

Abstract
Human exposure to particulate matter (PM) is a global environmental health concern. Zinc (Zn2+) is a ubiquitous respiratory toxicant that has been associated with PM health effects. However, the molecular mechanism of Zn2+ toxicity is not fully understood. H2O2 and Zn2+ have been shown to mediate signaling leading to adverse cellular responses in the lung and we have previously demonstrated Zn2+ to cause cellular H2O2 production. To determine the role of Zn2+-induced H2O2 production in the human airway epithelial cell response to Zn2+ exposure. BEAS-2B cells expressing the redox-sensitive fluorogenic sensors HyPer (H2O2) or roGFP2 (EGSH) in the cytosol or mitochondria were exposed to 50 µM Zn2+ for 5 min in the presence of 1 µM of the zinc ionophore pyrithione. Intracellular H2O2 levels were modulated using catalase expression either targeted to the cytosol or ectopically to the mitochondria. HO-1 mRNA expression was measured as a downstream marker of response to oxidative stress induced by Zn2+ exposure. Both cytosolic catalase overexpression and ectopic catalase expression in mitochondria were effective in ablating Zn2+-induced elevations in H2O2. Compartment-directed catalase expression blunted Zn2+-induced elevations in cytosolic EGSH and the increased expression of HO-1 mRNA levels. Zn2+ leads to multiple oxidative effects that are exerted through H2O2-dependent and independent mechanisms.
Keywords: Zinc, H2O2, Live cell imaging, Mitochondria, Catalase, Genetically encoded redox probes
Abbreviations: PM, particulate matter; Zn2+, zinc; HAEC, human airway epithelial cells; PTP, protein tyrosine phosphatases; Cyto, cytosolic; Mito, mitochondria; PYRI, sodium pyrithione; HO-1, heme oxygenase 1; ZnP, zinc pyrithione
Graphical abstract
Highlights
-
•
We used targeted catalase expression to examine the role of H2O2 in Zn2+-induced effects.
-
•
Cytosolic or mitochondrial catalase ablated Zn2+-induced mitochondrial H2O2 production.
-
•
Catalase expression blunted Zn2+-induced cytosolic EGSH and HO-1 mRNA.
-
•
Independently, decreasing GSHtotal or increasing EGSH failed to induce HO-1 mRNA.
-
•
Zn2+ causes multiple oxidative effects by H2O2-dependent and independent mechanisms.
Introduction
Human exposure to ambient particulate matter (PM) is a public health concern of global proportions. Observational studies demonstrate an association between exposure to PM and elevated rates of cardiovascular morbidity and mortality [1], [2], [3], [4], [5]. Despite the association between these adverse health effects and ambient PM levels, the constituents in PM responsible for its toxicity and the underlying mechanisms remain largely unknown. Epidemiological [6] and toxicological [7], [8] studies have specifically implicated the particle-associated transition metal zinc (Zn2+) as a contributor to PM health effects. Although zinc is an essential nutrient and vital to many physiological processes, inhalational exposure to zinc is associated with a number of adverse health outcomes [9].
The health effects of zinc inhalation are modeled by metal fume fever, an occupational disease characterized by a self-limited febrile flu-like condition with airway inflammation resulting from inhalation of ZnO particles generated during welding [10]. The mechanisms responsible for the pathophysiological effects of Zn2+ inhalation have been investigated in cultured human airway epithelial cells (HAECs) by our laboratory [11], [12], [13] and by other groups utilizing diverse in vitro models [14], [15], [16], [17]. Observations from these studies show that Zn2+ induces inflammatory and adaptive gene expression through processes that involve the deregulation of signaling cascades. Specifically, Zn2+ is thought to perturb multiple signaling pathways by direct interaction with thiol groups on key regulatory proteins, including protein tyrosine phosphatases (PTP) [18], [19], [20]. Zn2+ is a known mediator in signaling pathways, including the Keap1/Nrf2/ARE pathway [21], [22].
Unlike other transition metals associated with PM (e.g., Fe, Ni, Cu, V), Zn2+ lacks two adjacent valence states and, therefore, does not support single electron transfers to produce reactive oxygen species (ROS), meaning that ROS generated during Zn2+ exposure are derived from cellular metabolism. Zn2+ interferes with mitochondrial respiration at multiple points [9] and consistent with this, we recently reported that exposure of HAEC to Zn2+ results in increased intracellular generation of H2O2 of mitochondrial origin [23]. Physiologically, H2O2 serves as a second messenger that plays pivotal roles in the reversible inactivation of regulatory proteins, most notably PTP [24], [25], [26], [27]. Thus, there is evidence that toxicological Zn2+ exposure can induce gene expression through signaling mechanisms by direct interaction as well as through the generation of H2O2.
In order to determine the dependence of Zn2+-induced responses on H2O2, the present study expanded our previous live-cell imaging approach to monitor oxidative changes in the cytosol and mitochondria of HAEC exposed to Zn2+[28], [29] . We utilized cytosolic overexpression or ectopic mitochondrial expression of the H2O2 scavenging enzyme catalase in BEAS-2B cells bearing the genetically-encoded fluorogenic ratiometric sensors HyPer or roGFP2, which report on H2O2 and the glutathione redox potential (EGSH), respectively [30], [31], [32]. In this study we examined the link between oxidative events associated with Zn2+ exposure and signaling events, using the level of HO-1 gene expression as a downstream readout of the adaptive response to oxidant xenobiotic exposure [33], [34], [35]. This study reveals that exposure of HAEC to Zn2+ leads to multiple oxidative effects that are exerted through H2O2-dependent and independent mechanisms.
Materials and methods
Reagents
Tissue culture media and supplements were purchased from Lonza (Walkersville, MD, USA). Phenol red-free keratinocyte basal media (KBM) with or without glucose was acquired from Cell Applications, Inc. (San Diego, CA, USA). X-tremeGENE 9 DNA Transfection Reagent was obtained from Roche Applied Science (Indianapolis, IN, USA). Adenoviral vectors were obtained from the Gene Therapy Center Virus Vector Core Facility (University of North Carolina at Chapel Hill, USA). The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA): hydrogen peroxide (H2O2), aldrithiol-2 (A-2), dithiothreitol (DTT), 2-mercaptopyridine N-oxide sodium salt (Pyrithione, PYRI), zinc sulfate (Zn2+), 2-acetylamino-3-[4-(2-acetylamino-2-carboxyethylsufanylthio-carbonylamino) phenylthiocarbamoylsufanyl] propionic acid (2-AAPA), and buthionine sulfoximine (BSO). Basic laboratory supplies were obtained from Fisher Scientific (Raleigh, NC, USA).
Cell culture
SV40 large T antigen-transformed HAEC (BEAS-2B, subclone S6 [36]) were cultured as previously described [29], [37].
Viral transduction
Plasmids for the genetically encoded redox sensors roGFP2 and HyPer were the generous gift of S.J. Remington (University of Oregon, Eugene, OR, USA) and purchased from Evrogen (Axxora, Farmingdale, NY, USA), respectively. Cytosolic (Cyto) and mitochondrial (Mito) targeted versions of both plasmids were introduced into lentiviral vectors as described previously [28]. Human catalase targeted to the mitochondrial inter-membrane space was introduced into a lentiviral vector. Stable expression of Mito-catalase, Cyto-roGFP2, Mito-roGFP2, Cyto-HyPer, and Mito-HyPer was achieved using lentiviral transduction as previously described [29]. Mitochondrial localization of Mito-HyPer was verified by co-localizing its fluorescence with that of MitoTracker® Red CMXRos (Invitrogen, Grand Island, NY, USA) (Supplemental Fig. S1). The subcellular localization of Mito-roGFP2 has been validated previously [23]. Lentiviral mitochondrial fluorogenic sensor transduction efficiency was approximately 70%, while that of the cytosolic forms of the sensors was approximately 40% efficient, requiring enrichment by cell sorting, conducted by the UNC Flow Cytometry Core Facility utilizing an iCyt Reflection maintained under sterile conditions. Mitochondrial localization of catalase was confirmed immunocytochemically using an anti-catalase antibody (Santa Cruz Biotechnology, Dallas, TX, USA) and by immunoblot analysis with anti-GAPDH (Santa Cruz Biotechnology) and anti-Cytochrome C (Cell Signaling, Beverly, MA) antibodies as cytosolic- and mitochondrial-specific controls. Briefly, for immunoblot analysis mitochondria were isolated in a sucrose solution (250 mM sucrose, 15 mM NaCl) followed by centrifugations at 500g for 5 min and 20,000g for 10 min; the pellet containing the mitochondrial fraction was resuspended in RIPA (1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl, 150 mM Tris–HCl, pH 8.0) for immunoblotting. For cytosolic catalase overexpression, cells were transduced with an adenoviral vector encoding human catalase driven by a CMV promoter at a MOI of 500 for 4 h, followed by a 2-day incubation in KGM [38]. An empty vector driven by CMV was used as an experimental control. The pH-specific fluorogenic sensor pHred, created by the laboratory of Yellen [39], was obtained as a construct through Addgene (Cambridge, MA, USA) for expression into BEAS-2B cells via transient transfection of 1–2 µg plasmid DNA using the suggested X-tremeGENE 9 protocol.
Live cell imaging
Immediately before exposure, roGFP2 or HyPer expressing cells, were placed in KBM without phenol red and analyzed using a Nikon Eclipse C1si spectral confocal imaging system and 404, 488, and 561 nm primary laser lines (Nikon Instruments Corporation, Melville, NY, USA [29]). Sequential scans of each laser line were performed at a frequency of 60 s with at least 10 cells expressing the biosensor in the field of view, with results calculated as a ratio of the respective 525/30 nm emission for the 404 and 488 nm excitation of each sensor. Baseline data points were collected 10 min prior to the addition of 50 µM Zn2+ and 1 µM PYRI. To normalize for variability in the dynamic range of the sensors expressed in individual cells, HyPer expressing cells were treated with 50 µM H2O2 for 5 min followed by treatment with 1 mM H2O2 for 3 min and finally with DTT for 3 min, while roGFP2 expressing cells were treated with 1 mM H2O2 for 5 min followed by addition of 100 µM A-2 for 3 min and finally with DTT for 3 min to quantify the span of sensor responsiveness. Data were expressed normalized to the maximum sensor response (i.e., the maximum response elicited by H2O2) recorded during the experiment set as 100%, with the average starting baseline response set as 0%. Cell viability was determined using retention of intracellular Calcein-AM (Molecular Probes, Eugene, OR, USA).
Gene expression analysis
BEAS-2B cells with or without cytosolic catalase overexpression or ectopic expression of mitochondrial catalase were exposed to 0–50 µM Zn2+/1 µM PYRI for 5 min in KBM, washed and incubated at 37 °C, 5% CO2, 100% humidity for 2 h in KBM. Relative gene expression was quantified using the real-time PCR, ABI Prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) as previously described [28]. GAPDH mRNA was used for normalization. Oligonucleotide primer pairs and fluorescent probes for HO-1 were as follows: (5′CAGCAACAAAGTGCAAGATTCTG3′, 3′ACTGTAAGGACCCATCGGAGAAG5′); and for GAPDH: (5′GAAGGTGAAGGTCGGAGTC3′, 3′GAAGATGGTGATGGGATTTC5′). Oligonucleotides were designed using a primer design program (Primer Express, Applied Biosystems) and obtained from Integrated DNA Technologies (Coralville, IA). The catalase primer/probe set was a TaqMan Gene Expression Assay (Hs00156308_m1) obtained from Applied Biosystems.
Statistical analysis
All imaging data were quantified using NIS-Elements AR software (Nikon). Data are expressed as the mean±SEM of at least three separate experiments. Determination of statistical significance (p<0.05) of all imaging data were made using linear regression, comparing the slopes of linear portion of the time-course plots. Comparison of gene expression were conducted using Student's t-test with Bonferroni correction for multiple group comparisons. ANCOVA analysis was utilized when appropriate to test for multiple interactions, with p<0.05 considered statistically significant. PRISM (GraphPad Software, La Jolla, CA, USA) and R (R Core Team, Vienna, Austria) software packages were used for statistical tests.
Results
Catalase expression ablates Zn2+-induced H2O2 generation
The Zn2+-specific ionophore PYRI used at 1 µM, was found to reduce intercellular variability in the response of BEAS-2B cells to Zn2+ exposure [23] and was, therefore, included in all zinc exposure experiments in this study. Exposure to 1–100 µM Zn2+, 1 µM PYRI (ZnP) did not result in overt cytotoxicity in BEAS-2B cells over the time periods used in this study, as assessed by the cellular release of calcein-AM (data not shown). Exposure to 50 µM ZnP rapidly increased intracellular H2O2 levels in both the cytosol and the mitochondria, as reported by HyPer fluorescence in each of these cellular compartments (Fig. 1, Appendix A). In contrast PYRI alone did not induce a change in H2O2 (Fig. S2). The fluorescence emission intensity of the HyPer fluorophore is known to be affected by pH of its surroundings. Therefore, we monitored pH changes induced in cells exposed to ZnP using pHred [39]. As shown in Fig. 2, pH remained unchanged over the 10 min post ZnP exposure period during which the HyPer signals were monitored.
Fig. 1.
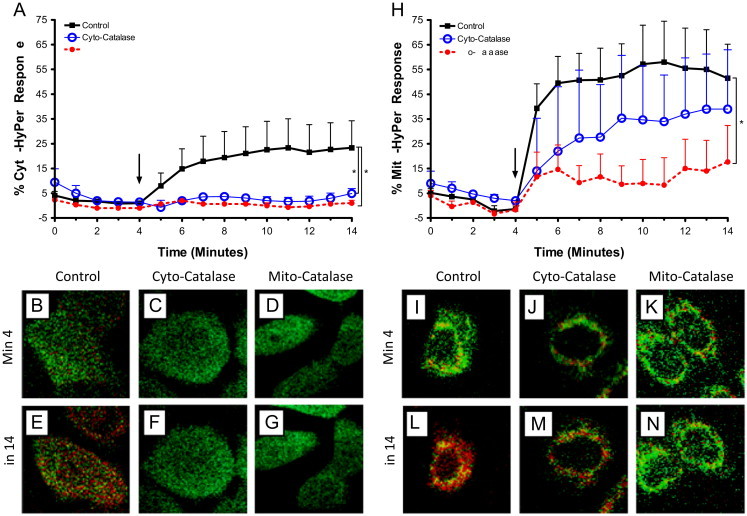
Targeted catalase expression abrogates Zn2+-induced cytosolic and mitochondrial increases in H2O2 levels in BEAS cells. BEAS-2B cells expressing HyPer in the cytosol (A–G) or mitochondria (H–N), and either over expressing catalase (Cyto-catalse) or targeted-ectopic catalase expression in the mitochondria (Mito-catalase) were exposed to 50 µM Zn2+/1 µM PYRI at the indicated time (arrow). Ratiometric fluorescence values normalized for sensor response utilizing the baseline and maximal response H2O2 as the minimum and maximum responses, respectively, in either the cytosol (A) or mitochondria (H) are shown. Values are presented as mean±SE (n≥3, where n consists of an average of 10 distinct cells' responses), ⁎ indicates statistical significant change compared to control by linear regression. Representative cells expressing basal catalase levels (B, E, I, L), cyto-catalase (C, F, J, M), or Mito-catalase (D, G, K, N) shown at baseline (min 4; B−D, I−K) and following Zn2+ stimulation (min 14; E–G, L–N). Fluorescence intensity color is shown as a function of increasing H2O2 levels: low H2O2 high H2O2.
high H2O2.
Fig. 2.
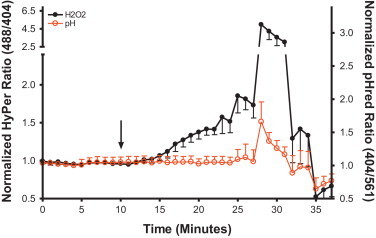
Zn2+-induced elevation in H2O2 reported by HyPer are not mediated by changes in pH. BEAS-2B cells co-expressing HyPer and the pH sensor pHred in the cytosol were exposed to 50 µM Zn2+/1 µM PYRI at the indicated time (arrow). Shown are ratiometric values calculated from the fluorescent 525/30 emission obtained through excitation of the 404, 488, and 561 nm laser excitation normalized to the baseline. HyPer sensor responsiveness was assessed using 50 µM H2O2 (min 25) and 5 mM DTT (min 35). pHred sensor response was verified by addition of 10 mM acetic acid (min 28) followed by addition of 10 mM ammonium chloride (min 33). Values are presented as mean±SE (n=3, where each n consists of an average of 10 distinct cells' responses).
We next tested the effectiveness of catalase expression as an interventional approach to suppress Zn2+-induced increases in H2O2 in a cell compartment-specific manner. Expression of adenoviral mediated expression of Cyto-catalase and lentiviral encoded expression of Mito-catalase resulted in similar increase levels of catalase mRNA compared to controls (Fig. 3A and B). Interestingly, expression of Mito-catalase was potentiated by exposure to 10–50 µM ZnP (Fig. 3B), possibly reflecting Zn2+-induced activation of the Sp1 transcription factor as reported previously in retinal pigment epithelial cells [40]. CMV-driven overexpression of catalase in the cytosol effectively prevented the elevation in cytosolic H2O2 concentrations induced by exposure to ZnP (Fig. 1A). Immunocytochemical analyses showed that the expression of catalase targeted to the mitochondria intermembrane space successfully delivered immunoreactive catalase to the mitochondria (Fig. 3D–K). Immunoblotting of cytosolic and mitochondrial fractions confirmed the presence of catalase in the mitochondria of Mito-catalase expressing BEAS cells but not in the mitochondria of control cells (Fig. 3C). The slight elevation in catalase found in the cytosol of cells transduced with Mito-catalase lentivirus presumably represents nascent catalase that has yet to reach the mitochondrial intermembrane space. Similar to the effect of overexpressing cytosolic catalase, cells bearing ectopic mitochondrial expression of catalase showed a marked reduction in Zn2+-induced increases in cytosolic H2O2 (Fig. 1A).
Fig. 3.
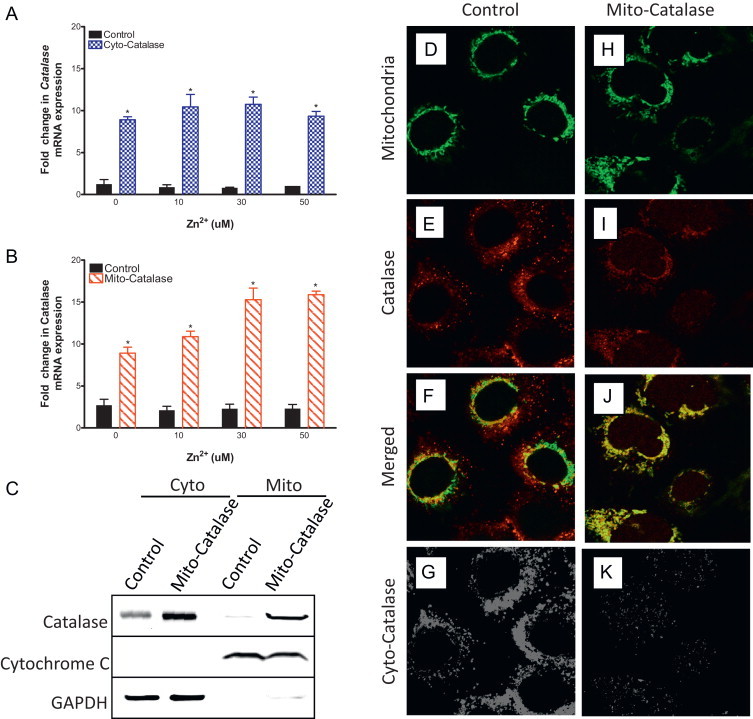
Ectopic expression of catalase in the mitochondria of BEAS-2B cells. BEAS overexpressing catalase (Cyto-catalase, A) or expressing ectopic mitochondrial catalase (Mito-catalase, B) were exposed to the indicated zinc concentrations and 1 µM PYRI for 5 min, washed, and then incubated for 2 h in fresh media before mRNA extraction. mRNA levels measured using TaqMan-basedRT-PCR were normalized to levels of GAPDH mRNA and expressed as fold increases over basal mRNA expression in cells not exposed to media control. Values are presented as mean±SE (n=3), ⁎ indicates p<0.05 as determined by Student's t-test compared to control. Mito-catalase expressing BEAS-2B cells were fractionated between cytosol (cyto) and mitochondria (mito) and immunoblotted against catalase, cytochrome C-a mitochondrial specific protein, and GAPDH—a cytosol specific protein (C). BEAS-2B cells expressing mitochondrial roGFP2 (D–K) with expression of the human catalase gene targeted to the mitochondria by a CMV promoter via lentiviral transduction (H–K) were fixed and immunostained using anti-catalase antibodies. Cells were stained with catalase primary antibody (E, I) and merged with the mitochondrial marker for colocalization analysis (F, J). For presence of cytosolic catalase localization analysis (G, K), the fluorescent intensity values across the images were converted to a binary value followed by subtraction of the mitochondria channel from the catalase channel leaving only catalase signal outside the mitochondria. Results shown are representative of three separate experiments.
Experiments in which the concentration of H2O2 in the mitochondria was monitored using HyPer targeted to the intermembrane space demonstrated that the presence of mitochondrial catalase also blunted Zn2+-induced increases in mitochondrial H2O2. In contrast, an excess of cytosolic catalase had only a minimal effect on Zn2+-induced elevations in mitochondrial H2O2 (Fig. 1H). Taken together, these findings confirm mitochondria as the principal source of Zn2+-induced increases in H2O2 and validated the use of targeted catalase expression as an efficient experimental intervention to suppress compartment-specific concentrations of H2O2 in cellular responses to Zn2+ exposure.
H2O2 contributes to Zn2+-induced oxidant stress
Next, we examined changes in EGSH as an objective measure of the oxidant stress presented to the cell by exposure to ZnP. Following the same approach used to monitor H2O2 with HyPer, cytosolic- and mitochondria-targeted versions of the EGSH-sensing fluorophore roGFP2 were stably expressed in BEAS-2B cells. Exposure to 50 µM ZnP induced a rapid rise in the cytosolic EGSH, which could be reduced substantially through overexpression of catalase in either the cytosol or the mitochondria (Fig. 4A). However, neither cytosolic nor mitochondrial catalase intervention affected the Zn2+-induced EGSH increase in the mitochondria (Fig. 4H). We also examined the possibility that roGFP2 fluorescence changes might arise from a direct interaction of Zn2+ with the sensor itself by using 2-AAPA to inhibit glutaredoxin, the enzyme through which roGFP2 senses EGSH changes [29]. Pretreatment of roGFP2-expressing BEAS-2B with 2-AAPA ablated Zn2+-induced increases in EGSH (Fig. 5) establishing that Zn2+-induced changes are not the result of a direct interaction of Zn2+ with roGFP2, since any Zn2+-direct effect on roGFP2 would not require glutaredoxin.
Fig. 4.
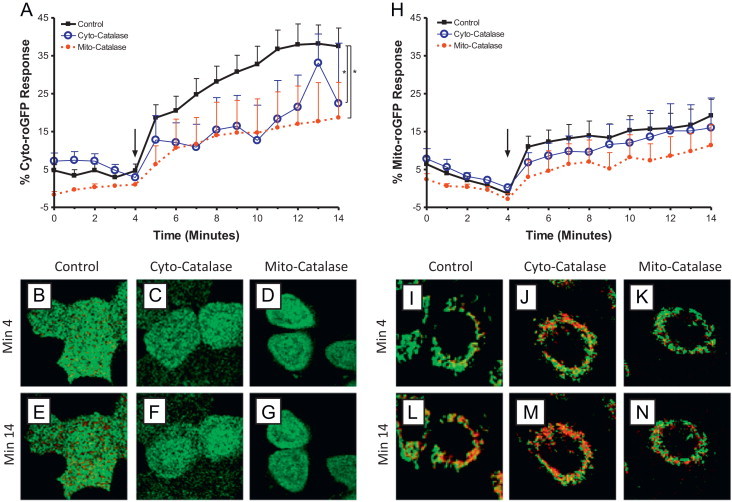
H2O2 mediates Zn2+-induced elevation in cytosolic but not mitochondrial glutathione redox potential. BEAS-2B cells expressing roGFP2 in the cytosol (A–G) or mitochondria (H–N), and either over expressing catalase (Cyto-catalse) or targeted-ectopic catalase expression in the mitochondria (Mito-catalase) were exposed to 50 µM Zn2+/1 µM PYRI at the indicated time (arrow). Ratiometric fluorescence values normalized as for sensor response utilizing the baseline and stimulus to 1 mM H2O2 as the minimum and maximum responses, respectively, in either the cytosol (A) or mitochondria (H) are shown. Values are presented as mean±SE (n≥3, where n consists of an average of 10 distinct cells' responses), ⁎ indicates statistical significant change compared to control by linear regression. Representative cells expressing basal catalase levels (B, E, I, L), Cyto-catalase (C, F, J, M), or Mito-catalase (D, G, K, N) shown at baseline (min 4; B–D, I–K) and following Zn2+ stimulation (min 14; E–G, L–N). Fluorescence intensity color is shown as a function of increasing EGSH, also interpreted as an increase in GSSG to GSH: lowEGSH highEGSH.
highEGSH.
Fig. 5.
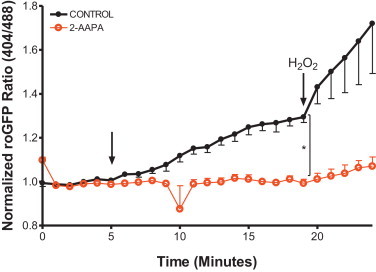
Zn2+ -induced changes in glutathione redox potential are properly reported by roGFP2. BEAS-2B cells stably expressing Cyto-roGFP2 were incubated with either 100 µM 2-AAPA, a glutaredoxin inhibitor, or DMSO as a vehicle control (CONTROL) for 2 h. Cells were exposed to 50 µM Zn2+/1 µM PYRI after 5 min of baseline measurements. Shown are ratiometric values (404/488) calculated from the fluorescent 525/30 emission of the 404 and 488 nm laser excitation normalized to baseline. To validate sensor response, cells were exposed to 1 mM H2O2 (min 20). Values are presented as mean±SE (n=4, where n consists of an average of 10 distinct cells' responses), ⁎ indicates statistical significant change compared to control by linear regression.
EGSH changes do not mediate HO-1 gene expression
In order to link the Zn2+-induced oxidative stress observed in the live cell imaging experiments to a downstream cellular response, we measured the level of expression of the HO-1 gene in BEAS-2B cells exposed to ZnP. In order to avoid potential cytotoxicity associated with exposure to Zn2+ for the prolonged periods required to observe changes in mRNA levels, BEAS-2B cells were exposed to ZnP for 5 min, a time point that coincides with the plateau of the observed Zn2+-induced H2O2 and EGSH increases (Fig. 1, Fig. 4). This short exposure to ZnP was followed by washing and a 2 h incubation in culture medium without added Zn2+ or PYRI. As seen in Fig. 6 (black bars), this “pulse” exposure to 0–10 µM ZnP resulted in concentration-dependent increases in HO-1 gene expression that were up to 45-fold over control levels exposed to 1 µM PYRI alone.
Fig. 6.
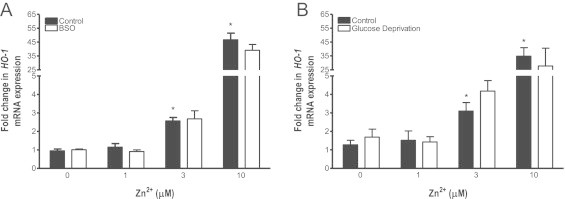
Depletion of glutathione levels does not alter Zn2+-induced HO-1 mRNA levels. BEAS-2B cells were incubated with 500 µM BSO for 24 h (A) or glucose deprived for 2 h (B) before exposure to the indicated Zn2+ concentrations and 1 µM PYRI for 5 min followed by 2 h in media alone. HO-1 mRNA levels were normalized to levels of GAPDH mRNA and expressed as fold increases over basal mRNA expression in control cells. Values are presented as mean±SE (n=3), * indicates p<0.05 as determined by ANCOVA compared to 0 µM Zn2+ exposure.
To examine the role of EGSH in mediating Zn2+ induced HO-1 expression, we next sought to sensitize the HAEC to ZnP exposure by decreasing the total glutathione content in the cells with the gamma-glutamyl synthetase inhibitor BSO. Treatment of BEAS-2B cells with 500 µM BSO alone for a 24-h period reduced the total glutathione pool by 80% and concomitantly increased GSSG by 40% (data not shown). Treatment of BEAS-2B cells with BSO alone produced only a small 1.61±0.06-fold increase in baseline HO-1 gene expression (Fig. S3), and had no effect on the subsequent HO-1 response to ZnP exposure (p=0.07, F=3.6, Fig. 6A).
Since impairing glutathione synthesis was relatively ineffective at inducing HO-1, we next determined the effect of decreasing intracellular stores of NADPH through glucose deprivation alone and followed by ZnP -exposure on HO-1 expression. A 2 h incubation in glucose-free media did not alter the total glutathione pool or change the GSH to GSSG ratio (data not shown) and induced a modest 3.08±1.29 fold increase in HO-1 expression (Fig. S3), but failed to alter HO-1 expression induced by Zn2+ (p=0.24, F=1.5, Fig. 6B).
H2O2 mediates Zn2+-induced adaptive gene expression
The role of H2O2 in Zn2+-induced HO-1 response was examined by utilizing the compartment-specific catalase expression strategy employed in the imaging studies. Catalase overexpression in the cytosol blunted Zn2+-induced HO-1 gene expression across the range of ZnP exposures tested (10–50 µM) by approximately 50% (p=0.04, F=4.5, Fig. 7A). Similarly, ectopic catalase expression in the mitochondria halved the HO-1 expression induced by the higher (30 and 50 µM) ZnP exposures (p=0.002, F=10.5, Fig. 7B)
Fig. 7.
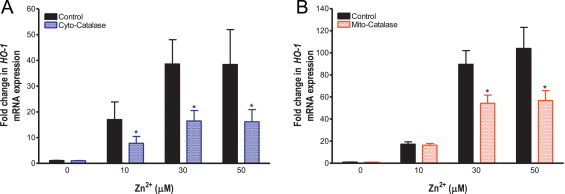
Targeted catalase expression blunts Zn2+-induced HO-1 mRNA levels in BEAS cells. BEAS-2B cells overexpressing catalase (A) or expressing ectopic mitochondrial catalase (B) were exposed to the indicated Zn2+ concentrations and 1 µM PYRI for 5 min followed by 2 h in media alone. HO-1 mRNA levels were normalized to levels of GAPDH mRNA and expressed as fold increases over basal mRNA expression in control cells. Values are presented as mean±SE (n=3), * indicates p<0.05 as determined by ANCOVA compared to control.
Discussion
The findings of this study add to previous work demonstrating the pathophysiological effects of Zn2+ exposure on HAEC [14], [16], [17], [41], [42]. Zn2+ can activate signaling through direct interaction with critical cysteine residues in regulatory proteins such as Keap1 [22] or PTP [18]. However, as described in our previous study [23] Zn2+ also acts as an oxidant stressor by inducing mitochondrial production of H2O2, a ROS implicated in multiple physiological signaling processes, including the induction of Keap1 and the redox regulation of PTP [43], [44], [45]. The present study investigated the role of H2O2 as a mediator of Zn2+-induced oxidant responses. We show that a brief “pulse” exposure to Zn2+ is sufficient to commit cells to activate signaling mechanisms which lead to the induction of adaptive gene expression. Using compartment-specific expression of catalase as an interventional approach we show that Zn2+ acts through both H2O2-dependent and -independent mechanisms to induce HO-1 expression and increases in cytosolic EGSH.
As recently reviewed [9], a number of studies demonstrate that Zn2+ exposure induces mitochondrial dysfunction through multiple mechanisms, including impairment of mitochondrial respiration through inhibition of the α-ketoglutarate dehydrogenase complex [46] and loss of mitochondrial membrane potential [47]. Of specific interest is the inhibition of cytochrome c oxidase activity, which can result in blocked electron transport and production of superoxide from complexes I and III [48]. In the present study, imaging-based experiments to monitor H2O2 levels and EGSH in real time, combined with a validated strategy in the form of catalase overexpression, provide a compartment-specific perspective on the oxidative effects of Zn2+ exposure. These data add to the weight of evidence supporting the notion that Zn2+ exposure impairs mitochondrial metabolic processes leading to increased production of H2O2 and changes in EGSH in HAEC.
Our observation that H2O2 scavenging with either cytosolic or mitochondrial catalase blunted Zn2+-induced cytosolic EGSH changes shows a H2O2-dependent effect of Zn2+ on the cytosolic glutathione pool. It is also noteworthy that Zn2+ is a known inhibitor of glutathione reductase [49], an effect that could potentially contribute to an accumulation of oxidized glutathione and increase EGSH. The fact that increasing the EGSH with the gamma-glutamyl synthetase inhibitor BSO does not result in significant elevations in HO-1 mRNA levels argues that the peroxide-dependent effect of Zn2+ on EGSH does not carry through to the activation of signaling pathways that regulate HO-1 expression. This interpretation is consistent with the fact that manipulation of EGSH with BSO failed to induce significant HO-1 expression. In this regard, it is interesting that a 2 h glucose deprivation, which was previously demonstrated to potentiate EGSH increases induced by ozone exposure in BEAS-2B cells [29], was ineffective in promoting HO-1 expression by itself. Thus, neither BSO pretreatment nor glucose deprivation potentiated Zn2+-induced HO-1 expression. Taken together, these findings argue that Zn2+-induced increase in EGSH is not sufficient to activate signaling leading to HO-1 expression (Fig. 8). This agrees with the previous proposed view that a change in EGSH should not be considered as an effector but rather as an indicator of intracellular redox metabolism [50]. On the other hand, the inability of catalase expression to ablate Zn2+-induced changes in EGSH completely, suggests the presence of a H2O2-independent mechanism that may involve direct Zn2+ reactivity with unknown protein thiol targets.
Fig. 8.
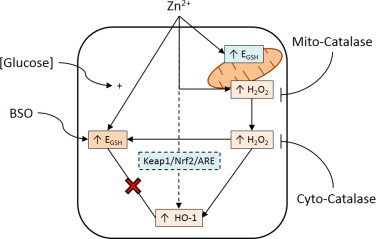
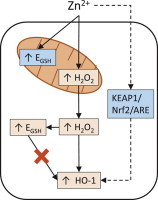
H2O2 mediates Zn2+-induced increases in cytosolic EGSH and HO-1 mRNA expression. Zn2+ exposure induces mitochondrial H2O2 production and elevates EGSH in both the cytosol and mitochondria. Cytosolic catalase overexpression (Cyto-catalase) ablates H2O2 levels in the cytosol, and reduces cytosolic EGSH and HO-1 mRNA expression. Ectopic catalase expression in mitochondria (Mito-catalase) ablates H2O2 levels in the mitochondria and cytosol as well as reduces cytosolic EGSH and HO-1 mRNA expression. Increased EGSH does not induce HO-1 mRNA expression as determined through BSO and glucose deprivation ([Glucose]) treatments. Dashed line indicates hypothesized mechanism accounting for H2O2-independent Zn2+-induced HO-1 mRNA expression.
The observation that HO-1 expression was only partially ablated by increased catalase expression in either the cytosol or mitochondria implies the presence of additional signaling mechanisms leading to Zn2+-induced adaptive responses. It is well established that the Keap1/Nrf2/ARE signaling pathway regulates HO-1 expression and, therefore, it is plausible that signaling intermediates along this pathway are involved in the response to Zn 2+[51], [52], [53]. One possibility is an effect on the nuclear transcription factor repressor Bach1, which is targeted for degradation in response to treatment with Zn-mesoporphyrin [54]. In this regard, the notion that the combination of Zn2+ and PYRI used in this study might be a structural mimic of Zn-mesoporphyrin is intriguing. However, utilizing the exposure conditions of the present study, experiments in our laboratory show ZnP does not affect Bach1 protein levels in HAEC (Speen, unpublished observations). Another possibility is presented by the presence of an active Zn2+-binding or broadly “metal(loid)”-specific sensor in Keap1 [22]. Preliminary studies in our laboratory using a version of Keap1 in which the amino acids that constitute the Zn2+ sensor have been substituted, suggest that direct Zn2+ binding of Keap1 is in fact a contributing mechanism leading to HO-1 expression in Zn2+-exposed BEAS-2B cells (Silbajoris, unpublished). Additional studies will be needed in order to further characterize H2O2-independent pathways leading to adaptive gene expression in HAEC exposed to Zn2+.
The data presented in this study demonstrate that H2O2 is a critical mediator of the oxidative effects induced by exposure to Zn2+ as schematized in Fig. 8, while also revealing the presence of multiple contributing pathways leading to adaptive responses by HAEC. These findings are relevant to understanding the mechanistic basis and potential mitigation of the adverse health effects of Zn2+ inhalation.
Acknowledgments
PAW was supported as a predoctoral candidate in part by NIEHS Toxicology Training Grant, T32 ES007126 and UNC-EPA Training Agreement, CR-83515201-0. The authors are grateful to Dr. A. Rappold for statistical consultation.
Footnotes
Disclaimer: The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. EPA, and approved for publication. The contents of this article should not be construed to represent Agency policy nor does mention of trade names or commercial products constitute endorsement or recommendation for use.The authors declare they have no actual or potential competing financial interests.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2014.10.005.
Appendix A. Supplementary material
Mito-HyPer is specifically localized in the mitochondria of BEAS cells. BEAS-2B cells transduced by lentivirus with mitochondrially-targeted HyPer (A) were colocalized with the mitochondria marker, MitoTracker Red (B). The two channels were merged, where the presence of yellow indicates colocalization of the HyPer construct and the mitochondria (C).
Exposure to Pyrithione alone does not induce intracellular H2O2 or EGSH responses. BEAS-2B cells stably expressing cytosolic (A) or roGFP2 (B) were exposed to vehicle (○), 1 µM PYRI (PYRI, ▲), or 5 µM Zn2+/1 µM PYRI (PYRI/Zn, ■) at the indicated time (arrow). Shown are ratiometric values calculated from the fluorescent 525/30 emission of the 404 and 488 nm laser excitation normalized as a percent of the maximum sensor response. Values are presented as mean±SE (n=3, where n consists of an average of 10 distinct cells' responses), * indicates statistical significant change compared to control by linear regression.
Depletion of glutathione levels does not lead to HO-1 mRNA expression in BEAS cells. BEAS-2B cells were incubated with 500 µM BSO for 24 h or starved of glucose for 2 h before, washed, and then incubated for 2 h in fresh media before mRNA extraction. BSO only imposed a 1.61±0.06(SEM) fold increase over untreated cells. Glucose deprivation induced a 3.08±1.29(SEM) fold increase in HO-1 gene expression. mRNA levels measured using TaqMan-based RT-PCR were normalized to levels of GAPDH mRNA and expressed as fold increases over basal mRNA expression.
References
- 1.Chahine T. Particulate air pollution, oxidative stress genes, and heart rate variability in an elderly cohort. Environmental Health Perspectives. 2007;115(11):1617–1622. doi: 10.1289/ehp.10318. 18007994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dockery D.W. Effect of air pollution control on mortality and hospital admissions in Ireland. Research Report (Health Effects Institute) 2013;176(176):3–109. 24024358 [PubMed] [Google Scholar]
- 3.Feng J., Yang W. Effects of particulate air pollution on cardiovascular health: a population health risk assessment. PloS One. 2012;7(3):e33385. doi: 10.1371/journal.pone.0033385. 22432017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Link M.S. Acute exposure to air pollution triggers atrial fibrillation. Journal of the American College of Cardiology. 2013;62(9):816–825. doi: 10.1016/j.jacc.2013.05.043. 23770178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pope C.A., 3rd Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation. 2006;114(23):2443–2448. doi: 10.1161/CIRCULATIONAHA.106.636977. 17101851 [DOI] [PubMed] [Google Scholar]
- 6.Claiborn C.S., Larson T., Sheppard L. Testing the metals hypothesis in Spokane, Washington. Environmental Health Perspectives. 2002;110(Suppl. 4):S547–S552. doi: 10.1289/ehp.02110s4547. 12194884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adamson I.Y. Zinc is the toxic factor in the lung response to an atmospheric particulate sample. Toxicology and Applied Pharmacology. 2000;166(2):111–119. doi: 10.1006/taap.2000.8955. 10896852 [DOI] [PubMed] [Google Scholar]
- 8.Xia T. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano. 2008;2(10):2121–2134. doi: 10.1021/nn800511k. 19206459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu W., Bromberg P.A., Samet J.M. Zinc ions as effectors of environmental oxidative lung injury. Free Radical Biology and Medicine. 2013;65:57–69. doi: 10.1016/j.freeradbiomed.2013.05.048. 23747928 [DOI] [PubMed] [Google Scholar]
- 10.El Safty A. Zinc toxicity among galvanization workers in the iron and steel industry. Annals of the New York Academy of Sciences. 2008;1140:256–262. doi: 10.1196/annals.1454.007. 18991923 [DOI] [PubMed] [Google Scholar]
- 11.Kim Y.M. Zn2+-induced IL-8 expression involves AP-1, JNK, and ERK activities in human airway epithelial cells. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2006;290(5):L1028–L1035. doi: 10.1152/ajplung.00479.2005. 16373669 [DOI] [PubMed] [Google Scholar]
- 12.Wu W. p38 and EGF receptor kinase-mediated activation of the phosphatidylinositol 3-kinase/Akt pathway is required for Zn2+-induced cyclooxygenase-2 expression. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2005;289(5):L883–L889. doi: 10.1152/ajplung.00197.2005. 15980035 [DOI] [PubMed] [Google Scholar]
- 13.Wu W. Regulation of cyclooxygenase-2 expression by cAMP response element and mRNA stability in a human airway epithelial cell line exposed to zinc. Toxicology and Applied Pharmacology. 2008;231(2):260–266. doi: 10.1016/j.taap.2008.04.012. 18513776 [DOI] [PubMed] [Google Scholar]
- 14.Gazaryan I.G. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. Journal of Biological Chemistry. 2007;282(33):24373–24380. doi: 10.1074/jbc.M611376200. 17565998 [DOI] [PubMed] [Google Scholar]
- 15.Guo D. Reactive oxygen species-induced cytotoxic effects of zinc oxide nanoparticles in rat retinal ganglion cells. Toxicology in Vitro: an International Journal Published in Association with BIBRA. 2013;27(2):731–738. doi: 10.1016/j.tiv.2012.12.001. 23232460 [DOI] [PubMed] [Google Scholar]
- 16.Rudolf E., Rudolf K., Cervinka M. Zinc induced apoptosis in HEP-2 cancer cells: The role of oxidative stress and mitochondria. BioFactors (Oxford, England) 2005;23(2):107–120. doi: 10.1002/biof.5520230206. 16179752 [DOI] [PubMed] [Google Scholar]
- 17.Xie Y. Aerosolized ZnO nanoparticles induce toxicity in alveolar type II epithelial cells at the air–liquid interface. Toxicological Sciences: an Official Journal of the Society of Toxicology. 2012;125(2):450–461. doi: 10.1093/toxsci/kfr251. 21964423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellomo E. Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics. 2014;6(7):1229–1239. doi: 10.1039/c4mt00086b. 24793162 [DOI] [PubMed] [Google Scholar]
- 19.Samet J.M. Tyrosine phosphatases as targets in metal-induced signaling in human airway epithelial cells. American Journal of Respiratory Cell and Molecular Biology. 1999;21(3):357–364. doi: 10.1165/ajrcmb.21.3.3656. 10460753 [DOI] [PubMed] [Google Scholar]
- 20.Tal T.L. Inhibition of protein tyrosine phosphatase activity mediates epidermal growth factor receptor signaling in human airway epithelial cells exposed to Zn2+ Toxicology and Applied Pharmacology. 2006;214(1):16–23. doi: 10.1016/j.taap.2005.11.011. 16410015 [DOI] [PubMed] [Google Scholar]
- 21.Dinkova-Kostova A.T., Holtzclaw W.D., Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry. 2005;44(18):6889–6899. doi: 10.1021/bi047434h. 15865434 [DOI] [PubMed] [Google Scholar]
- 22.McMahon M. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(44):18838–18843. doi: 10.1073/pnas.1007387107. 20956331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng W.Y. An integrated imaging approach to the study of oxidative stress generation by mitochondrial dysfunction in living cells. Environmental Health Perspectives. 2010;118(7):902–908. doi: 10.1289/ehp.0901811. 20413366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burgoyne J.R. Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system. Antioxidants and Redox Signaling. 2013;18(9):1042–1052. doi: 10.1089/ars.2012.4817. 22867279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giorgio M. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nature Reviews: Molecular Cell Biology. 2007;8(9):722–728. doi: 10.1038/nrm2240. 17700625 [DOI] [PubMed] [Google Scholar]
- 26.Rhee S.G. Cellular regulation by hydrogen peroxide. Journal of the American Society of Nephrology. 2003;14(8 Suppl. 3):S211–S215. doi: 10.1097/01.asn.0000077404.45564.7e. 12874433 [DOI] [PubMed] [Google Scholar]
- 27.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. Journal of Biological Chemistry. 2014;289(13):8735–8741. doi: 10.1074/jbc.R113.544635. 24515117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng W.Y. Linking oxidative events to inflammatory and adaptive gene expression induced by exposure to an organic particulate matter component. Environmental Health Perspectives. 2012;120(2):267–274. doi: 10.1289/ehp.1104055. 21997482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibbs-Flournoy E.A. Monitoring intracellular redox changes in ozone-exposed airway epithelial cells. Environmental Health Perspectives. 2013;121(3):312–317. doi: 10.1289/ehp.1206039. 23249900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belousov V.V. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3(4):281–286. doi: 10.1038/nmeth866. 16554833 [DOI] [PubMed] [Google Scholar]
- 31.Jiang K. Expression and characterization of a redox-sensing green fluorescent protein (reduction-oxidation-sensitive green fluorescent protein) in Arabidopsis. Plant Physiology. 2006;141(2):397–403. doi: 10.1104/pp.106.078246. 16760494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer A.J., Dick T.P. Fluorescent protein-based redox probes. Antioxidants and Redox Signaling. 2010;13(5):621–650. doi: 10.1089/ars.2009.2948. 20088706 [DOI] [PubMed] [Google Scholar]
- 33.Smith A.F., Loo G. Upregulation of haeme oxygenase-1 by zinc in HCT-116 cells. Free Radical Research. 2012;46(9):1099–1107. doi: 10.3109/10715762.2012.690872. 22564156 [DOI] [PubMed] [Google Scholar]
- 34.Wu M.L., Layne M.D., Yet S.F. Heme oxygenase-1 in environmental toxin-induced lung disease. Toxicology Mechanisms and Methods. 2012;22(5):323–329. doi: 10.3109/15376516.2012.666685. 22394342 [DOI] [PubMed] [Google Scholar]
- 35.Xue J. Zinc at sub-cytotoxic concentrations induces heme oxygenase-1 expression in human cancer cells. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2013;32(1):100–110. doi: 10.1159/000350128. 23868099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddel R.R. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Research. 1988;48(7):1904–1909. 2450641 [PubMed] [Google Scholar]
- 37.Tal T.L. Differential transcriptional regulation of IL-8 expression by human airway epithelial cells exposed to diesel exhaust particles. Toxicology and Applied Pharmacology. 2010;243(1):46–54. doi: 10.1016/j.taap.2009.11.011. 19914270 [DOI] [PubMed] [Google Scholar]
- 38.Erzurum S.C. Protection of human endothelial cells from oxidant injury by adenovirus-mediated transfer of the human catalase cDNA. Nucleic Acids Research. 1993;21(7):1607–1612. doi: 10.1093/nar/21.7.1607. 8479912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tantama M., Hung Y.P., Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. Journal of the American Chemical Society. 2011;133(26):10034–10037. doi: 10.1021/ja202902d. 21631110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tate J.D.J., Miceli M.V., Newsome D.A. Zinc induces catalase expression in cultured fetal human retinal pigment epithelial cells. Current Eye Research. 1997;16(10):1017–1023. doi: 10.1076/ceyr.16.10.1017.9011. 9330853 [DOI] [PubMed] [Google Scholar]
- 41.Bozym R.A. Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Experimental Biology and Medicine (Maywood, N.J.) 2010;235(6):741–750. doi: 10.1258/ebm.2010.009258. 20511678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson M., Hogstrand C., Maret W. Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase beta activity. Journal of Biological Chemistry. 2012;287(12):9322–9326. doi: 10.1074/jbc.C111.320796. 22275360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fourquet S. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. Journal of Biological Chemistry. 2010;285(11):8463–8471. doi: 10.1074/jbc.M109.051714. 20061377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee S.R. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. Journal of Biological Chemistry. 1998;273(25):15366–15372. doi: 10.1074/jbc.273.25.15366. 9624118 [DOI] [PubMed] [Google Scholar]
- 45.Mahadev K. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. Journal of Biological Chemistry. 2001;276(24):21938–21942. doi: 10.1074/jbc.C100109200. 11297536 [DOI] [PubMed] [Google Scholar]
- 46.Brown A.M. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. Journal of Biological Chemistry. 2000;275(18):13441–13447. doi: 10.1074/jbc.275.18.13441. 10788456 [DOI] [PubMed] [Google Scholar]
- 47.Dineley K.E. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion. 2005;5(1):55–65. doi: 10.1016/j.mito.2004.11.001. 16060292 [DOI] [PubMed] [Google Scholar]
- 48.Handy D.E., Loscalzo J. Redox regulation of mitochondrial function. Antioxidants and Redox Signaling. 2012;16(11):1323–1367. doi: 10.1089/ars.2011.4123. 22146081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bishop G.M., Dringen R., Robinson S.R. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radical Biology and Medicine. 2007;42(8):1222–1230. doi: 10.1016/j.freeradbiomed.2007.01.022. 17382203 [DOI] [PubMed] [Google Scholar]
- 50.Flohé L. The fairytale of the GSSG/GSH redox potential. Biochimica et Biophysica Acta. 2013;1830(5):3139–3142. doi: 10.1016/j.bbagen.2012.10.020. 23127894 [DOI] [PubMed] [Google Scholar]
- 51.Alam J. Nrf2, a cap’n’collar transcription factor, regulates induction of the heme oxygenase-1 gene. Journal of Biological Chemistry. 1999;274(37):26071–26078. doi: 10.1074/jbc.274.37.26071. 10473555 [DOI] [PubMed] [Google Scholar]
- 52.Osburn W.O., Kensler T.W. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutation Research. 2008;659(1–2):31–39. doi: 10.1016/j.mrrev.2007.11.006. 18164232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tkachev V.O., Menshchikova E.B., Zenkov N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry: Biokhimii͡a. 2011;76(4):407–422. doi: 10.1134/s0006297911040031. 21585316 [DOI] [PubMed] [Google Scholar]
- 54.Hou W. Zinc mesoporphyrin induces rapid and marked degradation of the transcription factor Bach1 and up-regulates HO-1. Biochimica et Biophysica Acta. 2008;1779(3):195–203. doi: 10.1016/j.bbagrm.2008.01.006. 18325350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mito-HyPer is specifically localized in the mitochondria of BEAS cells. BEAS-2B cells transduced by lentivirus with mitochondrially-targeted HyPer (A) were colocalized with the mitochondria marker, MitoTracker Red (B). The two channels were merged, where the presence of yellow indicates colocalization of the HyPer construct and the mitochondria (C).
Exposure to Pyrithione alone does not induce intracellular H2O2 or EGSH responses. BEAS-2B cells stably expressing cytosolic (A) or roGFP2 (B) were exposed to vehicle (○), 1 µM PYRI (PYRI, ▲), or 5 µM Zn2+/1 µM PYRI (PYRI/Zn, ■) at the indicated time (arrow). Shown are ratiometric values calculated from the fluorescent 525/30 emission of the 404 and 488 nm laser excitation normalized as a percent of the maximum sensor response. Values are presented as mean±SE (n=3, where n consists of an average of 10 distinct cells' responses), * indicates statistical significant change compared to control by linear regression.
Depletion of glutathione levels does not lead to HO-1 mRNA expression in BEAS cells. BEAS-2B cells were incubated with 500 µM BSO for 24 h or starved of glucose for 2 h before, washed, and then incubated for 2 h in fresh media before mRNA extraction. BSO only imposed a 1.61±0.06(SEM) fold increase over untreated cells. Glucose deprivation induced a 3.08±1.29(SEM) fold increase in HO-1 gene expression. mRNA levels measured using TaqMan-based RT-PCR were normalized to levels of GAPDH mRNA and expressed as fold increases over basal mRNA expression.