

Abstract
In this review, we describe research findings on the effects of alcohol exposure on two major catabolic systems in liver cells: the ubiquitin–proteasome system (UPS) and autophagy. These hydrolytic systems are not unique to liver cells; they exist in all eukaryotic tissues and cells. However, because the liver is the principal site of ethanol metabolism, it sustains the greatest damage from heavy drinking. Thus, the focus of this review is to specifically describe how ethanol oxidation modulates the activities of the UPS and autophagy and the mechanisms by which these changes contribute to the pathogenesis of alcohol-induced liver injury. Here, we describe the history and the importance of cellular hydrolytic systems, followed by a description of each catabolic pathway and the differential modulation of each by ethanol exposure. Overall, the evidence for an involvement of these catabolic systems in the pathogenesis of alcoholic liver disease is quite strong. It underscores their importance, not only as effective means of cellular recycling and eventual energy generation, but also as essential components of cellular defense.
Keywords: Autophagy, Ubiquitin proteasome, Ethanol, proteopathy, Steatosis
Graphical abstract
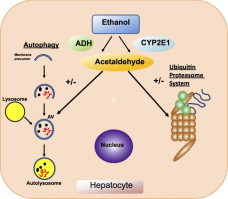
Highlights
-
•
Active intracellular catabolic systems are crucial to cell survival.
-
•
Autophagy and the ubiquitin–proteasome system (UPS) are predominant catabolic systems.
-
•
Alcohol oxidation to acetaldehyde modulates autophagy and the UPS in liver.
-
•
Continuous (chronic) ethanol oxidation disrupts hepatic autophagy and the UPS.
-
•
Such disruption contributes to alcoholic liver injury.
Introduction
Importance of intracellular catabolic systems
During the 20th century, the biomedical literature emphasized cellular anabolic processes, including, DNA replication, RNA transcription, protein synthesis, and the assembly of complex lipids and carbohydrates. In contrast, scientific interest in macromolecular catabolism was low, partly because of the erroneous notion that once such macromolecules are synthesized, they become permanent, irreplaceable cellular fixtures. Other scientists believed that one or more cellular degradation systems existed but it/they had minor physiological importance. In the late 1930s and early 1940s, definitive isotope studies by Schoenheimer and colleagues [1] demonstrated that cellular constituents are dynamic, as they continuously undergo breakdown and replenishment. This discovery prompted more research effort into catabolic systems. In the 1950s and 1960s, groundbreaking work by De Duve, using subcellular fractions from rat liver, revealed that lysosomes are distinct cellular organelles, containing acid hydrolases that catalyze the breakdown of all macromolecular forms [2–4]. De Duve and colleagues also found that liver cells use lysosomes to digest their own contents, a process he named autophagy or “self eating” [5,6]. Numerous studies of protein catabolism in the late 1960s and early 1970s, reported that the in vivo half-lives of individual proteins are essentially constant but are quite distinct from each other, ranging widely from several minutes to several days. From each protein's half-life, one can estimate its synthesis rate, which is balanced with its rate of degradation [7]. Investigators examined the reasons for such diverse catabolic rates. They revealed that the primary sequence, particularly a protein's NH2-terminal amino acid, its native conformation, and its size all strongly influence its rate of degradation [8,9]. These investigations laid the groundwork that led to the discovery in the late 1970s and early 1980s, of the soluble, proteolytic pathway now known as the ubiquitin–proteasome system (UPS) [10,11]. The UPS is now considered the principal proteolytic pathway in all eukaryotic cells. While the discoveries of lysosomes, autophagy and the UPS were independent events that, like other major biomedical discoveries, were first met with skepticism by other scientists, their impact has been far-reaching. It is now clear that disturbances of autophagy or the UPS are directly linked to the causes, exacerbation, and even the alleviation of disease. In fact, both catabolic pathways have become therapeutic targets. Liver disease that is caused by the hereditary disorder, alpha-1-antitrypsin (α-1AT) deficiency, in which the mutated form of α-1-AT accumulates and aggregates in liver cells, is ablated in animal models after treatment with the anti-seizure drug, carbamazepine, which reportedly accelerates autophagy [12]. Others report that in vivo treatment with a gene vector that expresses the transcription factor EB (TFEB), an important regulator of autophagy and lysosome biogenesis [13], also activates the autophagic pathway to enhance α-1AT removal. The proteasome inhibitor, bortezomib (Velcade®) is used with other anti-cancer drugs as an effective treatment for the hematological malignancy, multiple myeloma [14–16]. In the absence of disease, autophagy and the UPS maintain normal cell function by degrading larger molecules to smaller ones, which are further broken down to generate ATP. Both catabolic pathways are cytoprotective because they remove damaged proteins and dysfunctional organelles thereby preventing interference with normal cell function. While autophagy and the UPS occupy distinct cellular locations, they exhibit overlap in function by degrading some of the same protein substrates [17–19]. For example, when proteins aggregate, they become less recognizable substrates for and resistant to proteolysis by the proteasome. Such aggregates are more readily degraded by autophagy [20], probably because the acidic interior of the lysosome (~pH 4.7) [21] denatures such proteins for eventual digestion by the diverse array of proteases (cathepsins) that reside in that organelle. However, should the functions of the UPS and autophagy falter simultaneously, the potential for pathology increases significantly. In the liver, chronic, heavy alcohol consumption impedes both pathways. Such disturbances are linked to the pathogenesis of alcohol-induced liver injury.
Alcoholic liver disease (ALD)
Alcoholic beverages have been used and abused for centuries. It is likely that liver injury caused by heavy drinking is one of the oldest liver ailments known to humans [22]. Because the liver is the principal site of ethanol oxidation, it sustains the greatest injury after alcohol abuse. The severity of ALD ranges from steatosis (fatty liver) to decompensated cirrhosis. Despite many decades of investigation into the causes of and the treatments for ALD, the disease still remains difficult to manage in the clinic [22]. Current standards of care that include abstinence, nutrition therapy and corticosteroid treatment have had marginal success [23,24]. This is partly because patients who present with alcoholic hepatitis have end-stage or near end-stage liver disease after years of heavy drinking.
Hepatic ethanol oxidation proceeds by two major pathways: alcohol dehydrogenase (ADH), which resides in the cytosol and cytochrome P450 2E1 (CYP2E1), which is a component of the endoplasmic reticulum (a.k.a. the microsome fraction). Both enzymes oxidize ethanol to generate acetaldehyde. CYP2E1 is unique because it also catalyzes the oxidation of other compounds that are chemically and functionally distinct from ethanol. These include the industrial solvent, carbon tetrachloride [25], the antipyretic, acetaminophen [25,26], and the anesthetic, halothane [27]. Furthermore, heavy ethanol consumption consistently increases the hepatic content of the CYP2E1 apoenzyme. A major mechanism for its induction is that ethanol stabilizes CYP2E1 by protecting it from degradation by the proteasome [28]. From a toxicological standpoint, CYP2E1 induction is very important when one considers the dangers of heavy drinking combined with simultaneous exposure to any or all of the aforementioned substrates. For example, when hepatic CYP2E1 enzyme levels increase after excessive alcohol consumption, the hepatotoxicity of acetaminophen is intensified because the drug is more rapidly converted to its toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI) by elevated levels of CYP2E1.
Here, we document research on how ethanol-elicited oxidant stress, generated mostly by ethanol oxidation, affects the UPS and autophagy during alcohol-induced liver injury. It is clear that both catabolic pathways are vital for maintaining a healthy liver. Evidence strongly indicates that disruption of autophagy and UPS by oxidants derived from ethanol metabolism contribute significantly to the development of steatosis and proteopathy that occur during the course of ALD pathogenesis. Here, we describe both catabolic systems and the changes that occur in each after ethanol exposure.
Ethanol and intracellular catabolic systems
Basal macroautophagy in eukaryotic cells
During macroautophagy (i.e. autophagy), all macromolecules (proteins, nucleic acids, complex carbohydrates and triglycerides) and organelles (dysfunctional organelles, including damaged mitochondria) are degraded to smaller molecules (e.g. amino acids and glucose) to generate usable energy, and to eliminate potentially toxic cellular waste [29]. In the liver, enhanced autophagy is generally regarded as cytoprotective [30]. Autophagy in liver is normally activated by elevated glucagon levels, nutrient deprivation (e.g. fasting), metabolic (oxidant) stress, and exposure to pharmacologic agents such as rapamycin. Conversely, autophagy is suppressed by insulin or exposure to adequate nutrients. In well-nourished cells, the mammalian target of rapamycin complex 1 (MTORC1) suppresses macroautophagy. MTORC1 is a high molecular weight kinase, which is activated by nutrients or insulin and prevents the formation of the downstream Atg1–Atg17–Atg13 complex shown in Fig. 1[31], thereby blocking autophagy. When MTORC1 activity is suppressed by starvation or by rapamycin treatment, autophagy induction (step 1, Fig. 1) proceeds. It begins with autophagosome (AV) formation. Two ubiquitin-like proteins, Atg12 and Atg8 (a.k.a. pro-LC3 in Fig. 1) and their conjugation systems are active during the elongation and expansion of the AV membrane precursor called the phagophore. Atg12 is conjugated to Atg5 in a reaction catalyzed by Atg7 and Atg 10, which are E1- and E2-like enzymes, respectively. The Atg12–Atg5 conjugate then interacts with Atg16 to form a larger complex, Atg–12–5–16, that is localized to the phagophore. Through the participation of Beclin-1 and Atg14, the class III phosphoinositide-3-kinase (PI3K) complex directs the initiation of autophagosome formation. The C-terminal fragment of Atg8/pro-LC3 is removed by Atg4, generating the microtubule-associated light chain-3, type 1 (LC3-1), containing a C-terminal glycine, which is then conjugated to the phospholipid, phosphatidylethanolamine (PE) by the action of Atg7 and the E2-like enzyme, Atg3. The lipidated LC3, now known as LC3-II, is then attached to the phagophore membrane and completes its closure to form an autophagosome. LC3II is considered a standard biomarker for detection and quantification of AVs, which enclose parcels of cytoplasm containing particulate (organelles) and soluble substrates destined for degradation. After sequestering its cargo, each AV is trafficked to a lysosome via microtubules. AV–lysosome docking and fusion (step 3) is facilitated by the proteins VAMP 8 and Vti1 B [32]. Lysosome-associated membrane protein 2 (LAMP2) also participates in this fusion [33], to form an autolysosome, in which AV cargo breakdown (step 4) is catalyzed by lysosomal hydrolases. Lysosome biogenesis is crucial for maintaining an adequate number of these organelles for macromolecular turnover. The rate of such biogenesis varies in response to physiological conditions [34–37]. Lysosomes also have a key role in two accessory but important autophagy pathways, microautophagy and chaperone-mediated autophagy (CMA). These are described in greater detail in a separate review [38].
Fig. 1.
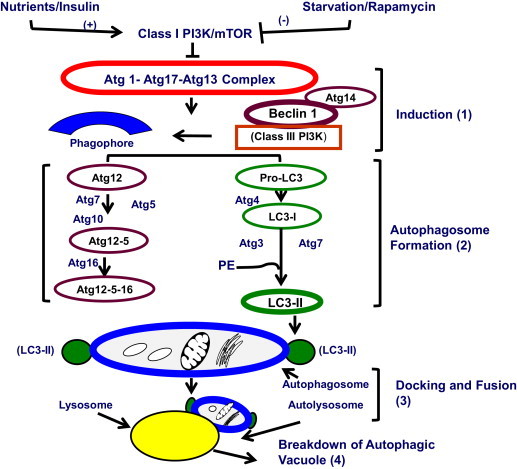
The macroautophagy pathway. Details are provided in the text.
Autophagy in cultured cells after ethanol exposure
In a previous investigation, we examined the effects of ethanol exposure on autophagy in parental HepG2 cells and their recombinants. HepG2 cells express neither ADH nor CYP2E1. Their recombinants were generated by transfecting HepG2 cells with vectors that stably express CYP2E1 and/or mouse ADH-1. VA-13 cells synthesize ADH but not CYP2E1 while E-47 cells express CYP2E1 but not ADH. VL17A cells produce both ADH and CYP2E1. Descriptions of the development and characterization of these cell lines are published [39,40].
Our autophagy studies revealed that exposure of VL-17A cells (ADH+/CyP2E1+) to ethanol increases the content of LC3B mRNA and the AV protein marker, LC3II. The magnitude of the rise in LC3II correlates positively with the ethanol concentration to which the cells are exposed. Furthermore, LC3II induction requires ethanol oxidation to acetaldehyde catalyzed by ADH, as it only occurs in VA-13 cells (ADH+/CYP2E1¯) and in VL-17A cells, both of which express the ADH enzyme. We confirmed this by blocking ethanol oxidation with 4-methylpyrazole and saw no enhancement of LC3II content. Conversely, we found LC3-II induction after direct exposure of all 4 cell lines to 100 or 300 µM acetaldehyde. Induction of LC3II by ethanol is closely associated with a nearly three-fold rise in AV numbers and a two-fold augmentation in their average size. These findings clearly indicate that acetaldehyde generated by ADH induces autophagosome content.
To assess whether the ethanol-induced rise in LC3II reflects an enhancement of autophagosome formation or an inhibition of their transit to lysosomes, we conducted autophagic flux measurements using bafilomycin, which blocks lysosomal proteolysis. These measurements [41] revealed that 24 h of ethanol exposure both enhances LC3-II synthesis and decreases LC3 degradation in VL-17A cells. We confirmed these findings in subsequent experiments which demonstrated that the ethanol-induced rise in LC3-II is similar to that induced by the microtubule inhibitor, nocodazole, which blocks trafficking of AVs to lysosomes. Additionally, ethanol exposure causes a significant rise in intracellular P62/SQSTM1 (P62), a scaffolding protein that is also a marker of lysosomal proteolysis. Elevated P62 levels usually indicate its accumulation due to obstruction of P62 degradation [42,43]. To confirm this finding we also observed that ethanol-exposure inhibits the activities of lysosomal cathepsins B and L. The latter finding is linked to a two-fold reduction in lysosome numbers. These results indicate that ethanol exposure decreases lysosome biogenesis in ethanol-oxidizing cultured cells, a finding we reported previously in vivo [37]. Thus, in recombinant HepG2 cells that express ADH, the rise in LC3-II following ethanol exposure results from both enhanced AV biogenesis, and decreased destruction of AVs by a depleted population of lysosomes.
Interestingly, ethanol-elicited induction of LC3II (AV content) is higher in VL-17A cells than in VA-13 cells. The latter findings correlate with higher levels of lipid peroxides in the form of malondialdehyde (MDA) in VL-17A cells than in identically-treated VA-13 cells. These data suggest that the higher level of MDA in VL-17A cells (presumably generated by oxidants derived from both ADH and CYP2E1 catalysis) is associated with a higher LC3II content than in VA-13 cells, which express ADH only. These results indicate that ethanol oxidation by ADH generates acetaldehyde, which enhances autophagosome formation and that primary and secondary oxidants produced by CYP2E1 somehow augment this induction. We surmise that MDA and other secondary oxidants may contribute to the slowdown in autophagic flux mentioned above. It is noteworthy that CYP2E1 catalysis alone does not initiate a rise in LC3II, at least in these experiments, most of which were 24 h in duration. It is also worth noting that a 24 h exposure produces characteristics of both acute and chronic ethanol exposure[44]. Others report AV induction in VL-17A cells after 6 h of ethanol treatment, which is predominantly acute ethanol exposure that enhances AV content and accelerates autophagic flux [45]. Fig. 2 presents our interpretation of the aforementioned results with parental and recombinant HepG2 cells and our view of how autophagy is affected after acute and chronic ethanol exposure.
Fig. 2.
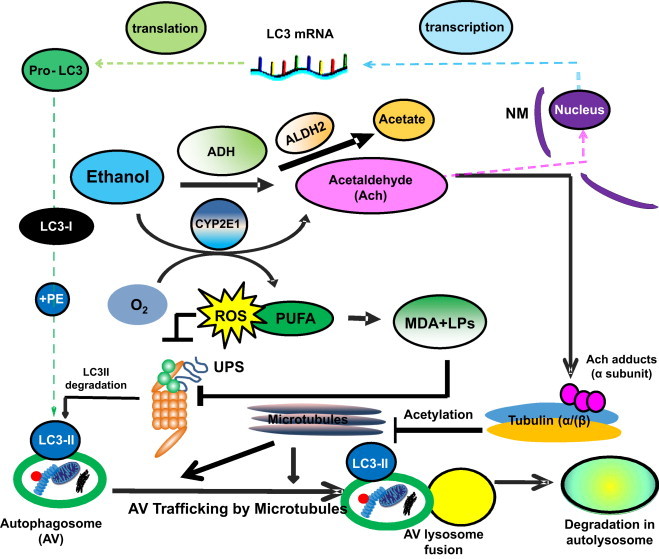
Multilevel regulation of autophagosome content by ethanol oxidation liver cells. During acute (or early) ethanol administration, metabolically-derived acetaldehyde (Ach) enhances AV formation in liver cells by increasing the level of LC3B mRNA, presumably by enhanced transcription. LC3B mRNA is then translated into pro-LC3, which matures to LC3-I. The latter is lipidated with phosphatidylethanolamine (PE) to form LC3-II, which attaches to the AV membrane. The AV is trafficked by microtubules to the lysosome for degradation. During habitual (chronic) ethanol exposure, CYP2E1 is induced, generating MDA and other lipid peroxides (LPs), which inhibit proteasome activity (depicted as UPS) and which also stabilizes LC3-II from degradation. Acetaldehyde (Ach) generated by ethanol oxidation forms adducts with proteins, including the α tubulin subunit. Acetaldehyde production is also closely associated with tubulin acetylation [100]. We hypothesize that formation of Ach-α-tubulin adducts or acetylated tubulin may block the intracellular polymerization of microtubules and prevent AV–lysosome fusion, thereby causing AV (LC3-II) and protein accumulation (proteopathy), which, with steatosis (fatty liver), contributes to alcohol-induced hepatomegaly. Reproduced and modified with permission from Ref. [44].
Evidence that ethanol decreases lipophagy
Alcohol-induced steatosis (fatty liver) develops in 90% of heavy drinkers [22]. Steatosis was formerly believed to develop exclusively from a decreased cellular redox (NAD/NADH ratio) generated during sequential oxidations of ethanol and acetaldehyde by ADH and aldehyde dehydrogenase 2 (ALDH2) in the cytosol and mitochondria, respectively [46]. The latter redox change caused accelerated fatty acid synthesis and decelerated fatty acid oxidation. Subsequent investigations have since revealed that such changes are governed by specific transcription factors [47–49]. However, the seminal report that autophagy regulates lipid metabolism [29], stimulated interest in ascertaining whether alcohol exposure influences lipophagy, the autophagosomal engulfment and lysosomal degradation of lipid droplets. Wu, et al. examined whether CYP2E1 expression influences lipid accumulation and autophagy in ethanol-exposed CYP2E1-expressing E47 cells [50]. They detected higher levels of steatosis in ethanol-treated E-47 cells than in identically-treated CYP2E1-deficient C34 cells. Interestingly, these workers observed that ethanol induces autophagy in C34 cells but not in E-47 cells. In fact, in E47 cells, ethanol exposure elevates the content of P62/SQSTM1, a sign of obstructed autophagy. Thus, in alcohol-treated E-47 cells, elevated CYP2E1 expression enhances oxidant production, which impairs autophagy, causing lipid accumulation, which, likely reflects decelerated lipophagy. The latter results are consistent with other reports demonstrating that the accumulation of certain fatty acids impedes autophagy, which likely also reflects slower degradation of lipid droplet cargo [29].
Ethanol and autophagy in vivo
In experimental alcohol-fed rats, hepatic proteopathy (protein accumulation) occurs within 12 days after the commencement of chronic ethanol feeding [51]. Proteopathy reflects disrupted protein metabolism. As described earlier, intracellular breakdown of long-lived proteins occurs principally by autophagy in lysosomes [29,51,52]. We demonstrated that chronic ethanol administration slows the degradation of such proteins by inhibiting lysosome function [52] caused in part by a disruption of lysosome biogenesis [21,37,53]. Lysosomes degrade the contents of AVs during autophagy (Fig. 1). A detailed electron microscopy study confirmed that, compared with perfused livers of control rats, autophagy was impeded in ethanol-fed rats, showing lower numbers of autolysosomes (i.e. the products of AV–lysosome fusions) and lower rates of valine release, a sign of reduced proteolysis [54].
Other reports on the status of hepatic autophagy after ethanol exposure are controversial. Some conflict with each other and with the aforementioned findings. Ding et al. showed that acute (binge) ethanol administration, (using four equally-divided gavages in 20-min intervals) enhances autophagy [45], while Wu et al used a longer ethanol binge regimen (i.e. twice daily for 4 days) and reported decreased liver autophagy [55]. After 4 weeks of chronic ethanol feeding to mice, Lin et al. described enhanced hepatic autophagy in these animals [56]. Similarly, a recent immuno-electron microscopy (IEM) study concluded that livers of 10-week ethanol-fed rats had elevated AVs and a higher incidence of AV–cathepsin co-localization. They concluded that livers of ethanol-fed rats had enhanced autophagy [57]. Thus, the aforementioned chronic studies show contrasting findings to suggest that that this area of investigation is still unsettled. In recent unpublished work, we compared autophagy in livers of mice subjected to both acute and chronic ethanol administration. In addition to measuring standard autophagy parameters, we tested whether the nuclear content of transcription factor EB (TFEB), which controls autophagy, is differentially affected by acute versus chronic ethanol administration. In acute studies we gavaged transgenic mice that express green fluorescent protein (GFP) that is fused with LC3 (GFP–LC3) with a single dose of ethanol or PBS and measured autophagic parameters 3 or 12 h later. In chronic studies, we fed GFP–LC3 mice control or ethanol liquid diet (29.2%) ethanol as calories for several weeks. Compared with PBS-gavaged control mice, acute ethanol-treated mice exhibited greater AV numbers, a higher frequency of AV–lysosome co-localization and elevated levels of free GFP (a measure of endogenous GFP–LC3 hydrolysis). All the latter parameters indicated enhanced autophagy and they correlated closely with higher TFEB levels in hepatic nuclear fractions of acutely-treated, ethanol-gavaged mice. Livers from chronically ethanol-fed mice also exhibited higher AV numbers than controls. However, their livers exhibited significantly lower lysosome numbers, a lower frequency autophagosome–lysosome co-localization, higher P62/SQSTM1 levels and lower free GFP levels than pair-fed controls. The latter findings were associated with lower nuclear TFEB levels in ethanol-fed mice. Thus, acute ethanol gavage enhanced autophagy, associated with a higher nuclear content of TFEB. In contrast, chronic ethanol feeding impeded hepatic autophagy and was closely associated with lower levels of nuclear TFEB. Our findings indicate that acute and chronic ethanol administration exert differential effects on autophagy in the liver, as judged not only by autophagic markers but also by the nuclear content of transcription factor EB, which controls both autophagy and lysosome biogenesis. These findings not only confirmed our earlier reports [51] but they were also consistent with the properties of TFEB [58–61].
The ubiquitin–proteasome system
The 26S proteasome is a large (2000 kDa) non-lysosomal, multicatalytic (i.e. it possesses multiple catalytic sites) protease that acts in concert with a small (8.5 kDa) polypeptide called ubiquitin (Ub) and three enzymes, E1, E2, and E3 that catalyze attachment of Ub to proteins by a stepwise process called ubiquitylation. E1 is the Ub activating enzyme. E2 is the Ub conjugating enzyme and E3 is the Ub–protein ligase. In mammalian cells there is only one or two forms of E1 that catalyze the activation step, which is relatively nonspecific and requires E1, Ub, and ATP to form an Ub–adenylate intermediate. E1 transfers the activated (adenylated) Ub to an E2 isoenzyme, which then carries activated Ub to the protein substrate. Together with an E3 isozyme, (which has the greatest substrate specificity) the E2–E3 scaffold covalently joins the COOH-terminal glycine of Ub with the ε-NH2 group of an internal lysine of the substrate protein, forming an isopeptide linkage. Alternatively, Ub can form a peptide bond with the α-NH2 group at the substrate protein's NH2-terminus. Thus, the three ubiquitylation enzymes act sequentially and specifically to covalently join multiple ubiquitin moieties into a chain, (most often linked together via the lysine 48 residue of each ubiquitin molecule in the chain; called the “K48 polyubiquitin configuration”), which is recognized as a substrate by the 26S proteasome. Prior to its degradation, the polyubiquitylated protein undergoes de-ubiquitylation (ubiquitin removal) by the 19S regulatory complex of the 26S proteasome. The de-ubiquitylated protein substrate then enters the cylindrical 20S core, where it is hydrolyzed by multiple peptidases located on the β subunits of the core, generating peptides, which are subsequently degraded to their constituent amino acids. The UPS is depicted in Fig. 3, which also illustrates the equilibrium between the 26S and the 20S forms of the proteasome.
Fig. 3.
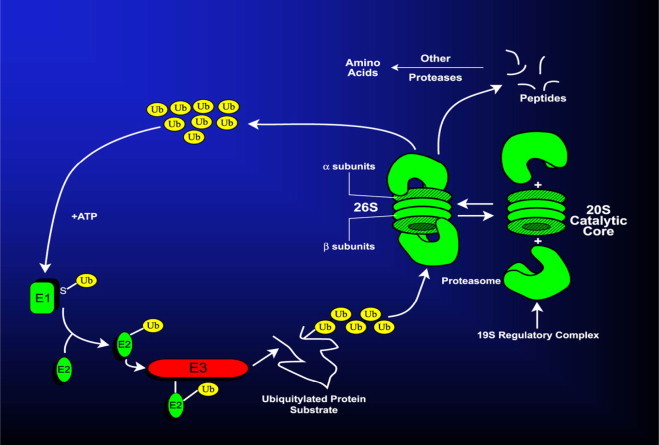
The ubiquitin–proteasome system. Details are provided in the text.
Proteins degraded by the UPS are typically characterized by their relatively short half-lives. Many of them have critical functions in cell cycle regulation, apoptosis, tumor suppression, signal transduction, and transcription [62,63]. It is also noteworthy that the cylinder-shaped 20S catalytic core of the 26S proteasome can dissociate from the larger complex and is itself catalytically active, degrading proteins in the absence of ATP and ubiquitin. In fact, the 20S proteasome is the predominant enzyme form in most mammalian cells [64] and rather selectively degrades oxidized, misfolded and covalently modified proteins [65,66]. Such degradation is critical as a means of cellular detoxification, as damaged proteins are potentially harmful because they can become entangled into protein aggregates. Such aggregates are resistant to degradation by the proteasome and require removal by autophagy [20,67]. Accumulation of such aggregates into cellular compartments called aggresomes is a survival mechanism in cells, but their over-accumulation in cells is believed to compromise cell survival. In liver, such aggresomes are known as alcoholic hyaline or Mallory-Denk (M-D) bodies. These are considered by some to be histological signatures of ALD, but are reportedly associated with other types of liver disease as well [20,68].
Ethanol consumption and the UPS
Heavy alcohol consumption by humans affects the levels of ubiquitin and its protein conjugates. Serum concentrations of both free ubiquitin (the unconjugated 8.5 kDa polypeptide) and polyubiquitin chains are higher in patients with cirrhosis than in normal subjects or in patients with milder forms of alcoholic liver disease [69]. This is related to the finding that in hepatocytes of alcoholics, M-D bodies contain large amounts of ubiquitin as well as the cytokeratins 8 and/or 18, and P62. The latter proteins and polypeptides are believed to be essential components for M-D formation [70]. M-D bodies also contain the heat shock proteins 70, 90 and 25 as well as specific subunits of the 26S proteasome [71], to suggest that M-D body formation represents a failed attempt by the UPS to degrade the protein components of M-D bodies. Evidence also indicates that in ethanol-exposed HepG2 (E-47) cells, that over-express CYP2E1, oxidant generation by CYP2E1 stimulates formation of M-D body-like aggresomes that contain immunoreactive cytokeratin. Formation of such aggregates in these cells correlates with an ethanol-elicited decline in proteasome activity [72].
When ethanol, as part of a liquid diet, is continuously infused into rats by intragastric intubation, it causes greater liver damage than that produced after chronic oral ethanol administration. Both methods of ethanol feeding induce the levels of immunoreactive free ubiquitin and ubiquitin–protein conjugates in the liver. When ethanol is orally administered for one or five weeks, ubiquitin levels rise to a greater extent after 1 week than after 5 weeks of oral feeding, to suggest that part of the early rise in ubiquitin is a stress response akin to heat shock, which enhances hepatic ubiquitin production [73]. Five weeks of oral ethanol administration also causes ubiquitin to rise, probably due to reduced proteolysis by the 26S proteasome. Chronic oral ethanol administration does not affect the activities of hepatic ubiquitylation enzymes [73].
Ethanol exposure and the 20S proteasome
Compared with rats intragastrically-fed with control diet, intragastric ethanol administration causes a 35–40% reduction of three major peptidases, the chymotrypsin-like, the trypsin-like and the peptidyl-glutamyl peptide hydrolase activities of the 20S proteasome [74]. The reduction in proteasome activity after such treatment correlates inversely with increased hepatic content of lipid peroxides, indicating that ethanol-induced oxidant stress negatively influences proteasome activity [75]. Furthermore, reduced proteasome activity causes accumulation of both native and damaged (e.g. oxidized) proteins in the liver. The loss in proteasome activity and to some degree, the extent of such protein accumulation are associated with the severity of ethanol-induced liver injury [76].
The aforementioned studies clearly show that 20S proteasome peptidase activities are significantly reduced after intragastric ethanol administration. However, our initial studies compared intragastric with oral ethanol feeding. They showed that oral ethanol feeding causes no such decline in proteasome activity. In fact, the chymotrypsin-like and the peptidyl-glutamyl-peptide hydrolase activities of the 20S enzyme in livers of orally ethanol-fed rats tend to be higher than those of pair-fed controls [74]. However, after we compiled the results of several subsequent oral feeding studies, we found that oral ethanol feeding indeed increases the chymotrypsin-like peptidase activity of the proteasome, but it also decreases this peptidase activity, depending upon the serum ethanol concentration at the time of sacrifice. Thus, proteasome activity correlates inversely with the serum ethanol concentration (Fig. 4). However, we believe that ethanol oxidation in the liver, rather than ethanol itself, causes this dose-dependent decline in enzyme activity. This is because separate in vitro studies revealed that proteasome activity is elevated after exposure to relatively low concentrations (10–100 µM) of peroxynitrite (OONO−; PN), which is a secondary metabolite of ethanol, generated by the reaction between superoxide (O2−), derived from mitochondrial oxidation and CYP2E1 catalysis and nitric oxide (NO), generated by the inducible nitric oxide synthase. Conversely, exposure of the proteasome to higher (1 mM) PN inhibits or even abolishes its chymotrypsin-like activity. The latter in vitro data were confirmed by in vivo experiments in which we injected animals with molsidomine, an agent that is metabolized to SIN-1, a donor of NO, which subsequently reacts with O2− to form PN [77]. In other work, we made similar observations of differential sensitivity of the proteasome to oxidants. Proteasome activity increases in cultured Huh-7 cells that constitutively express the hepatitis C virus core protein, which itself generates low levels of oxidants. Proteasome chymotrypsin-like activity is elevated by CYP2E1-derived oxidant generation in L-14 cells, but its activity decreases when the same cells are exposed to ethanol, indicating that proteasome activity is elevated by low levels of HCV core-induced oxidants but is inhibited by higher levels of oxidants generated by ethanol metabolism [78]. The implications of these latter findings are important, as liver and hepatoma cells express the immunoproteasome (IP), allowing these cells to process antigens [79,80]. The IP contains distinct β-subunits that enable the enzyme to cleave antigens to peptides of more uniform sizes than those produced by the constitutive proteasome [81]. Such peptides interact with the major histocompatibility complex class 1 (MHC class 1), and are then presented on the cell surface. There, they are recognized by cytotoxic T cells, which kill antigen-bearing hepatocytes. Work in our laboratory using VL-17A (ADH+/CYP2E1+) cells demonstrated that ethanol-induced oxidant stress suppressed proteasome activity, thereby impeding the cleavage of peptides for presentation by these cells in the context of MHC class 1 [82]. These same cells respond well to interferon gamma (IFN-Ɣ) treatment, which enhances proteasome activity. However, ethanol exposure blocks both IFN-Ɣ signaling and proteasome induction [83]. We observed similar results in hepatocytes and in HepB5 cells, an immortalized line of cells stably transfected with CYP2E1 plasmid and in which the ethanol-elicited reduction in proteasome activity also suppresses MHC-1 restricted antigen presentation on their surfaces [84].
Fig. 4.
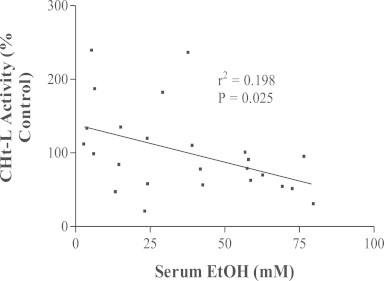
Inverse correlation between hepatic proteasome activity and serum ethanol levels in ethanol-fed rats. Data are derived from four separate studies in which rats were fed liquid control or ethanol diets for four to six weeks. Each data point is the proteasome chymotrypsin-like activity in one ethanol-fed rat, expressed as percent of the same activity of its pair-fed control rat. Data points are plotted as a function of the serum ethanol concentration in each animal. The plot was drawn by linear regression and statistically analyzed.
Proteasome activity and alcoholic-induced steatosis
The UPS and the 20S proteasome each degrade native, damaged and regulatory proteins. The latter group includes transcription factors and cytokines that enhance hepatic lipogenesis. These include the sterol regulatory element binding protein 1C (SREBP-1C), early growth response-1 (Egr-1) and tumor necrosis factor-alpha (TNF-α). We demonstrated in vivo that Egr-1 partially regulates ethanol-induced steatosis after acute alcohol administration[85].
We also examined Egr-1 regulation in ethanol-exposed VL-17A cells. Egr-1 promoter activity and mRNA levels increase after 1 h of ethanol exposure, followed by a rise in Egr-1 protein, which precedes an ethanol-induced rise in triglyceride accumulation. Elevated Egr-1 protein is sustained in part by accelerated Egr-1 synthesis and by an ethanol-induced decrease in proteasome activity, the latter of which stabilizes Egr-1. Knockdown of Egr-1 expression with siRNA only partially blocks ethanol-induced Egr-1 and triglyceride accumulation, providing further evidence that reduced proteasome activity stabilizes Egr-1. Such stabilization maintains Egr-1 at significantly higher levels even without its accelerated synthesis. This results in sustained cellular steatosis. The decline in proteasome activity is associated with higher CYP2E1 levels that would generate more oxidants to inhibit the proteasome [71,72,86]. However, we found that ethanol-exposed E-47 cells, which express CYP2E1 but not ADH, exhibit no Egr-1 induction, even though their CYP2E1 levels rise nearly two-fold. In contrast, Egr-1 levels increase 1.6-fold in ethanol-exposed VA-13 cells, which express ADH but not CYP2E1. These latter findings clearly show that ADH has a larger role than CYP2E1 in both the acceleration of Egr-1 synthesis and the oxidant-induced decline in proteasome activity, which causes Egr-1 stabilization. Thus ethanol-induced inhibition of proteasome activity requires ethanol oxidation, implying that acetaldehyde and oxidants derived from CYP2E1 catalysis are involved in Egr-1 stabilization. It is noteworthy that other genes involved in alcoholic liver injury, including platelet-derived growth factor (PDGF), transforming growth factor-beta (TGF-β), fibroblast growth factor (FGF), TNF-α and SREBP-1C each has an Egr-1 binding site on its promoter region [87,88–90]. Thus, elevated levels of Egr-1, which are sustained by proteasome inhibition, likely function in sustaining alcohol-induced lipid accumulation. In addition, the temporal regulation of the aforementioned genes by Egr-1 is a molecular basis for the progression from steatosis to more severe forms of alcoholic liver injury.
Summary and future work
The foregoing review has described the effects of ethanol exposure on two major catabolic pathways, autophagy and the UPS in the liver. From the results described, it is clear that ethanol oxidation by both ADH and CYP2E1 is the principal means by which ethanol modulates the activity of each catabolic pathway. Furthermore, it is noteworthy that the end result of ethanol exposure on these pathways is not always their down-regulation, as acute ethanol treatment enhances autophagy but has no effect on the proteasome [45,85]. These and other results described herein, strongly imply that autophagy is the more sensitive of the two hydrolytic pathways. The simplest explanation for this difference is that autophagy involves a greater number of “moving parts”, which are more sensitive than those of the UPS (Fig. 1). Furthermore, besides Atg proteins, autophagy depends on other protein factors and molecular motors, including tubulin, which is sensitive to ethanol metabolites because it readily reacts with and forms adducts with acetaldehyde [91]. Thus, the same oxidant levels derived from ethanol metabolism that activate the proteasome may inhibit autophagy. However, with continued heavy drinking it is likely that both pathways are negatively affected. As always, the findings summarized here prompt more questions, such as: why does acute ethanol stimulate autophagy, while chronic ethanol suppresses it? One explanation is that acute ethanol administration (i.e. a single ethanol gavage) causes rapid, (>90%) hepatic mitochondrial depolarization, which depends on ethanol oxidation in hepatocytes. Such an occurrence likely stimulates an equally rapid autophagic (perhaps mitophagic) response to the burst of superoxide release from depolarized mitochondria, most of which are repolarized within 24 h after ethanol administration [92]. In contrast, chronic ethanol exposure produces nearly constant ethanol oxidation, during which reactive oxygen/nitrogen species are continuously produced and eventually overwhelm the liver cell's ability to remove damaged proteins and dysfunctional organelles. While further investigation is necessary to validate this hypothesis, existing data mentioned herein, strongly support it. Another critical question is: What is the status of autophagy and the UPS during the more severe stages of alcoholic liver disease, including fibrosis and cirrhosis? Animal studies would suggest that both catabolic pathways are down-regulated. However, such studies are limited because rodents are generally resistant to liver damage beyond steatosis and rather heroic experimental manipulations (e.g. intragastric feeding) are required to achieve severe alcohol-induced liver injury in rats and mice. Thus, carefully conducted studies are warranted, using surgically- or biopsy-derived liver tissue from human donors to determine the status of these hydrolytic systems in alcoholic patients. If the activities of both the UPS and autophagy are indeed down-regulated in livers of such patients, there are safe and approved compounds currently available that reportedly accelerate autophagy. Two of these are carbamazepine and caffeine [12,93,94]. In fact, coffee, which contains caffeine, and is classically regarded as an antidote for a hangover, is now recommended for patients with fibrotic liver disease [95]. However, it should be emphasized that autophagy and the UPS, while very important in cellular waste management, are only two parts of a larger picture of defensive maneuvers employed by liver cells to maintain their viability. Nevertheless, these hydrolytic pathways appear to have evolved mostly for the purpose of liver cell survival. Their burgeoning importance in disease processes continues to be an active area of investigation.
Finally, it is important to note that alcohol abuse damages extrahepatic tissues, most notably the brain. Are the UPS and autophagy similarly affected in neural tissue as they are in hepatic tissue? Brain ethanol metabolism differs from that in liver, as the majority of neural ethanol oxidation is catalyzed by the peroxisomal enzyme, catalase which uses hydrogen peroxide and ethanol as the substrates to form acetaldehyde and water. Additionally, CYP2E1 is expressed in brain and contributes to neural ethanol oxidation [96,97]. Acute studies with ethanol-treated developing mice revealed that the brains of these animals exhibit enhanced autophagy. This response is replicated in cultured neuroblastoma cells exposed to ethanol in vitro [98]. Investigators believe that autophagy induction in the brain is a protective response to the sudden burst of ethanol-elicited oxidant stress, similar to that which occurs in the liver. Recent studies also revealed that chronic ethanol administration causes significant accumulation of polyubiquitylated proteins in the mouse cerebral cortex. The latter findings are associated with a decline in the levels of the constitutive 20S proteasome subunits, α2 and β5, to suggest an ethanol-induced down-regulation of proteasome activity. Concurrently, in the same ethanol-fed animals, there is an activation of brain MTORC1, the negative regulator of autophagy. This is associated with a decline in autophagy components, including the lysosomal protease, cathepsin B and Atg5, and Atg12, two proteins involved in autophagosome formation, (see Fig. 1) [99]. These results imply that, despite differences in the enzymes that catalyze ethanol oxidation in the liver and brain, autophagy and the UPS are similarly affected in both organs by acute and chronic ethanol administration. Acute ethanol treatment activates brain autophagy, while chronic ethanol suppresses it as well as the proteasome. Thus, it is likely that these effects are initially caused by the formation of acetaldehyde and are subsequently exacerbated by CYP2E1 catalysis, which forms Ach but also participates in the production of secondary oxidants.
Disclaimer
The contents of this paper do not represent the views of the Department of Veterans Affairs or the United States Government.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Acknowledgments
Financial support for results presented or mentioned herein was provided by institutional funds from the Section of Gastroenterology and Hepatology, UNMC Department of Internal Medicine, a Dean's Reviewed Research Grant from the University of Nebraska Medical Center, by Grant no. 1-R01-AA16546 and previous grants from the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health and with Biomedical Research Funds from the United States, Department of Veterans Affairs. Work was supported with resources and facilities, in the Research Service of the Nebraska-Western Iowa Health Care System, part of the United States, Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development.
References
- 1.Schoenheimer R. The Dynamic State of Body Constituents. Harvard University Press; Cambridge, MA: 1942. [Google Scholar]
- 2.De Duve C., Wattiaux R. Functions of lysosomes. Annual Review of Physiology. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. 5322983 [DOI] [PubMed] [Google Scholar]
- 3.Bowers W.E. Christian de Duve and the discovery of lysosomes and peroxisomes. Trends in Cell Biology. 1998;8(8):330–333. doi: 10.1016/s0962-8924(98)01314-2. 9704410 [DOI] [PubMed] [Google Scholar]
- 4.De Duve C. Lysosomes revisited. European Journal of Biochemistry. 1983;137(3):391–397. doi: 10.1111/j.1432-1033.1983.tb07841.x. 6319122 [DOI] [PubMed] [Google Scholar]
- 5.Deter R.L., Baudhuin P., De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. Journal of Cell Biology. 1967;35(2):C11–C16. doi: 10.1083/jcb.35.2.c11. 6055998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deter R.L., De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. Journal of Cell Biology. 1967;33(2):437–449. doi: 10.1083/jcb.33.2.437. 4292315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schimke R.T., Doyle D. Control of enzyme levels in animal tissues. Annual Review of Biochemistry. 1970;39:929–976. doi: 10.1146/annurev.bi.39.070170.004433. 4394639 [DOI] [PubMed] [Google Scholar]
- 8.Lee K.L., Darke P.L., Kenney F.T. Role of coenzyme in aminotransferase turnover. Journal of Biological Chemistry. 1977;252(14):4958–4961. 17609 [PubMed] [Google Scholar]
- 9.Bachmair A., Finley D., Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234(4773):179–186. doi: 10.1126/science.3018930. 3018930 [DOI] [PubMed] [Google Scholar]
- 10.Hershko A., Ciechanover A., Heller H., Haas A.L., Rose I.A. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(4):1783–1786. doi: 10.1073/pnas.77.4.1783. 6990414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciechanover A., Elias S., Heller H., Ferber S., Hershko A. Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes. Journal of Biological Chemistry. 1980;255(16):7525–7528. 6249802 [PubMed] [Google Scholar]
- 12.Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherjee A., Goldbach C., Watkins S., Michalopoulos G., Perlmutter D.H. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229–232. doi: 10.1126/science.1190354. 20522742 [DOI] [PubMed] [Google Scholar]
- 13.Pastore N., Blomenkamp K., Annunziata F., Piccolo P., Mithbaokar P., Maria Sepe R., Vetrini F., Palmer D., Ng P., Polishchuk E., Iacobacci S., Polishchuk R., Teckman J., Ballabio A., Brunetti-Pierri N. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Molecular Medicine. 2013;5(3):397–412. doi: 10.1002/emmm.201202046. 23381957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burger A.M., Seth A.K. The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. European Journal of Cancer. 2004;40(15):2217–2229. doi: 10.1016/j.ejca.2004.07.006. 15454246 [DOI] [PubMed] [Google Scholar]
- 15.Orlowski R.Z., Stinchcombe T.E., Mitchell B.S., Shea T.C., Baldwin A.S., Stahl S., Adams J., Esseltine D.L., Elliott P.J., Pien C.S., Guerciolini R., Anderson J.K., Depcik-Smith N.D., Bhagat R., Lehman M.J., Novick S.C., O’Connor O.A., Soignet S.L. Phase I trial of the proteasome inhibitor PS−341 in patients with refractory hematologic malignancies. Journal of Clinical Oncology. 2002;20(22):4420–4427. doi: 10.1200/JCO.2002.01.133. 12431963 [DOI] [PubMed] [Google Scholar]
- 16.Xu Q., Farah M., Webster J.M., Wojcikiewicz R.J. Bortezomib rapidly suppresses ubiquitin thiolesterification to ubiquitin-conjugating enzymes and inhibits ubiquitination of histones and type I inositol 1,4,5-trisphosphate receptor. Molecular Cancer Therapeutics. 2004;3(10):1263–1269. 15486193 [PubMed] [Google Scholar]
- 17.Gao Z., Gammoh N., Wong P.M., Erdjument-Bromage H., Tempst P., Jiang X. Processing of autophagic protein LC3 by the 20S proteasome. Autophagy. 2010;6(1):126–137. doi: 10.4161/auto.6.1.10928. 20061800 [DOI] [PubMed] [Google Scholar]
- 18.Dolganiuc A., Thomes P.G., Ding W.X., Lemasters J.J., Donohue T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcoholism: Clinical and Experimental Research. 2012;36:1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. 22551004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doherty F.J., Osborn N.U., Wassell J.A., Heggie P.E., Laszlo L., Mayer R.J. Ubiquitin-protein conjugates accumulate in the lysosomal system of fibroblasts treated with cysteine proteinase inhibitors. Biochemical Journal. 1989;263(1):47–55. doi: 10.1042/bj2630047. 2557825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harada M., Hanada S., Toivola D.M., Ghori N., Omary M.B. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47(6):2026–2035. doi: 10.1002/hep.22294. 18454506 [DOI] [PubMed] [Google Scholar]
- 21.Kharbanda K.K., McVicker D.L., Zetterman R.K., MacDonald R.G., Donohue T.M. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology. 1997;26(4):929–933. doi: 10.1002/hep.510260419. 9328315 [DOI] [PubMed] [Google Scholar]
- 22.O’Shea R.S., Dasarathy S., McCullough A.J. Alcoholic liver disease. Hepatology. 2010;51(1):307–328. doi: 10.1002/hep.23258. 20034030 [DOI] [PubMed] [Google Scholar]
- 23.Orman E.S., Odena G., Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. Journal of Gastroenterology and Hepatology. 2013;28(Suppl. 1):S77–S84. doi: 10.1111/jgh.12030. 23855300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altamirano J., Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nature Reviews Gastroenterology and Hepatology. 2011;8(9):491–501. doi: 10.1038/nrgastro.2011.134. 21826088 [DOI] [PubMed] [Google Scholar]
- 25.Coon M.J., Koop D.R. Alcohol-inducible cytochrome P-450 (P-450ALC) Archives of Toxicology. 1987;60(1–3):16–21. doi: 10.1007/BF00296940. 3304205 [DOI] [PubMed] [Google Scholar]
- 26.Dai Y., Cederbaum A.I. Cytotoxicity of acetaminophen in human cytochrome P4502E1-transfected HepG2 cells. Journal of Pharmacology and Experimental Therapeutics. 1995;273(3):1497–1505. 7791125 [PubMed] [Google Scholar]
- 27.Spracklin D.K., Hankins D.C., Fisher J.M., Thummel K.E., Kharasch E.D. Cytochrome P450 2E1 is the principal catalyst of human oxidative halothane metabolism in vitro. Journal of Pharmacology and Experimental Therapeutics. 1997;281(1):400–411. 9103523 [PubMed] [Google Scholar]
- 28.Roberts B.J., Song B.J., Soh Y., Park S.S., Shoaf S.E. Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. Journal of Biological Chemistry. 1995;270(50):29632–29635. doi: 10.1074/jbc.270.50.29632. 8530344 [DOI] [PubMed] [Google Scholar]
- 29.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976. 19339967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czaja M.J., Ding W.X., Donohue T.M., Jr., Friedman S.L., Kim J.S., Komatsu M., Lemasters J.J., Lemoine A., Lin J.D., Ou J.H., Perlmutter D.H., Randall G., Ray R.B., Tsung A., Yin X.M. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9(8):1131–1158. doi: 10.4161/auto.25063. 23774882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizushima N., Levine B. Autophagy in mammalian development and differentiation. Nature Cell Biology. 2010;12(9):823–830. doi: 10.1038/ncb0910-823. 20811354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furuta N., Amano A. Cellular machinery to fuse antimicrobial autophagosome with lysosome. Communicative and Integrative Biology. 2010;3(4):385–387. doi: 10.4161/cib.3.4.12030. 20798834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fortunato F., Bürgers H., Bergmann F., Rieger P., Büchler M.W., Kroemer G., Werner J. Impaired autolysosome formation correlates with lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137(1):350–360. doi: 10.1053/j.gastro.2009.04.003. 19362087 [DOI] [PubMed] [Google Scholar]
- 34.Eskelinen E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Molecular Aspects of Medicine. 2006;27(5–6):495–502. doi: 10.1016/j.mam.2006.08.005. 16973206 [DOI] [PubMed] [Google Scholar]
- 35.Ma X., Godar R.J., Liu H., Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy. 2012;8(3):297–309. doi: 10.4161/auto.18658. 22302006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saftig P., Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nature Reviews Molecular Cell Biology. 2009;10(9):623–635. doi: 10.1038/nrm2745. 19672277 [DOI] [PubMed] [Google Scholar]
- 37.Kharbanda K.K., McVicker D.L., Zetterman R.K., Donohue T.M., Jr. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochimica et Biophysica Acta. 1996;1291(1):45–52. doi: 10.1016/0304-4165(96)00043-8. 8781524 [DOI] [PubMed] [Google Scholar]
- 38.Donohue T.M., Jr. Autophagy and ethanol-induced liver injury. World Journal of Gastroenterology. 2009;15(10):1178–1185. doi: 10.3748/wjg.15.1178. 19291817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu D., Cederbaum A.I. Ethanol cytotoxicity to a transfected HepG2 cell line expressing human cytochrome P4502E1. Journal of Biological Chemistry. 1996;271(39):23914–23919. doi: 10.1074/jbc.271.39.23914. 8798623 [DOI] [PubMed] [Google Scholar]
- 40.Donohue T.M., Osna N.A., Clemens D.L. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. International Journal of Biochemistry and Cell Biology. 2006;38(1):92–101. doi: 10.1016/j.biocel.2005.07.010. 16181800 [DOI] [PubMed] [Google Scholar]
- 41.Rubinsztein D.C., Cuervo A.M., Ravikumar B., Sarkar S., Korolchuk V., Kaushik S., Klionsky D.J. In search of an “autophagomometer”. Autophagy. 2009;5(5):585–589. doi: 10.4161/auto.5.5.8823. 19411822 [DOI] [PubMed] [Google Scholar]
- 42.Korolchuk V.I., Mansilla A., Menzies F.M., Rubinsztein D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Molecular Cell. 2009;33(4):517–527. doi: 10.1016/j.molcel.2009.01.021. 19250912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korolchuk V.I., Menzies F.M., Rubinsztein D.C. A novel link between autophagy and the ubiquitin–proteasome system. Autophagy. 2009;5(6):862–863. doi: 10.4161/auto.8840. 19458478 [DOI] [PubMed] [Google Scholar]
- 44.Thomes P.G., Ehlers R.A., Trambly C.S., Clemens D.L., Fox H.S., Tuma D.J., Donohue T.M. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy. 2013;9(1):63–73. doi: 10.4161/auto.22490. 23090141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding W.X., Li M., Chen X., Ni H.M., Lin C.W., Gao W., Lu B., Stolz D.B., Clemens D.L., Yin X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139(5):1740–1752. doi: 10.1053/j.gastro.2010.07.041. 20659474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zakhari S., Li T.K. Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46(6):2032–2039. doi: 10.1002/hep.22010. 18046720 [DOI] [PubMed] [Google Scholar]
- 47.You M., Fischer M., Deeg M.A., Crabb D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) Journal of Biological Chemistry. 2002;277(32):29342–29347. doi: 10.1074/jbc.M202411200. 12036955 [DOI] [PubMed] [Google Scholar]
- 48.Fischer M., You M., Matsumoto M., Crabb D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. Journal of Biological Chemistry. 2003;278(30):27997–28004. doi: 10.1074/jbc.M302140200. 12791698 [DOI] [PubMed] [Google Scholar]
- 49.You M., Crabb D.W. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2004;287(1):G1–G6. doi: 10.1152/ajpgi.00056.2004. 15194557 [DOI] [PubMed] [Google Scholar]
- 50.Wu D., Wang X., Zhou R., Cederbaum A. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochemical and Biophysical Research Communications. 2010;402(1):116–122. doi: 10.1016/j.bbrc.2010.09.127. 20932821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donohue T.M., Jr., Zetterman R.K., Tuma D.J. Effect of chronic ethanol administration on protein catabolism in rat liver. Alcoholism: Clinical and Experimental Research. 1989;13(1):49–57. doi: 10.1111/j.1530-0277.1989.tb00283.x. 2646978 [DOI] [PubMed] [Google Scholar]
- 52.Donohue T.M., Jr., McVicker D.L., Kharbanda K.K., Chaisson M.L., Zetterman R.K. Ethanol administration alters the proteolytic activity of hepatic lysosomes. Alcoholism: Clinical and Experimental Research. 1994;18(3):536–541. doi: 10.1111/j.1530-0277.1994.tb00906.x. 7943651 [DOI] [PubMed] [Google Scholar]
- 53.Kharbanda K.K., McVicker D.L., Zetterman R.K., Donohue T.M., Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochimica et Biophysica Acta. 1995;1245(3):421–429. doi: 10.1016/0304-4165(95)00121-2. 8541322 [DOI] [PubMed] [Google Scholar]
- 54.Pösö A.R., Hirsimäki P. Inhibition of proteolysis in the liver by chronic ethanol feeding. Biochemical Journal. 1991;273(1):149–152. doi: 10.1042/bj2730149. 1989576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu D., Wang X., Zhou R., Yang L., Cederbaum A.I. Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy. Free Radical Biology and Medicine. 2012;53(6):1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. 22819980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin C.W., Zhang H., Li M., Xiong X., Chen X., Chen X., Dong X.C., Yin X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. Journal of Hepatology. 2013;58(5):993–999. doi: 10.1016/j.jhep.2013.01.011. 23339953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eid N., Ito Y., Maemura K., Otsuki Y. Elevated autophagic sequestration of mitochondria and lipiddroplets in steatotic hepatocytes of chronic ethanol-treated rats:an immunohistochemical and electron microscopic study. Journal of Molecular Histology. 2013;44:311–326. doi: 10.1007/s10735-013-9483-x. 23371376 [DOI] [PubMed] [Google Scholar]
- 58.Settembre C., Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7(11):1379–1381. doi: 10.4161/auto.7.11.17166. 21785263 [DOI] [PubMed] [Google Scholar]
- 59.Settembre C., De Cegli R., Mansueto G., Saha P.K., Vetrini F., Visvikis O., Huynh T., Carissimo A., Palmer D., Klisch T.J., Wollenberg A.C., Di Bernardo D., Chan L., Irazoqui J.E., Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature Cell Biology. 2013;15(6):647–658. doi: 10.1038/ncb2718. 23604321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., Sardiello M., Rubinsztein D.C., Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. 21617040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C., Facchinetti V., Sabatini D.M. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO Journal. 2012;31(5):1095–1108. doi: 10.1038/emboj.2012.32. 22343943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X.L., Hassel B.A. Involvement of proteasomes in gene induction by interferon and double-stranded RNA. Cytokine. 2001;14(5):247–252. doi: 10.1006/cyto.2001.0887. 11444904 [DOI] [PubMed] [Google Scholar]
- 63.Park D.J., Lenz H.J. The role of proteasome inhibitors in solid tumors. Annals of Medicine. 2004;36(4):296–303. doi: 10.1080/07853890410029031. 15224656 [DOI] [PubMed] [Google Scholar]
- 64.Brooks P., Fuertes G., Murray R.Z., Bose S., Knecht E., Rechsteiner M.C., Hendil K.B., Tanaka K., Dyson J., Rivett J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochemical Journal. 2000;346(1):155–161. 10657252 [PMC free article] [PubMed] [Google Scholar]
- 65.Shringarpure R., Grune T., Mehlhase J., Davies K.J. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. Journal of Biological Chemistry. 2003;278(1):311–318. doi: 10.1074/jbc.M206279200. 12401807 [DOI] [PubMed] [Google Scholar]
- 66.Curry-McCoy T.V., Osna N.A., Donohue T.M., Jr. Modulation of lysozyme function and degradation after nitration with peroxynitrite. Biochimica et Biophysica Acta. 2009;1790(8):778–786. doi: 10.1016/j.bbagen.2009.04.008. 19376194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bence N.F., Sampat R.M., Kopito R.R. Impairment of the ubiquitin–proteasome system by protein aggregation. Science. 2001;292(5521):1552–1555. doi: 10.1126/science.292.5521.1552. 11375494 [DOI] [PubMed] [Google Scholar]
- 68.Rautou P.E., Mansouri A., Lebrec D., Durand F., Valla D., Moreau R. Autophagy in liver diseases. Journal of Hepatology. 2010;53(6):1123–1134. doi: 10.1016/j.jhep.2010.07.006. 20810185 [DOI] [PubMed] [Google Scholar]
- 69.Takagi M., Yamauchi M., Takada K., Ohkawa K. Serum ubiquitin–protein conjugates in normal subjects and patients with alcoholic liver diseases: immunoaffinity isolation and electrophoretic mobility. Alcoholism: Clinical and Experimental Research. 2002;26(11):1692–1696. doi: 10.1097/01.ALC.0000036924.31671.0E. 12436058 [DOI] [PubMed] [Google Scholar]
- 70.Stumptner C., Fuchsbichler A., Zatloukal K., Denk H. In vitro production of Mallory bodies and intracellular hyaline bodies: the central role of sequestosome 1/p62. Hepatology. 2007;46(3):851–860. doi: 10.1002/hep.21744. 17685470 [DOI] [PubMed] [Google Scholar]
- 71.Bardag-Gorce F., van Leeuwen F.W., Nguyen V., French B.A., Li J., Riley N., McPhaul L.W., Lue Y.H., French S.W. The role of the ubiquitin-proteasome pathway in the formation of Mallory bodies. Experimental and Molecular Pathology. 2002;73(2):75–83. doi: 10.1006/exmp.2002.2451. 12231209 [DOI] [PubMed] [Google Scholar]
- 72.Bardag-Gorce F., French B.A., Nan L., Song H., Nguyen S.K., Yong H., Dede J., French S.W. CYP2E1 induced by ethanol causes oxidative stress, proteasome inhibition and cytokeratin aggresome (Mallory body-like) formation. Experimental and Molecular Pathology. 2006;81(3):191–201. doi: 10.1016/j.yexmp.2006.07.007. 17034788 [DOI] [PubMed] [Google Scholar]
- 73.Born L.J., Kharbanda K.K., McVicker D.L., Zetterman R.K., Donohue T.M., Jr. Effects of ethanol administration on components of the ubiquitin proteolytic pathway in rat liver. Hepatology. 1996;23(6):1556–1563. doi: 10.1002/hep.510230636. 8675177 [DOI] [PubMed] [Google Scholar]
- 74.Donohue T.M., Jr., Zetterman R.K., Zhang-Gouillon Z.Q., French S.W. Peptidase activities of the multicatalytic protease in rat liver after voluntary and intragastric ethanol administration. Hepatology. 1998;28(2):486–491. doi: 10.1002/hep.510280228. 9696015 [DOI] [PubMed] [Google Scholar]
- 75.Donohue T.M., Jr., Kharbanda K.K., Casey C.A., Nanji A.A. Decreased proteasome activity is associated with increased severity of liver pathology and oxidative stress in experimental alcoholic liver disease. Alcoholism: Clinical and Experimental Research. 2004;28(8):1257–1263. doi: 10.1097/01.alc.0000134233.89896.19. 15318126 [DOI] [PubMed] [Google Scholar]
- 76.Fataccioli V., Andraud E., Gentil M., French S.W., Rouach H. Effects of chronic ethanol administration on rat liver proteasome activities: relationship with oxidative stress. Hepatology. 1999;29(1):14–20. doi: 10.1002/hep.510290106. 9862843 [DOI] [PubMed] [Google Scholar]
- 77.Osna N.A., Haorah J., Krutik V.M., Donohue T.M., Jr. Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology. 2004;40(3):574–582. doi: 10.1002/hep.20352. 15349895 [DOI] [PubMed] [Google Scholar]
- 78.Osna N.A., White R.L., Krutik V.M., Wang T., Weinman S.A., Donohue T.M., Jr. Proteasome activation by hepatitis C core protein is reversed by ethanol-induced oxidative stress. Gastroenterology. 2008;134(7):2144–2152. doi: 10.1053/j.gastro.2008.02.063. 18549882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cascio P., Hilton C., Kisselev A.F., Rock K.L., Goldberg A.L. 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO Journal. 2001;20:2357–2366. doi: 10.1093/emboj/20.10.2357. 11350924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khan S., van den Broek M., Schwarz K., de Giuli R., Diener P.A., Groettrup M. Immunoproteasomes largely replace constitutive proteasomes during an antiviral and antibacterial immune response in the liver. Journal of Immunology. 2001;167(12):6859–6868. doi: 10.4049/jimmunol.167.12.6859. 11739503 [DOI] [PubMed] [Google Scholar]
- 81.Griffin T.A., Nandi D., Cruz M., Fehling H.J., Kaer L.V., Monaco J.J., Colbert R.A. Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. Journal of Experimental Medicine. 1998;187(1):97–104. doi: 10.1084/jem.187.1.97. 9419215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Osna N.A., White R.L., Todero S., McVicker B.L., Thiele G.M., Clemens D.L., Tuma D.J., Donohue T.M., Jr. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology. 2007;45(1):53–61. doi: 10.1002/hep.21442. 17187415 [DOI] [PubMed] [Google Scholar]
- 83.Osna N.A., Clemens D.L., Donohue T.M. Interferon gamma enhances proteasome activity in recombinant Hep G2 cells that express cytochrome P4502E1: modulation by ethanol. Biochemical Pharmacology. 2003;66(5):697–710. doi: 10.1016/s0006-2952(03)00252-1. 12948850 [DOI] [PubMed] [Google Scholar]
- 84.Osna N.A., White R.L., Thiele G.M., Donohue T.M., Jr. Ethanol metabolism alters major histocompatibility complex class I-restricted antigen presentation in liver cells. Hepatology. 2009;49(4):1308–1315. doi: 10.1002/hep.22787. 19195028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Donohue T.M., Jr., Osna N.A., Trambly C.S., Whitaker N.P., Thomes P.G., Todero S.L., Davis J.S. Early growth response-1 contributes to steatosis development after acute ethanol administration. Alcoholism: Clinical and Experimental Research. 2012;36(5):759–767. doi: 10.1111/j.1530-0277.2011.01681.x. 22141421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bardag-Gorce F., Li J., French B.A., French S.W. The effect of ethanol-induced CYP2E1 on proteasome activity: the role of 4-hydroxynonenal. Experimental and Molecular Pathology. 2005;78(2):109–115. doi: 10.1016/j.yexmp.2004.10.005. 15713435 [DOI] [PubMed] [Google Scholar]
- 87.Derdak Z., Villegas K.A., Wands J.R. Early growth response-1 transcription factor promotes hepatic fibrosis and steatosis in long-term ethanol-fed Long-Evans rats. Liver International. 2012;32:761–770. doi: 10.1111/j.1478-3231.2012.02752.x. 22292946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McMullen M.R., Pritchard M.T., Wang Q., Millward C.A., Croniger C.M., Nagy L.E. Early growth response-1 transcription factor is essential for ethanol-induced fatty liver injury in mice. Gastroenterology. 2005;128(7):2066–2076. doi: 10.1053/j.gastro.2005.02.065. 15940638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pritchard M.T., Nagy L.E. Ethanol-induced liver injury: potential roles for egr-1. Alcoholism: Clinical and Experimental Research. 2005;29(Suppl. 11):S146S–S150S. doi: 10.1097/01.alc.0000189286.81943.51. 16344600 [DOI] [PubMed] [Google Scholar]
- 90.Yan S.F., Fujita T., Lu J., Okada K., Shan Zou Y., Mackman N., Pinsky D.J., Stern D.M. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nature Medicine. 2000;6(12):1355–1361. doi: 10.1038/82168. 11100120 [DOI] [PubMed] [Google Scholar]
- 91.Smith S.L., Jennett R.B., Sorrell M.F., Tuma D.J. Acetaldehyde substoichiometrically inhibits bovine neurotubulin polymerization. Journal of Clinical Investigation. 1989;84(1):337–341. doi: 10.1172/JCI114159. 2500458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhong Z., Ramshesh V.K., Rehman H., Liu Q., Theruvath T.P., Krishnasamy Y., Lemasters J.J. Acute ethanol causes hepatic mitochondrial depolarization in mice: role of ethanol metabolism. PLoS One. 2014;9(3):e91308. doi: 10.1371/journal.pone.0091308. 24618581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ding W.X. Drinking coffee burns hepatic fat by inducing lipophagy coupled with mitochondrial beta-oxidation. Hepatology. 2014;59(4):1235–1238. doi: 10.1002/hep.26736. 24114874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sinha R.A., Farah B.L., Singh B.K., Siddique M.M., Li Y., Wu Y., Ilkayeva O.R., Gooding J., Ching J., Zhou J., Martinez L., Xie S., Bay B.H., Summers S.A., Newgard C.B., Yen P.M. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59(4):1366–1380. doi: 10.1002/hep.26667. 23929677 [DOI] [PubMed] [Google Scholar]
- 95.Dranoff J.A., Feld J.J., Lavoie E.G., Fausther M. How does coffee prevent liver fibrosis? Biological plausibility for recent epidemiological observations. Hepatology. 2014;60(2):464–467. doi: 10.1002/hep.27032. 24464631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gill K., Menez J.F., Lucas D., Deitrich R.A. Enzymatic production of acetaldehyde from ethanol in rat brain tissue. Alcoholism: Clinical and Experimental Research. 1992;16(5):910–915. doi: 10.1111/j.1530-0277.1992.tb01892.x. 1443429 [DOI] [PubMed] [Google Scholar]
- 97.Zimatkin S.M., Deitrich R.A. Ethanol metabolism in the brain. Addiction Biology. 1997;2(4):387–399. doi: 10.1080/13556219772444. [DOI] [PubMed] [Google Scholar]
- 98.Chen G., Ke Z., Xu M., Liao M., Wang X., Qi Y., Zhang T., Frank J.A., Bower K.A., Shi X., Luo J. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8(11):1577–1589. doi: 10.4161/auto.21376. 22874567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pla A., Pascual M., Renau-Piqueras J., Guerri C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain. Cell Death and Disease. 2014;5:e1066. doi: 10.1038/cddis.2014.46. 24556681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joseph R.A., Shepard B.D., Kannarkat G.T., Rutledge T.M., Tuma D.J., Tuma P.L. Microtubule acetylation and stability may explain alcohol-induced alterations in hepatic protein trafficking. Hepatology. 2008;47(5):1745–1753. doi: 10.1002/hep.22014. 18161881 [DOI] [PMC free article] [PubMed] [Google Scholar]