Fig. 2.
Fig. 1.
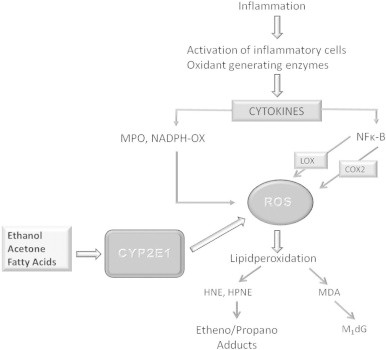
Simplified pathophysiology of reactive oxygen species (ROS) and etheno-DNA adduct formation. Inflammation driven cytokine secretion results among others in NFκB activation and in the activation of NADPH oxidase (NADPH-Ox) as well as myeloperoxidase (MPO). NFκB is also activated by acetaldehyde, the first metabolite of ethanol oxidation. NFκB stimulates lipoxigenase (LOX), cyclooxygenase 2 (COX2), and inducible nitric oxide synthase (iNOS). As a result ROS and reactive nitrogen species (RNS) are generated, which lead to lipidperoxidation with the occurrence of lipidperoxidation products such as 4-hydroxynonenal (4-HNE), 4-hydroxyhydroperoxy-2-nonenal (HPNE), and malondialdehyde (MDA). These adducts react with DNA bases to form exocyclic etheno/propane-DNA adducts. Chronic alcohol consumption results in the induction of cytochrome P-4502 E1 which is involved in ethanol oxidation through the microsomal ethanol oxidizing pathway. During this reaction ROS is generated without inflammation. Other compounds such as free fatty acids or acetone also induce CYP2E1 which is especially relevant in nonalcoholic fatty liver disease (NAFLD), when the liver is loaded with fat and in patients with diabetes mellitus when acetone is generated in the liver.
Fig. 2.
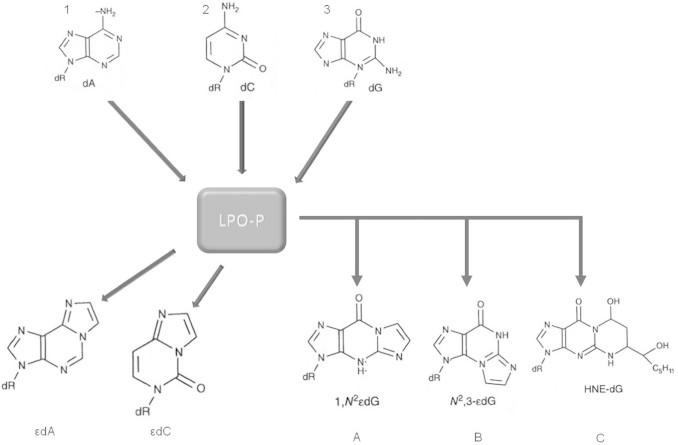
Generation of various etheno DNA-adducts. Deoxyadenine, deoxycytosine, and deoxyguanosine react with lipidperoxidation products to form 1N6-ethano-2′-deoxyadenosine (εdA), 3,N4-etheno-2′-deoxycytidine (εdC), and 1,N2-etheno-2′-deoxyguanosine (1,N2εdG), N2,3-etheno-2′-deoxyguanosine (N2,3-εdG), and HNE-derived 1,N2-propano-2′-deoxyguanosine adduct (HNE-dG).
LPO products derived from y-linoleic acid, including HNE, a major LPO product and its electrophilic epoxy-, hydroperoxy-, and oxo-enal intermediates react with the DNA bases A, C, and G to yield inter alia the unsubstituted etheno-DNA adducts 1,N6-etheno-2′-deoxyadenosine (εdA), 3, N4-etheno-2′-deoxycytidine (εdC), 1,N2-etheno-2′-deoxyguanosine (1,N2εdG), and N2,3-etheno-2′-deoxyguanosine (N2,3εdG). In addition, also substituted base adducts are formed such as HNE-dG carrying a fatty acid chain residue (Fig. 2). 2,N4-etheno-5-methyl-2′-deoxycytidine (ε5mdC), an endogenous, hitherto unknown LPO-derived adduct was identified in the DNA of human tissue which could play a role in epigenetic mechanisms of carcinogenesis [10,19–27].
In addition, DNA can also be modified directly by ROS and RNS to 8-nitro-dG and 8-Oxo-dG [28]. All of these DNA changes have been detected in human specimens [29–34].
The importance of etheno DNA adducts in carcinogenesis
Exocyclic etheno-DNA adducts exhibit strong mutagenic properties producing various types of base pair substitution mutations and other types of genetic damage in all organisms tested so far [35,36]. εdA can lead to AT→GC transition and AT→TA and AT→CG transversions [37,38]. εdC can cause CG→AT transversions and CG→TA transition [39,40], and N2,3εdG can lead to GC→AT transition [40]. Incorporation of a single εdA in either DNA strand of HeLa cells showed a similar miscoding frequency and was more mutagenic than 8-oxo-dG [41].
Some etheno-adducts are poorly repaired in some tissues and cells supporting their biological relevance [42]. Strong support that etheno-DNA adducts play a causal role in the initiation and progression of liver carcinogenesis comes from the formation of εdA and εdC in vivo by the human liver carcinogen vinyl chloride [43] and by the potent multiorgan, multispecies carcinogen urethane via their reactive epoxy-intermediates [44]. The biological importance of etheno-DNA adducts is further stressed as they are preferentially formed in codon 249 of TP53 (which encodes p53), leading to a mutation that renders cells more resistant to apoptosis and provides them some growth advantage [45].
LPO-derived reactive products and their macromolecular interactions have been so far characterized primarily by in vitro studies, making it difficult, to pinpoint the main precursors and pathways involved in the generation of cancer-relevant DNA damage in human in vivo. For this reason earlier studies analysed in human specimens εdA and εdC as maker lesions for several other exocyclic adducts that could be formed with DNA in vivo, for which sensitive detection methods were not yet available.
Using ultrasensitive and specific detection methods [46], two miscoding etheno-DNA adducts εdA and εdC and also ε5mdC were unequivocally identified in humans. Samples were collected from “at-risk” patients affected by chronic inflammatory processes, persistent viral infections, iron storage- and alcohol-related diseases or exposed to inherited/acquired cancer risk factors. Adduct levels increased 10–100-fold progressively in human cancer-prone organs including liver, bile duct, oesophagus, colon and pancreas. Consistent results were also observed in rodent tumour models, that mimick human disease (for review [47]). Taken together these data incriminate LPO-derived adducts as strongly mutagenic cancer-causing lesions.
Alcohol and oxidative stress
Chronic ethanol consumption may results in the development of alcoholic liver disease (ALD) and cancer of various sites including the liver and the upper aerodigestive tract [2,48]. One mechanism by which alcohol exerts its deleterious effects is the generation of ROS. As already pointed out the formation of ROS such as superoxide anion (O2−) and hydrogen peroxide (H2O2) causes oxidative injury [1,2]. ROS as well as acetaldehyde, the first metabolite of ethanol oxidation, both activate NFκB an important transcription factor involved in carcinogenesis [48]. Inflammation driven oxidative stress including activated hepatic macrophages as observed in alcoholic hepatitis (AH) is predominantly responsible for the generation of ROS in AH [49]. Also hepatic iron overload as observed in the alcoholic increases ROS [50,51]. Furthermore, ethanol also results in the increase of iNOS with an increased production of nitric oxide and the generation of the highly reactive peroxynitrite (ONOO−) [52].
In addition, several enzyme systems are capable to produce ROS including CYP2E1 as part of the microsomal ethanol oxidizing system (MEOS) which metabolizes ethanol to acetaldehyde in the presence of oxygen and NADPH [53,54]. This system is of major importance, since it can be induced by chronic consumption of ethanol. It has been shown that CYP2E1 induction occurs already at a daily ethanol dose of 40 g and already at 1 week of consumption, which is further enhanced with time. However, an interindividual intensity of CP2E1 increase has been observed. Some individuals react to alcohol consumption with a striking CYP2E1 induction, while others reveal only a weak induction of CYP2E1 [55]. It is noteworthy that CYP2E1 induction by ethanol may also depend on dietary factors since medium chain triglycerides diminish CYP2E1 induction as compared to the application of long chain triglycerides in animal experiments [56].
CYP2E1 has a high rate of NADPH oxidase activity, resulting in the generation of large quantities of O2−, H2O2 and hydroxyethyl radicals [1,2,57]. Thus, CYP2E1 dependent microsomal ethanol oxidation produces ROS leading to lipidperoxidation with the generation of lipidperoxidation products such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) [1,2].
The role of CYP2E1 in the formation of ROS, in the progression of ALD and in ethanol mediated carcinogenesis has been clearly demonstrated [2,58–62]. Thus, the severity of ALD was significantly enhanced in CYP2E1 overexpressing mice [63], and reduced in CYP2E1 knockout mice [62]. When chlormethiazole (CMZ), a strong and specific CYP2E1 inhibitor was given in addition to an ethanol containing diet which induces ALD a significant reduction of ROS and RNS was noted in the liver of these animals [62] associated with a striking improvement of the liver disease [64]. Subsequently, oxidized DNA lesions have been found to be lower in CYP2E1 knockout mice as compared to wild type mice following chronic alcohol administration [65].
CYP2E1 induction and etheno-DNA adduct generation in the liver
Effect of ethanol
We have recently investigated the effect of CYP2E1 on lipidperoxidation products and etheno-DNA lesions in HepG2 cells overexpressing CYP2E1, and in humans with alcoholic liver disease [66]. When HepG2 cells overexpressing CYP2E1 were incubated with increasing concentrations of ethanol up to 50 mM, an increasing load of εdA and εdC could be detected as compared to control cells. This was not only a concentration dependent, but also a time dependent process. However, when 20 µM CMZ were added to the cell culture a highly significant inhibition of the generation of etheno-DNA adducts was observed [66].
Since increased levels of εdA adducts have been observed in the hepatic nuclei of patients with ALD (Fig. 3) [30], we extended our experiments and studied liver biopsies from alcoholic patients with various severities of ALD by using immunohistology for the detection of CYP2E1 and etheno-DNA adducts. Again there was a significant correlation between CYP2E1, the lipidperoxidation product 4-HNE and εdA, as well as εdC [66].
Fig. 3.
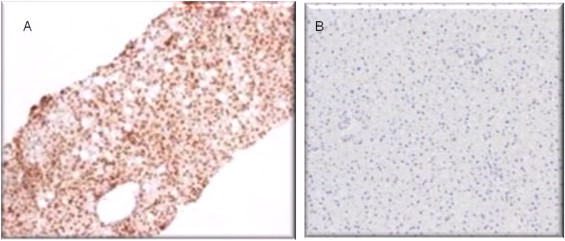
Immunohistology of εdA in two liver biopsies from patients with alcoholic liver disease (A) and control patients with a normal liver (B). The brownish colour shows εdA. This adduct occurs in the nuclei of the hepatocytes and the percentage of nuclei positive hepatocytes can be counted. A significant load of etheno adducts is observed in ALD, while the control healthy liver reveals background activity only.
Most recently, we have investigated CYP2E1 and exocyclic etheno-DNA adducts in a large cohort study of 97 alcoholics with non-cirrhotic ALD. All patients were liver biopsied, histologically evaluated and CYP2E1 as well as etheno-DNA adducts were immunohistologically determined. As a result a strong significant correlation between CYp2E1 and εdA (p=0.0001) has been observed (Seitz, personal observation).
Non-alcoholic fatty liver disease (NAFLD)
CYP2E1 is not only induced by chronic ethanol ingestion, but also in NAFLD [67–69] possibly by free fatty acids and acetone [54]. Although, this induction is less pronounced as in ALD, it also has severe consequences with respect to the generation of oxidative stress. Indeed, inflammation driven oxidative stress may be an important mechanism in the progression of NAFLD [70]. We therefore determined CYP2E1 as well as εdA in liver biopsies from patients with pure non-alcoholic fatty liver and patients with NASH using immunohistology. εdA was detected in a broad range of intensity and correlated significantly with the severity of inflammation, but not with CYP2E1 (Linhart and Seitz, personal communication).
NAFLD also is an increasing health problem in children [71–73]. As reported recently, oxidative stress as measured by the hepatic expression of 8-hydroxy-2-deoxyguanosine (8-OHG), serum protein carbonyls, and circulating antibody against malondialdehyde adducted human serum albumin has been frequently found in children with NAFLD and was also found to be associated with an increased severity of steatohepatitis [74]. Therefore, we determined εdA, and CYP2E1 in liver biopsies of children with NASH. In these studies we also could show for the first time that not only CYP2E1 was found to be increased, but also that εdA occurs. In a few of these children at an age below 15 years and the diagnosis of diabetes mellitus a striking load of εdA was found in the nuclei of their hepatocytes [75]. In contrast to ALD these adducts did not significantly correlate with CYP2E1, but rather with the state of inflammation. Thus, inflammatory driven ROS production may be predominant to explain εdA formation in patients with NAFLD.
In animal experiments, the progression of NASH is influenced by the concomitant administration of ethanol [76,77]. This has been shown in dietary induced NASH, which could be due among others to oxidative, nitrosative, and mitochondrial stress, as well as increased inflammation and cellular apoptosis [76,77].
Furthermore, in the Zucker rat, a leptin deficiency and insulin resistance genetic NASH model the administration of ethanol not only increased CYP2E1, but also εdA in a linear way [66].
The fact that ethanol consumption in patients with NASH enhances oxidative stress possibly predominantly by a further increase in CYP2E1 associated with the generation of highly carcinogenic etheno-DNA lesions may of special interest in the context that patients with NASH have significant higher HCC risk and develop HCC in a much shorter time frame when they consume alcohol even at social levels [78].
CYP2E1 induction and etheno-DNA adduct generation in the oesophagus and in the colorectal mucosa
Chronic alcohol consumption is a major risk factor for undefined cancer [2,79]. Various mechanisms may mediate carcinogenesis including the genotoxic effect of acetaldehyde and oxidative stress [2,79–81]. As discussed above for the liver, ethanol may also exert its carcinogenic effect in other tissues among others via the induction of CYP2E1 and the generation of carcinogenic etheno-DNA adducts. Therefore, we investigated if such effects can also be observed in the human oesophagus [82]. We studied undefined biopsies of 37 patients with upper aerodigestive tract cancer and heavy alcohol consumption of more than 100 g on average per day as well as 16 controls without tumours (12 teetotallers and 4 subjects with a maximum of 25 g ethanol/day). CYP2E1, etheno-DNA adducts and Ki67 as a marker for cell proliferation were determined immunohistologically in the undefined mucosa adjacent to the tumour. Chronic alcohol ingestion resulted in a significant induction of CYP2E1 which correlated with the amount of alcohol consumed. Furthermore, a significant correlation between CYP2E1 and the generation of the carcinogenic exocyclic etheno-DNA adducts εdA and εdC was observed. Etheno-DNA adducts also correlated significantly with cell proliferation, which was especially enhanced in patients who both drank and smoked. The results showed clearly again a correlation between CYP2E1 and etheno-DNA adducts. In contrast to the liver this induction correlated significantly with the amount of ethanol ingested [82].
More recently, we also investigated immunohistologically the effect of ethanol on colorectal CYP2E1 and etheno-DNA adducts in colorectal biopsies from 31 alcoholics and 15 non-drinking controls (Linhart and Seitz, personal communication). Again we found a significant correlation between the two parameters. It is interesting that chronic ethanol consumption using a Lieber DeCarli diet resulted in a hyperproliferation of the colorectal mucosa with an extension of the proliferative compartment towards the lumen of the crypt, which is a first step in carcinogenesis of this tissue [83]. A similar observation was made in patients with heavy alcohol consumption [84]. The administration of vitamin E, a radical scavenger, however, reduced the proliferative rate significantly emphasizing indirectly that most likely oxidative stress induced by CYP2E1 may be responsible for this regenerative behaviour [85].
CYP2E1 and experimental hepatocarcinogenesis
It has been believed for a long time that ethanol by itself is not a carcinogen rather than a co-carcinogen or a tumour promoter. However, meanwhile various animal studies have demonstrated that the administration of ethanol alone without any chemical carcinogen can result in tumours of the liver [86], the upper aerodigestive tract [87], the mammary gland [88] and the intestine [89]. Besides the fact that DNA lesions induced either by the binding of acetaldehyde to DNA [2,48] or by the reaction of DNA with ROS do occur during chronic ethanol consumption, the role of CYP2E1 in this process has not intensively investigated.
In a series of experiments we used an animal model in which a small amount of diethylnitrosamine (20 mg/kg b.wt.) was administered once to initiate hepatocarcinogenesis [90,91]. CYP2E1, inflammatory proteins, cell proliferation, protein bound 4-HNE, etheno-DNA adducts as well as 8-hydroxy-2′-deoxyguanosine (8-OHdG), retinoid concentrations and hepatic carcinogenesis were examined. Chronic ethanol ingestion for 1 month resulted in increased CYP2E1 levels and an increased nuclear accumulation of NFκB protein. In addition, TNFα expression was also enhanced associated with increased cyclin D1 expression and p-GST positive altered hepatic foci. All these changes were significantly inhibited by the concomitant administration of CMZ. Following 10 months of ethanol feeding hepatocellular adenoma were detected in ethanol fed rats only, but not in control rats. The administration of CMZ inhibited completely the formation of hepatic adenomas. In addition, 8-OHdG formation was found to be significantly increased after alcohol and almost normalized with CMZ. Although, etheno-DNA adduct formation increased following ethanol ingestion and decreased with CMZ, this effect was not significant.
More recently, Tsuchishima and co-workers produced hepatocellular carcinoma in mice without any additional insult. This process was significantly associated with the expression of CYP2E1 [92].
Summary
The most important mechanism associated with oxidative stress and the generation of ROS is chronic inflammation. During inflammation cytokines are liberated resulting in the activation of oxidant generation enzymes such as NADPH oxidase, and NFκB with the activation of LOX, Cox-2 and iNOS finally leading to the formation of ROS. In addition, ROS can also be generated through CYP2E1 which is induced by chronic alcohol consumption as well as in NASH where free fatty acids as well as acetone (mostly in diabetics) induce CYP2E1. ROS leads to lipidperoxidation with the occurrence of lipidperoxidation products such as 4-HNE and MDA. Both compounds can bind to DNA forming highly carcinogenic etheno-DNA adducts. In a series of experiments we could show that a significant correlation exists between CYP2E1 levels and etheno-DNA adduct formation in cell culture, animal experiments and biopsies from patients with ALD. Since in NASH the inflammatory process predominates as compared to the induction of CYP2E1 the etheno-DNA adduct levels do not correlate with CYP2E1 but rather with the intensity of the inflammatory process.
Conclusion
Cell culture and animal experiments as well as clinical biopsy studies in patients with ALD emphasize an important role of CYP2E1 in alcohol mediated carcinogenesis in the liver, but also in other tissues. In addition, CYP2E1 seems to be a driving force in the progression of ALD. Inhibition of CYP2E1 by a nontoxic inhibitor may be a successful approach in the treatment of ALD and alcohol mediated carcinogenesis.
Acknowledgements
The authors want to thank Ms. Grönebaum for typing the manuscript. Original studies were supported by the Dietmar Hopp Foundation and by the Manfred Lautenschläger Foundation.
References
- 1.Albano E. Alcohol, oxidative stress and free radical damage. Proceedings of the Nutrition Society. 2006;65:278–290. doi: 10.1079/pns2006496. 16923312 [DOI] [PubMed] [Google Scholar]
- 2.Seitz H.K., Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nature Reviews: Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. 17646865 [DOI] [PubMed] [Google Scholar]
- 3.Coussens L.M., Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. 12490959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hussain S.P., Hofseth L.J., Harris C.C. Radical causes of cancer. Nature Reviews: Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. 12671666 [DOI] [PubMed] [Google Scholar]
- 5.Kawanishi S., Hiraku Y. Oxidative and nitrative DNA damage as biomarker for carcinogenesis with special reference to inflammation. Antioxidants and Redox Signaling. 2006;8:1047–1058. doi: 10.1089/ars.2006.8.1047. 16771694 [DOI] [PubMed] [Google Scholar]
- 6.Mantovani A., Allavena P., Sica A., Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. 18650914 [DOI] [PubMed] [Google Scholar]
- 7.Ohshima H., Bartsch H. Chronic infections and inflammatory processes as cancer risk factors: possible role of nitric oxide in carcinogenesis. Mutation Research. 1994;305:253–264. doi: 10.1016/0027-5107(94)90245-3. 7510036 [DOI] [PubMed] [Google Scholar]
- 8.Karin M., Lawrence T., Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. 16497591 [DOI] [PubMed] [Google Scholar]
- 9.Lonkar P., Dedon P.C. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. International Journal of Cancer: Journal International du Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. 21387284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartsch H., Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbeck's Archives of Surgery/Deutsche Gesellschaft für Chirurgie. 2006;391:499–510. doi: 10.1007/s00423-006-0073-1. 16909291 [DOI] [PubMed] [Google Scholar]
- 11.Karin M., Greten F.R. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nature Reviews: Immunology. 2005;5:749–759. doi: 10.1038/nri1703. 16175180 [DOI] [PubMed] [Google Scholar]
- 12.Zha S., Yegnasubramanian V., Nelson W.G., Isaacs W.B., De Marzo A.M. Cyclooxygenases in cancer: progress and perspective. Cancer Letters. 2004;215:1–20. doi: 10.1016/j.canlet.2004.06.014. 15374627 [DOI] [PubMed] [Google Scholar]
- 13.Prescott S.M., White R.L. Self-promotion? Intimate connections between APC and prostaglandin H synthase-2. Cell. 1996;87:783–786. doi: 10.1016/s0092-8674(00)81983-2. 8945503 [DOI] [PubMed] [Google Scholar]
- 14.Schmid K., Nair J., Winde G., Velic I., Bartsch H. Increased levels of promutagenic etheno-DNA adducts in colonic polyps of FAP patients. International Journal of Cancer: Journal International du Cancer. 2000;87:1–4. doi: 10.1002/1097-0215(20000701)87:1<1::aid-ijc1>3.0.co;2-c. 10861445 [DOI] [PubMed] [Google Scholar]
- 15.Williams C.S., Luongo C., Radhika A., Zhang T., Lamps L.W., Nanney L.B., Beauchamp R.D. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996;111:1134–1140. doi: 10.1016/s0016-5085(96)70083-5. 8831610 [DOI] [PubMed] [Google Scholar]
- 16.Williams M.V., Lee S.H., Pollack M., Blair I.A. Endogenous lipid hydroperoxide-mediated DNA-adduct formation in min mice. Journal of Biological Chemistry. 2006;281:10127–10133. doi: 10.1074/jbc.M600178200. 16449227 [DOI] [PubMed] [Google Scholar]
- 17.Marks F., Müller-Decker K., Fürstenberger G. A causal relationship between unscheduled eicosanoid signaling and tumor development: cancer chemoprevention by inhibitors of arachidonic acid metabolism. Toxicology. 2000;153:11–26. doi: 10.1016/s0300-483x(00)00301-2. 11090944 [DOI] [PubMed] [Google Scholar]
- 18.Nair J., Fürstenberger G., Bürger F., Marks F., Bartsch H. Promutagenic etheno-DNA adducts in multistage mouse skin carcinogenesis: correlation with lipoxygenase-catalyzed arachidonic acid metabolism. Chemical Research in Toxicology. 2000;13:703–709. doi: 10.1021/tx000045d. 10956057 [DOI] [PubMed] [Google Scholar]
- 19.Winter C.K., Segall H.J., Haddon W.F. Formation of cyclic adducts of deoxyguanosine with the aldehydes trans-4-hydroxy-2-hexenal and trans-4-hydroxy-2-nonenal in vitro. Cancer Research. 1986;46:5682–5686. 3756915 [PubMed] [Google Scholar]
- 20.Chung F.L., Chen H.J., Nath R.G. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. 8895475 [DOI] [PubMed] [Google Scholar]
- 21.El Ghissassi F., Barbin A., Nair J. Formation of 1,N6-ethenoadenine and 3,N4-ethenocytosine by lipid peroxidation products and nucleic acid bases. Chemical Research in Toxicology. 1995;8:278–283. doi: 10.1021/tx00044a013. 7766812 [DOI] [PubMed] [Google Scholar]
- 22.Vaca C.E., Wilhelm J., Harms-Ringdahl M. Interaction of lipid peroxidation products with DNA. A review. Mutation Research. 1988;195:137–149. doi: 10.1016/0165-1110(88)90022-x. 3277035 [DOI] [PubMed] [Google Scholar]
- 23.Pryor W.A., Porter N.A. Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free Radical Biology and Medicine. 1990;8:541–543. doi: 10.1016/0891-5849(90)90153-a. 2193853 [DOI] [PubMed] [Google Scholar]
- 24.Blair I.A. DNA adducts with lipid peroxidation products. Journal of Biological Chemistry. 2008;283:15545–15549. doi: 10.1074/jbc.R700051200. 18285329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sodum R.S., Chung F.L. Stereoselective formation of in vitro nucleic acid adducts by 2,3-epoxy-4-hydroxynonanal. Cancer Research. 1991;51:137–143. 1703030 [PubMed] [Google Scholar]
- 26.Nair U., Bartsch H., Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: a review of published adduct types and levels in humans. Free Radical Biology and Medicine. 2007;43:1109–1120. doi: 10.1016/j.freeradbiomed.2007.07.012. 17854706 [DOI] [PubMed] [Google Scholar]
- 27.Chung F.L., Chen H.J., Nath R.G. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. 8895475 [DOI] [PubMed] [Google Scholar]
- 28.Hiraku Y., Kawanishi S. Role of nitrative DNA damage in inflammation related carcinogenesis. In: Hiraku Y., Kawanishi S., Ohshima H., editors. Cancer and Inflammation Mechanisms: Chemical, Biological, and Chemical Aspects. John Wiley & Sons; Hoboken, NJ: 2014. pp. 41–59. [Google Scholar]
- 29.Eberle G., Barbin A., Laib R.J. N6-etheno-2′-deoxyadenosine and 3,N4-etheno-2′-deoxycytidine detected by monoclonal antibodies in lung and liver DNA of rats exposed to vinyl chloride. Carcinogenesis. 1989;10:209–212. doi: 10.1093/carcin/10.1.209. 2783395 [DOI] [PubMed] [Google Scholar]
- 30.Frank A., Seitz H.K., Bartsch H. Immunohistochemical detection of 1,N6-ethenodeoxyadenosine in nuclei of human liver affected by diseases predisposing to hepato-carcinogenesis. Carcinogenesis. 2004;25:1027–1031. doi: 10.1093/carcin/bgh089. 14742317 [DOI] [PubMed] [Google Scholar]
- 31.Nair J., Nair U.J., Sun X. Quantifying etheno-DNA adducts in human tissues, white blood cells, and urine by ultrasensitive (32)P-postlabeling and immunohistochemistry. Methods in Molecular Biology (Clifton, N.J.) 2011;682:189–205. doi: 10.1007/978-1-60327-409-8_14. 21057929 [DOI] [PubMed] [Google Scholar]
- 32.Nair J., Barbin A., Guichard Y. N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytine in liver DNA from humans and untreated rodents detected by immunoaffinity/32P-postlabeling. Carcinogenesis. 1995;16:613–617. doi: 10.1093/carcin/16.3.613. 7697821 [DOI] [PubMed] [Google Scholar]
- 33.Nair J., Godschalk R.W., Nair U. Identification of 3,N(4)-etheno-5-methyl-2′-deoxycytidine in human DNA: A new modified nucleoside which may perturb genome methylation. Chemical Research in Toxicology. 2012;25:162–169. doi: 10.1021/tx200392a. 22148471 [DOI] [PubMed] [Google Scholar]
- 34.Nair J., Srivatanakul P., Haas C. High urinary excretion of lipid peroxidation-derived DNA damage in patients with cancer-prone liver diseases. Mutation Research. 2010;683:23–28. doi: 10.1016/j.mrfmmm.2009.10.002. 19822158 [DOI] [PubMed] [Google Scholar]
- 35.Barbin A. Etheno-adduct-forming chemicals: from mutagenicity testing to tumor mutation spectra. Mutation Research. 2000;462:55–69. doi: 10.1016/s1383-5742(00)00014-4. 10767618 [DOI] [PubMed] [Google Scholar]
- 36.Bartsch H., Barbin A., Marion M.J., Nair J., Guichard Y. Formation, detection, and role in carcinogenesis of ethenobases in DNA. Drug Metabolism Reviews. 1994;26:349–371. doi: 10.3109/03602539409029802. 8082574 [DOI] [PubMed] [Google Scholar]
- 37.Basu A.K., Wood M.L., Niedernhofer L.J., Ramos L.A., Essigmann J.M. Mutagenic and genotoxic effects of three vinyl chloride-induced DNA lesions: 1,N6-ethenoadenine, 3,N4-ethenocytosine, and 4-amino-5-(imidazol-2-yl)imidazole. Biochemistry. 1993;32:12793–12801. doi: 10.1021/bi00210a031. 8251500 [DOI] [PubMed] [Google Scholar]
- 38.Pandya G.A., Moriya M. N6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry. 1996;35:11487–11492. doi: 10.1021/bi960170h. 8784204 [DOI] [PubMed] [Google Scholar]
- 39.Palejwala V.A., Rzepka R.W., Simha D., Humayun M.Z. Quantitative multiplex sequence analysis of mutational hot spots. Frequency and specificity of mutations induced by a site-specific ethenocytosine in M13 viral DNA. Biochemistry. 1993;32:4105–4111. doi: 10.1021/bi00066a036. 8471617 [DOI] [PubMed] [Google Scholar]
- 40.Moriya M., Zhang W., Johnson F., Grollman A.P. Mutagenic potency of exocyclic DNA adducts: marked differences between Escherichia coli and simian kidney cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:11899–11903. doi: 10.1073/pnas.91.25.11899. 7991554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine R.L., Yang I.Y., Hossain M., Pandya G.A., Grollman A.P., Moriya M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Research. 2000;60:4098–4104. 10945616 [PubMed] [Google Scholar]
- 42.Swenberg J.A., Fedtke N., Ciroussel F., Barbin A., Bartsch H. Etheno adducts formed in DNA of vinyl chloride-exposed rats are highly persistent in liver. Carcinogenesis. 1992;13:727–729. doi: 10.1093/carcin/13.4.727. 1576725 [DOI] [PubMed] [Google Scholar]
- 43.Cheng K.C., Preston B.D., Cahill D.S., Dosanjh M.K., Singer B., Loeb L.A. The vinyl chloride DNA derivative N2,3-ethenoguanine produces G–A transitions in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:9974–9978. doi: 10.1073/pnas.88.22.9974. 1946466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller J.A., Miller E.C. The metabolic activation and nucleic acid adducts of naturally-occurring carcinogens: recent results with ethyl carbamate and the spice flavors safrole and estragole. British Journal of Cancer. 1983;48:1–15. doi: 10.1038/bjc.1983.151. 6191767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu W., Feng Z., Eveleigh J., Iyer G., Pan J., Amin S., Chung F.L. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis. 2002;23:1781–1789. doi: 10.1093/carcin/23.11.1781. 12419825 [DOI] [PubMed] [Google Scholar]
- 46.Nair J., Nair U.J., Sun X., Wang Y., Arab K., Bartsch H. Quantifying etheno-DNA adducts in human tissues, white blood cells, and urine by ultrasensitive (32)P-postlabeling and immunohistochemistry. Methods in Molecular Biology (Clifton, N.J.) 2011;682:189–205. doi: 10.1007/978-1-60327-409-8_14. 21057929 [DOI] [PubMed] [Google Scholar]
- 47.Bartsch H., Nair J. Lipid peroxidation-derived DNA adduts and the role in inflammation-related carcinogenesis. In: Hiraku Y., Kawanishi S., Ohshima H., editors. Cancer and Inflammation Mechanisms: Chemical, Biological and Clinical Aspects. John Wiley & Sons; Hoboken, NJ: 2014. pp. 61–74. [Google Scholar]
- 48.Seitz H.K., Stickel F. Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biological Chemistry. 2006;387:349–360. doi: 10.1515/BC.2006.047. 16606331 [DOI] [PubMed] [Google Scholar]
- 49.Bautista A.P. Neutrophilic infiltration in alcoholic hepatitis. Alcohol (Fayetteville, N.Y.) 2002;27:17–21. doi: 10.1016/s0741-8329(02)00206-9. 12062632 [DOI] [PubMed] [Google Scholar]
- 50.Millonig G., Ganzleben I., Peccerella T., Casanovas G., Brodziak-Jarosz L., Breitkopf-Heinlein K., Dick T.P., Seitz H.K., Muckenthaler M.U. Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3) Journal of Biological Chemistry. 2012;287:37472–37482. doi: 10.1074/jbc.M112.358911. 22932892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujita N., Miyachi H., Tanaka H. Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiology, Biomarkers and Prevention: a Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology. 2009;18:424–432. doi: 10.1158/1055-9965.EPI-08-0725. 19190144 [DOI] [PubMed] [Google Scholar]
- 52.Chamulitrat W., Spitzer J.J. Nitric oxide and liver injury in alcohol-fed rats after lipopolysaccharide administration. Alcoholism, Clinical and Experimental Research. 1996;20:1065–1070. doi: 10.1111/j.1530-0277.1996.tb01947.x. 8892528 [DOI] [PubMed] [Google Scholar]
- 53.Lieber C.S., DeCarli L.M. Hepatic microsomal ethanol-oxidizing system. In vitro characteristics and adaptive properties in vivo. Journal of Biological Chemistry. 1970;245(10):2505–2512. 4315645 [PubMed] [Google Scholar]
- 54.Lieber C.S. CYP2E1: from ASH to Nash. Hepatology Research: the Official Journal of the Japan Society of Hepatology. 2004;28(1):1–11. doi: 10.1016/j.hepres.2003.08.001. 14734144 [DOI] [PubMed] [Google Scholar]
- 55.Oneta C.M., Lieber C.S., Li J.J., Rüttimann S., Schmid B., Lattmann J., Rosman A.S., Seitz H.K. Dynamics of cytochrome P4502E1 activity in man: induction by ethanol and disappearance during withdrawal phase. Journal of Hepatology. 2002;36(1):47–52. doi: 10.1016/s0168-8278(01)00223-9. 11804663 [DOI] [PubMed] [Google Scholar]
- 56.Lieber C.S., Cao Q., DeCarli L.M., Leo M.A., Mak K.M., Ponomarenko A., Ren C. Role of medium-chain triglycerides in the alcohol-mediated cytochrome P450 2E1 induction of mitochondria. Alcoholism, Clinical and Experimental Research. 2007;31:1660–1668. doi: 10.1111/j.1530-0277.2007.00475.x. 17681033 [DOI] [PubMed] [Google Scholar]
- 57.Albano E., Clot P., Morimoto M., Tomasi A., Ingelman-Sundberg M., French S.W. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology (Baltimore, Md.) 1996;23:155–163. doi: 10.1002/hep.510230121. 8550035 [DOI] [PubMed] [Google Scholar]
- 58.Leung T.M., Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of Hepatology. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. 22940046 [DOI] [PubMed] [Google Scholar]
- 59.Dey A., Cederbaum A.I. Induction of cytochrome P450 2E1 promotes liver injury in ob/ob mice. Hepatology (Baltimore, Md.) 2007;45:1355–1365. doi: 10.1002/hep.21603. 17538970 [DOI] [PubMed] [Google Scholar]
- 60.Wang X., Lu Y., Cederbaum A.I. Induction of cytochrome P450 2E1 increases hepatotoxicity caused by Fas agonistic Jo2 antibody in mice. Hepatology (Baltimore, Md.) 2005;42:400–410. doi: 10.1002/hep.20792. 16025513 [DOI] [PubMed] [Google Scholar]
- 61.Pérez M.J., Cederbaum A.I. Proteasome inhibition potentiates CYP2E1-mediated toxicity in HepG2 cells. Hepatology (Baltimore, Md.) 2003;37(6):1395–1404. doi: 10.1053/jhep.2003.50228. 12774019 [DOI] [PubMed] [Google Scholar]
- 62.Lu Y., Zhuge J., Wang X., Bai J., Cederbaum A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology (Baltimore, Md.) 2008;47(5):1483–1494. doi: 10.1002/hep.22222. 18393316 [DOI] [PubMed] [Google Scholar]
- 63.Morgan K., French S.W., Morgan T.R. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology (Baltimore, Md.) 2002;36:122–134. doi: 10.1053/jhep.2002.33720. 12085356 [DOI] [PubMed] [Google Scholar]
- 64.Gouillon Z., Lucas D., Li J., Hagbjork A.L., French B.A., Fu P., Fang C. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine (New York, N.Y.) 2000;224:302–308. doi: 10.1046/j.1525-1373.2000.22435.x. 10964266 [DOI] [PubMed] [Google Scholar]
- 65.Bradford B.U., Kono H., Isayama F., Kosyk O., Wheeler M.D., Akiyama T.E., Bleye L. Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology (Baltimore, Md.) 2005;41:336–344. doi: 10.1002/hep.20532. 15660387 [DOI] [PubMed] [Google Scholar]
- 66.Wang Y., Millonig G., Nair J., Patsenker E., Stickel F., Mueller S., Bartsch H., Seitz H.K. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology (Baltimore, Md.) 2009;50(2):453–461. doi: 10.1002/hep.22978. 19489076 [DOI] [PubMed] [Google Scholar]
- 67.Weltman M.D., Farrell G.C., Hall P., Ingelman-Sundberg M., Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md.) 1998;27:128–133. doi: 10.1002/hep.510270121. 9425928 [DOI] [PubMed] [Google Scholar]
- 68.Abdelmegeed M.A., Banerjee A., Yoo S.H. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. Journal of Hepatology. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. 22668639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chalasani N., Gorski J.C., Asghar M.S. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md.) 2003;37:544–550. doi: 10.1053/jhep.2003.50095. 12601351 [DOI] [PubMed] [Google Scholar]
- 70.Fujii H., Kawada N. Inflammation and fibrogenesis in steatohepatitis. Journal of Gastroenterology. 2012;47:215–225. doi: 10.1007/s00535-012-0527-x. 22310735 [DOI] [PubMed] [Google Scholar]
- 71.Roberts E.A. Pediatric nonalcoholic fatty liver disease (NAFLD): a “growing” problem? Journal of Hepatology. 2007;46:1133–1142. doi: 10.1016/j.jhep.2007.03.003. 17445934 [DOI] [PubMed] [Google Scholar]
- 72.Ruiz-Extremera Á, Carazo Á, Salmerón Á. Factors associated with hepatic steatosis in obese children and adolescents. Journal of Pediatric Gastroenterology and Nutrition. 2011;53:196–201. doi: 10.1097/MPG.0b013e3182185ac4. 21788762 [DOI] [PubMed] [Google Scholar]
- 73.Lerret S.M., Garcia-Rodriguez L., Skelton J. Predictors of nonalcoholic steatohepatitis in obese children. Gastroenterology Nursing: the Official Journal of the Society of Gastroenterology Nurses and Associates. 2011;34:434–437. doi: 10.1097/SGA.0b013e3182371356. 22129796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nobili V., Parola M., Alisi A., Marra F., Piemonte F., Mombello C., Sutti S. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. International Journal of Molecular Medicine. 2010;26:471–476. doi: 10.3892/ijmm_00000487. 20818484 [DOI] [PubMed] [Google Scholar]
- 75.Qin H., Teufel U., Engelmann G. Detection of hepatic highly carcinogenic, exocyclic etheno-DNA-adducts in patients with alcoholic and non-alcoholic fatty liver disease and in children with non-alcoholic steatoheopatitis. Journal of Hepatology. 2012;56(Suppl. 2):S520. [Google Scholar]
- 76.Xu J., Lai K.K., Verlinsky A., Lugea A., French S.W., Cooper M.P., Ji C. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. Journal of Hepatology. 2011;55:673–682. doi: 10.1016/j.jhep.2010.12.034. 21256905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y., Seitz H.K., Wang X.D. Moderate alcohol consumption aggravates high-fat diet induced steatohepatitis in rats. Alcoholism, Clinical and Experimental Research. 2010;34(3):567–573. doi: 10.1111/j.1530-0277.2009.01122.x. 20028348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ascha M.S., Hanouneh I.A., Lopez R. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md.) 2010;51:1972–1978. doi: 10.1002/hep.23527. 20209604 [DOI] [PubMed] [Google Scholar]
- 79.Baan R., Straif K., Grosse Y., Secretan B., El Ghissassi F., Bouvard V., Altieri A., Cogliano V., WHO International Agency for Research on Cancer Monograph Working Group Carcinogenicity of alcoholic beverages. Lancet: Oncology. 2007;8:292–293. doi: 10.1016/s1470-2045(07)70099-2. 17431955 [DOI] [PubMed] [Google Scholar]
- 80.Visapää J.P., Götte K., Benesova M., Li J., Homann N., Conradt C., Inoue H. Increased cancer risk in heavy drinkers with the alcohol dehydrogenase 1C*1 allele, possibly due to salivary acetaldehyde. Gut. 2004;53:871–876. doi: 10.1136/gut.2003.018994. 15138216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Homann N., König I.R., Marks M., Benesova M., Stickel F., Millonig G., Mueller S. Alcohol and colorectal cancer: the role of alcohol dehydrogenase 1C polymorphism. Alcoholism, Clinical and Experimental Research. 2009;33:551–556. doi: 10.1111/j.1530-0277.2008.00868.x. 19120062 [DOI] [PubMed] [Google Scholar]
- 82.Millonig G., Wang Y., Homann N., Bernhardt F., Qin H., Mueller S., Bartsch H. Ethanol-mediated carcinogenesis in the human esophagus implicates CYP2E1 induction and the generation of carcinogenic DNA-lesions. International Journal of Cancer: Journal International du Cancer. 2011;128(3):533–540. doi: 10.1002/ijc.25604. 20715111 [DOI] [PubMed] [Google Scholar]
- 83.Simanowski U.A., Suter P., Russell R.M., Heller M., Waldherr R., Ward R., Peters T.J. Enhancement of ethanol induced rectal mucosal hyper regeneration with age in F344 rats. Gut. 1994;35:1102–1106. doi: 10.1136/gut.35.8.1102. 7926914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simanowski U.A., Homann N., Knühl M., Arce L., Waldherr R., Conradt C., Bosch F.X. Increased rectal cell proliferation following alcohol abuse. Gut. 2001;49:418–422. doi: 10.1136/gut.49.3.418. 11511565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vincon P., Wunderer J., Simanowski U.A., Koll M., Preedy V.R., Peters T.J., Werner J. Inhibition of alcohol-associated colonic hyperregeneration by alpha-tocopherol in the rat. Alcoholism, Clinical and Experimental Research. 2003;27:100–106. doi: 10.1097/01.ALC.0000046341.31828.A4. 12544013 [DOI] [PubMed] [Google Scholar]
- 86.Beland F.A., Benson R.W., Mellick P.W., Kovatch R.M., Roberts D.W., Fang J.L., Doerge D.R. Effect of ethanol on the tumorigenicity of urethane (ethyl carbamate) in B6C3f1 mice. Food and Chemical Toxicology: an International Journal Published for the British Industrial Biological Research Association. 2005;43:1–19. doi: 10.1016/j.fct.2004.07.018. 15582191 [DOI] [PubMed] [Google Scholar]
- 87.Soffritti M., Belpoggi F., Cevolani D., Guarino M., Padovani M., Maltoni C. Results of long-term experimental studies on the carcinogenicity of methyl alcohol and ethyl alcohol in rats. Annals of the New York Academy of Sciences. 2002;982:46–69. doi: 10.1111/j.1749-6632.2002.tb04924.x. 12562628 [DOI] [PubMed] [Google Scholar]
- 88.Watabiki T., Okii Y., Tokiyasu T., Yoshimura S., Yoshida M., Akane A., Shikata N. Long-term ethanol consumption in ICR mice causes mammary tumor in females and liver fibrosis in males. Alcoholism, Clinical and Experimental Research. 2000;24:117S–122S. 10803793 [PubMed] [Google Scholar]
- 89.Roy H.K., Gulizia J.M., Karolski W.J., Ratashak A., Sorrell M.F., Tuma D. Ethanol promotes intestinal tumorigenesis in the MIN mouse. Multiple intestinal neoplasia. Cancer Epidemiology, Biomarkers and Prevention: a Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology. 2002;11:1499–1502. 12433735 [PubMed] [Google Scholar]
- 90.Chavez P.R., Lian F., Chung J. Long-term ethanol consumption promotes hepatic tumorigenesis but impairs normal hepatocyte proliferation in rats. Journal of Nutrition. 2011;141:1049–1055. doi: 10.3945/jn.110.136531. 21490289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ye Q., Lian F., Chavez P.R. Cytochrome P450 2E1 inhibition prevents hepatic carcinogenesis induced by diethylnitrosamine in alcohol-fed rats. Hepatobiliary Surgery and Nutrition. 2012;1:5–18. doi: 10.3978/j.issn.2304-3881.2012.11.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsuchishima M., George J., Shiroeda H. Chronic ingestion of ethanol induces hepatocellular carcinoma in mice without additional hepatic insult. Digestive Diseases and Sciences. 2013;58:1923–1933. doi: 10.1007/s10620-013-2574-4. 23371017 [DOI] [PubMed] [Google Scholar]