

Abstract
Efavirenz (EFV) is an anti-retroviral drug frequently combined with isoniazid (INH) to treat HIV-1/tuberculosis co-infected patients. Both drugs have been associated with idiosyncratic liver injury (DILI), but combined anti-retroviral and anti-tubercular therapy can increase the risk for DILI as compared to either drug class alone. Because both EFV and INH have been implicated in targeting mitochondria, we aimed at exploring whether the two drugs might cause synergistic effects on the electron transport chain. We found that EFV inhibited complex I activity in isolated mouse liver mitochondria (IC50 ˜30 μM), whereas hydrazine, a major metabolite of INH generated by acylamidase-mediated hydrolytic cleavage, inhibited complex II activity (IC50 ˜30 μM). Neither INH alone (≤1000 μM) nor EFV alone (≤30 μM) was able to induce cell injury in cultured mouse hepatocytes. However, combined EFV/INH exposure resulted in increased superoxide formation and peroxynitrite stress, leading to the opening of the cyclosporine A-insensitive mode of the mitochondrial permeability transition (mPT), and necrotic cell death. The peroxynitrite scavengers, CBA or Fe-TMPyP, protected against mPT induction and alleviated cell injury. The acylamidase inhibitor bis-p-nitrophenyl phosphate prevented cell injury, suggesting that hydrazine greatly contributed to the toxicity. Methylene blue, a redox-active alternative electron acceptor/donor that bypasses complex I/II, effectively protected against EFV/INH-induced toxicity. These data demonstrate that, in murine hepatocytes, the mitochondrial electron transport chain is a critical target of combined EFV/INH exposure, and that this drug combination can lead to peroxynitrite stress-induced mPT and hepatocellular necrosis. These results are compatible with the concept that underlying silent mitochondrial dysfunction may be a key susceptibility factor contributing to idiosyncratic drug-induced liver injury.
Keywords: Efavirenz, Isoniazid, Mitochondria, Complex I, Drug-induced liver injury (DILI), Hepatotoxicity, Methylene blue, Peroxynitrite
Abbreviations: 1-ABT, 1-aminobenzotriazole; BNPP, bis-p-nitrophenyl phosphate; CBA, coumarin-7-boronic acid; CYP, cytochrome P450; EFV, efavirenz; ETC, electron transport chain; Fe-TMPyP, 5,10,15,20-tetrakis(N-methyl-4′-pyridyl)porphyrinato iron(III); INH, isoniazid; MB, methylene blue (methylthioninium chloride, 3,7-bis(dimethylamino)phenazathionium chloride); NAT, N-acetyltransferase; ROS, reactive oxygen species
Graphical abstract
Highlights
-
•
We model efavirenz (EFV) and isoniazid (INH) hepatotoxicity in mouse hepatocytes.
-
•
We find that EFV inhibits mitochondrial complex I, hydrazine inhibits complex II.
-
•
Co-exposure to EFV/INH causes peroxynitrite stress and induces the mPT.
-
•
Methylene blue bypasses complex I/II and protects from cell death.
-
•
Mitochondrial ETC impairment is critical in EFV/INH liver injury.
Introduction
An increasing number of therapeutic drugs have been implicated in targeting mitochondria and causing mitochondrial dysfunction, which likely contributes to some of the adverse effects and organ toxicity associated with these drugs [1–4]. Specifically, inhibition of mitochondrial electron transport at one or several sites of the electron transport chain (ETC) is one common mechanism by which drugs can interfere with energy homeostasis and the redox balance in mitochondria [5]. Minor impairment of ETC function normally does not entail biologically significant effects, due to the large inherent reserve capacity of the mitochondrion, and because of certain functional threshold effects for respiratory complexes I through IV [6–8]. However, this may dramatically change in the presence of an inherited or acquired mitochondrial deficiency, which can greatly amplify superimposed drug effects and severely impair energy production and mitochondrial function [9]. For example, underlying pharmacologic or genetic complex I dysfunction has been implicated in augmenting and potentiating the mitochondrial and cellular toxicity of mitochondria-targeting drugs [10–13].
Consistent with this concept, we have recently demonstrated that selective inhibition of complex I with rotenone or piericidin A was able to trigger lethal cell injury induced by otherwise non-toxic concentrations of the anti-tubercular drug, isoniazid (INH) in cultured mouse hepatocytes [14]. The mechanism of this synergistic effect is not completely clear, but we have shown that hydrazine, a major hydrolytic metabolite of INH, inhibited mitochondrial complex II and caused increased leakage of superoxide from the electron transport chain [14]. The resulting joint inhibition of complexes I and II caused massive ATP depletion and necrotic cell death in hepatocytes.
In an attempt to translate these findings into a clinically more relevant situation, we have exposed hepatocytes to a combination of efavirenz (EFV) and INH. Efavirenz is an anti-retroviral drug frequently combined with INH to treat HIV-1/tuberculosis co-infected patients [15,16]. Efavirenz has recently been associated with liver injury in susceptible patients, and a series of elegant mechanistic studies have revealed that EFV causes mitochondrial stress in murine hepatocytes [17–20]. On the other hand, INH has been used therapeutically for many decades, and the risk for inducing liver injury in susceptible patients has been well known [21,22], but the susceptibility factors are largely unknown. Interestingly, combined anti-retroviral/anti-tubercular therapy significantly increases the risk for developing liver injury as compared to anti-retroviral therapy alone [23], but it is not known whether mitochondrial dysfunction may be involved. The aim of this study was to explore whether EFV in combination with INH will precipitate cell injury in mouse hepatocytes via joint inhibition of the respiratory complexes I and II. Furthermore, we sought to explore whether pharmacologic intervention with methylene blue, an alternative electron carrier that can bypass the proximal ETC, can prevent the energy crisis and protect against lethal cell injury associated with the mitochondria-targeting drugs.
Materials and methods
Chemicals
Isoniazid (INH), efavirenz (EFV), and hydrazine (HzN) were purchased from Sigma (St. Louis, MO). All chemicals were obtained at the highest grade available.
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Connecticut. Young adult male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Prior to use, the mice were acclimatized for >1 week and kept on a 14/10-h light/dark cycle under controlled environmental conditions. They had free access to mouse chow (Teklad Global Rodent Diet; Harlan Laboratories, Boston, MA) and water.
Isolation of hepatic mitochondria and complexes I and II activity measurement
Mitochondria were isolated from untreated mice according to standard procedures as previously described [14]. Protein content was determined with the BCR protein assay using albumin as the reference protein. The mitochondria-enriched fractions were kept at −80 °C until analysis. Complexes I and II activities were determined in freeze–thawed (2×) mitochondria according to standard methods [24]. Briefly, complex I was measured as NADH: ubiquinone oxidoreductase activity in 25 mM potassium phosphate buffer containing 5 mM MgCl2, pH 7.2, and 2.5 mg/ml BSA, 0.13 mM NADH, 2 μg/ml antimycin A, and 65 μM ubiquinone (Q1). NADH oxidation was monitored as decrease in absorbance at 340 nm. Complex II was measured as succinate: ubiquinone oxidoreductase activity, linked to the artificial electron acceptor, 2,6-dichlorophenolindophenol (DCPIP), in phosphate buffer without BSA, containing 20 mM sodium succinate, 50 μM DCPIP, 2 μg/ml antimycin A, 2 μg/ml rotenone, and 65 μM Q1. DCPIP reduction was monitored at 600 nm.
Primary mouse hepatocyte culture and exposure to drugs
Hepatocytes were isolated from mice by retrograde collagenase perfusion, and subsequently cultured in supplemented Williams’ Medium E as described [14]. Briefly, the cells were plated in 48-well plates (8.0 × 104 cells per well) coated with 50 μg/ml rat tail collagen. The hepatocytes were allowed to attach for 3 h in a humidified atmosphere of 5% CO2/95% air at 37 °C. Subsequently, the cells were washed and then incubated in the same medium. After overnight pre-culture, the medium was replaced by fresh serum- and antibiotic-free medium to which the drugs were added from stock solutions. DMSO was used as solvent for EFV and other lipophilic compounds (final concentrations not exceeding 0.1%), and culture medium was used to dissolve INH. In some experiments, the cells were post-treated with methylene blue (MB) 20 min after exposure to EFV and/or INH.
Determination of cell injury
Release of cytosolic lactate dehydrogenase (LDH) into the extracellular medium (CytoTox-One Homogeneous Membrane Integrity Assay, Promega, Madison, WI) was used as an indicator of cytotoxicity. The data were expressed as percentage of activity present in the medium as compared to the total intra- and extracellular LDH activity. Total cellular ATP content was measured by luminescence techniques (Cell Titer-Glo Luminescent Cell Viability Assay, Promega). Chemiluminescence was determined in black 96-well plates, and ATP content was calculated from a standard curve. INH, EFV, or MB did not interfere with the luciferin/luciferase reaction. The nuclear fluorescence of propidium iodide, which can permeate into cells with compromised plasma membrane only, was taken as an indicator of necrotic cell death.
Assessment of the mitochondrial permeability transition
To demonstrate the opening of the mitochondrial permeability transition (mPT) pore, we used the fluorogenic marker, calcein acetoxymethylester (AM). To selectively label mitochondria with the probe, the cells were loaded with 1 μM calcein-AM in the presence of CoCl2 (1 mM) for 15 min at 4 °C, followed by 6 h incubation at 37 °C [25]. By loading the cells in the cold, the calcein-AM can cross both the cell membrane and the mitochondrial membranes, because the cytosolic esterases are not active. Upon recovery at 37 °C, calcein-AM in mitochondria is cleaved and calcein remains trapped. Because the added Co2+, which is a high-affinity ligand for calcein, quenches the fluorescence upon binding, the cytosol readily loses its fluorescence, whereas intact mitochondria retain their bright fluorescence because Co2+ is not readily taken up by mitochondria. However, following drug-induced opening of the mPT pore, the intramitochondrial calcein (Mr = 622) will leak into the cytosol, and the mitochondrial staining will be lost. The green fluorescence was imaged with an Olympus Bx51 fluorescence microscope (40× objective).
Assessment of mitochondrial transmembrane potential in hepatocytes
The mitochondrial inner transmembrane potential (Δψm) was measured with tetramethyl rhodamine methylester (TMRM, Molecular Probes/Invitrogen). Hepatocytes were loaded with TMRM (100 nM; non-quenching mode) [26] for 20 min at 37 °C, and drug-induced changes in fluorescence were recorded with an Olympus Bx51 fluorescence microscope.
Measurement of mitochondrial ROS/RNS generation in hepatocytes
Mitochondrial generation of superoxide was estimated with the cell-permeable and mitochondria-targeted fluorogenic probe, hydroethidine (HE) linked to triphenylphosphonium (MitoSOX Red, Invitrogen). The drug-pretreated cells were loaded with MitoSOX Red (25 nM) for 10 min at 37 °C, washed with fresh culture medium, and the mitochondrial 2-hydroxy ethidium-derived fluorescence was determined at 396/580 nm (excitation/emission), respectively, in a Safire2 microplate reader (Tecan, Maennedorf, Switzerland). Hepatocellular formation of peroxynitrite was determined with the fluorogenic probe, coumarin-7-boronic acid (CBA, Cayman, Ann Arbor, MI), which reacts stochiometrically and rapidly with ONOO− several orders of magnitude faster than with H2O2 [27]. Hepatocytes were preloaded with 100 µM CBA for 20 min at 37 °C, and the generation of hydroxycoumarin was determined at 332/450 nm (excitation/emission) in a Safire2 microplate reader.
Statistical analysis
All data were expressed as mean ± SD. If there was normal distribution, a standard analysis of variance (ANOVA) was used, followed by Dunnett’s test for multiple comparisons versus the control group. When normality failed, a Kruskal–Wallis one-way analysis of variance on ranks was used followed by Dunn’s test for multiple comparison versus the control group. A P value of ≤0.05 was considered significant.
Results
Co-exposure to efavirenz/isoniazid, but not to the individual drugs alone, induces cell injury in cultured mouse hepatocytes
To determine the concentration-dependent toxic response to EFV or INH, we first exposed cultured mouse hepatocytes to both of the individual drugs alone. We found that EFV (up to 30 μM) or INH (up to 3000 μM) did not induce increased LDH release from hepatocytes or cause decreases in intracellular ATP concentrations, as compared to the solvent controls (Fig. 1). However, because EFV and INH are often combined clinically, and because combined anti-retroviral/anti-tubercular therapy can significantly increase the risk for developing liver injury as compared to anti-retroviral therapy alone [23], we next determined the effect of EFV/INH co-treatment on hepatocellular viability. We found that the combination of efavirenz (30 μM) and INH (1000 μM) caused marked cell injury, as evidenced by a time-dependent increase in LDH release, reaching >40% leakage after 24 h (Fig. 1A). Furthermore, during EFV/INH co-exposure, intracellular ATP levels decreased rapidly to <20% of solvent control levels during the first 6 h and were nearly undetectable after 12 h of exposure (Fig. 1B). These data suggest that efavirenz and isoniazid may act synergistically to trigger hepatocellular injury at otherwise non-toxic concentrations.
Fig. 1.
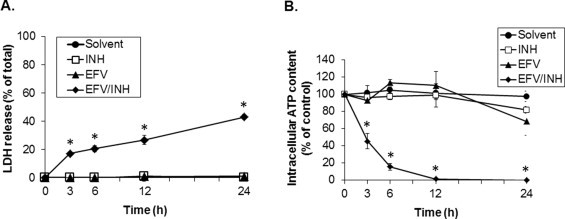
Synergistic effects of non-toxic concentrations of efavirenz (EFV) and isoniazid (INH) on cell injury and intracellular ATP concentrations during exposure of cultured mouse hepatocytes to combined EFV/INH. Time course of (A) LDH release and (B) intracellular ATP content after treatment with solvent (DMSO, 0.1%), INH alone (1000 μM), EFV alone (30 μM), or combined EFV/INH. Data are mean ± SD of three independent hepatocyte preparations using quadruplicate wells. *P < 0.05 versus solvent control.
Efavirenz/isoniazid toxicity is BNPP-sensitive, indicating a key role for hydrazine
To ascertain whether a metabolite(s) of INH was involved in causing the observed toxicity, we used selective inhibitors of metabolic pathways responsible for INH biotransformation. First, to explore a possible role of CYPs, we pretreated some hepatocytes with the pan-CYP inhibitor, 1-aminobenzotriazole (1-ABT). We found that 1-ABT (0–300 μM) did not significantly alter the toxic response to combined EFV/INH (data not shown). As 1-ABT is also an N-acetyltransferase (NAT) inhibitor [28], these data indicate that it is unlikely that an acetylated or oxidative metabolite of either INH or EFV was a major contributor to the cell injury. Because hydrazine, a major hydrolytic metabolite of isoniazid, has been implicated in the hepatic toxicity of isoniazid [29–32], we hypothesized that hydrazine was the primary toxic species contributing to the cytotoxicity in this model. To test this hypothesis, we used bis-p-nitrophenyl phosphate (BNPP), a specific acylamidase inhibitor, to prevent hydrazine formation in cells exposed to INH. We found that BNPP indeed attenuated the toxicity of EFV/INH (30 μM/1000 μM, respectively) in a concentration-dependent manner (Fig. 2A) and that the loss of intracellular ATP was largely prevented (Fig. 2B). Because the metabolism of EFV does not involve any acylamidase activity [33], these data suggest that hydrazine is a key contributor to the lethal cell injury provoked by EFV/INH co-treatment.
Fig. 2.
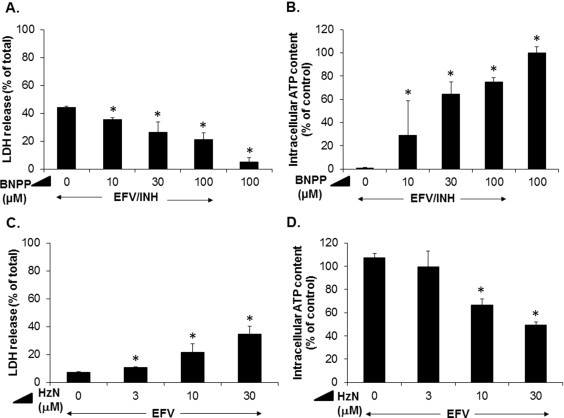
Role of hydrazine (HzN) in EFV/INH-induced hepatocellular injury. (A and B) Concentration-dependent effects of bis-p-nitrophenyl phosphate (BNPP, an inhibitor of acylamidase-mediated hydrolysis of INH to HzN), on cell injury and intracellular ATP concentrations caused by exposure of cultured mouse hepatocytes to a combination of EFV (30 μM) and INH (1000 μM) for 24 h. Data are mean ± SD of three independent hepatocyte preparations using quadruplicate wells. *P < 0.05 versus no addition of BNPP. (C and D) Concentration-dependent effects of HzN in combination with a fixed concentration (30 μM) of EFV on LDH release and intracellular ATP content after 24 h. Data are mean ± SD of three independent hepatocyte preparations using quadruplicate wells. *P < 0.05 versus EFV alone.
Next, we determined the lowest concentration at which hydrazine was able to cause toxicity in combination with a fixed nontoxic concentration of EFV. We found that there was a concentration-dependent potentiation of cell injury and loss of ATP that became significant at 10 μM hydrazine, while hydrazine alone did not induce cell injury at these low concentrations (Fig. 2C and D). Thus, the hydrazine concentrations that can trigger cell injury in vitro are in the same range or even lower than the hepatic concentrations of hydrazine formed after a single dose of 50 mg/kg INH in mice [34].
Efavirenz inhibits mitochondrial complex I activity, and hydrazine inhibits complex II activity—a potentially dangerous combination that hits the ETC at two sites
We have previously demonstrated that non-toxic concentrations of pharmacologic inhibitors of complex I (rotenone, piericidin A) can trigger massive cell injury induced by otherwise non-toxic concentrations of isoniazid [14]. Because indirect evidence has suggested that EFV might also be a complex I inhibitor [17], we hypothesized that combined treatment of cells with INH and EFV will precipitate cell injury via a similar mechanism. Therefore, to ascertain whether EFV directly inhibits complex I activity, we first determined NADH: ubiquinone oxidoreductase activity in isolated mouse liver mitochondria. We found that EFV indeed caused a concentration-dependent inhibition of complex I activity, with an IC50 of approx. 30 μM (Fig. 3A). The solvent (DMSO, 0.3% final) did not inhibit complex I function (not shown). Rotenone (20 μM) was used as a positive control for the assay and was found to inhibit total complex I activity by approximately 50%, which is typical for liver mitochondria [35]. As an expected consequence of complex I dysfunction, we found that the mitochondrial inner transmembrane potential (Δψm) was decreased in cultured hepatocytes exposed to EFV. This was evidenced by using the fluorescent probe TMRM, which normally accumulates in the mitochondrial matrix due to the inside negative potential but which is lost from the matrix if the Δψm decreases (Fig. 3B). Furthermore, we found that hepatocytes exposed to EFV exhibited a concentration- and time-dependent increase in MitoSOX Red-derived fluorescence, indicating net increases in mitochondrial superoxide which, again, is likely a consequence of complex I inhibition ( Fig. 3C).
Fig. 3.
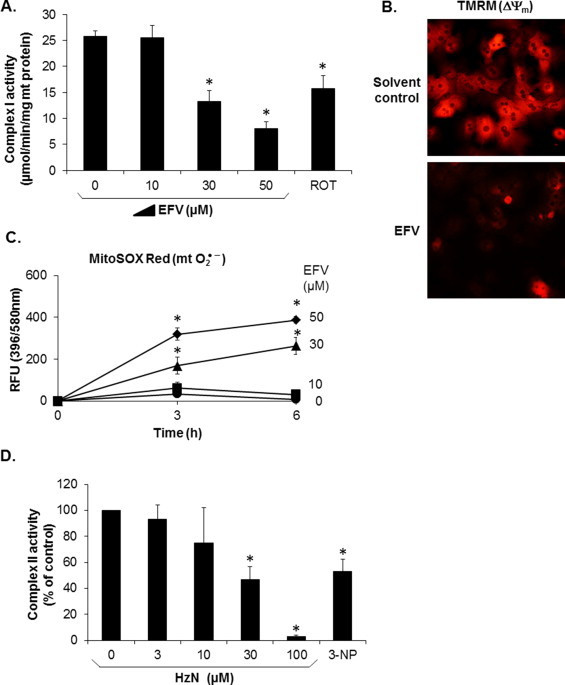
Effects of EFV on complex I activity, mitochondrial inner transmembrane potential (Δψm), and mitochondrial superoxide generation, and effect of HzN on complex II activity. (A) Isolated mouse hepatic mitochondria were used to determine the concentration-dependent effects of EFV on complex I activity. Rotenone (ROT, 20 μM) was used as a positive control. Data are mean ± SD of three independent mitochondrial preparations using duplicate determinations. *P < 0.05 versus solvent control. (B) Cultured mouse hepatocytes were loaded with TMRM, washed, and exposed to EFV (30 μM) for 1 h. The photomicrograph shows the results from one cell preparation typical of three independent experiments. The bright (punctate) fluorescence indicates the mitochondrial accumulation of TMRM, driven by an intact Δψm, and the loss of the mitochondrial fluorescence after EFV exposure. (C) MitoSOX Red-derived fluorescence in cultured hepatocytes, indicative of increased superoxide generation. Data are mean ± SD of three independent hepatocyte preparations using quadruplicate wells. *P < 0.05 versus solvent control. (D) Isolated mouse hepatic mitochondria were used to determine the concentration-dependent effects of HzN on complex II activity. 3-Nitropropionic acid (3-NP, 3 mM) was used as a positive control. Data are mean ± SD of three independent mitochondrial preparations using duplicate determinations. *P < 0.05 versus solvent control.
Because hydrazine has been implicated as a key player in isoniazid toxicity to hepatocytes, and because we have previously demonstrated that hydrazine inhibited complex II activity in isolated and solubilized yeast mitochondria [14], we sought to confirm these findings in mammalian mitochondria. We found that, in isolated mouse liver mitochondria, hydrazine inhibited complex II activity, with an IC50 of approximately 30 μM (Fig. 3D). These data suggest that EFV and INH together might severely inhibit the proximal mitochondrial electron flow by jointly blocking ubiquinone reduction, providing a working hypothesis to explain the hepatocellular toxicity induced by the drugs used in combination.
Efavirenz/isoniazid co-treatment causes peroxynitrite stress in hepatocytes resulting in the induction of the mitochondrial permeability transition
Superoxide generated from complex I/II inhibition can be enzymatically converted to hydrogen peroxide, but this reaction is outcompeted by the ultrarapid non-enzymatic reaction of superoxide with nitric oxide to form peroxynitrite. Because this latter reaction is kinetically controlled by superoxide levels [36], we hypothesized that the observed increased superoxide signal following exposure to efavirenz/isoniazid might result in peroxynitrite stress. With the advent of novel and highly selective peroxynitrite probes, including coumarin-7-boronic acid (CBA) [27], it has become possible to monitor treatment-induced peroxynitrite formation in intact cells. Cells preloaded with CBA followed by exposure to non-toxic concentrations of EFV (≤30 μM) alone developed increases in fluorescence due to 7-hydroxycoumarin, whereas cells exposed to INH alone did not exhibit increases in this marker as compared to solvent controls (Fig. 4A). In contrast, combined EFV/INH treatment caused further increases in CBA-derived fluorescence over six hours, suggesting enhanced peroxynitrite formation. The addition of Fe-TMPyP, a cell-permeable metalloporphyrin which acts as a peroxynitrite decomposition catalyst [37,38] effectively attenuated the fluorescence, confirming that the signal was selective for peroxynitrite Fig. 4B).
Fig. 4.
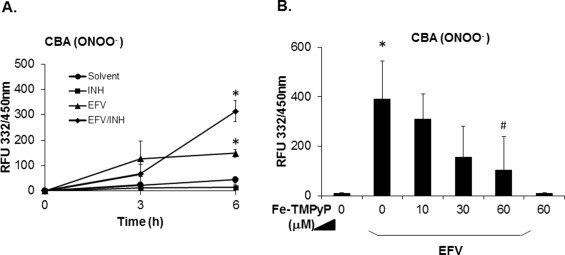
Time course of EFV and/or INH-induced peroxynitrite (ONOO−) generation in hepatocytes. (A) Hepatocytes were preloaded with coumarin-7-boronic acid (CBA, 100 μM) for 3 h and exposed to EFV alone (30 μM), INH alone (1000 μM), or a combination of the two. Hydroxycoumarin fluorescence generated from peroxynitrite-mediated CBA oxidation was serially recorded with a plate reader. (B) Fe-TMPyP, a peroxynitrite decomposition catalyst, was used to demonstrate the selectivity of the assay for peroxynitrite in hepatocytes exposed to EFV (50 μM) for 6 h. Data are mean ± SD of three independent hepatocyte preparations using quadruplicate wells. *P < 0.05 versus solvent control; #P < 0.05 versus EFV alone.
Peroxynitrite has been implicated in oxidizing/nitrating critical mitochondrial targets that are involved in mediating cell death, including proteins regulating the mitochondrial permeability transition (mPT) [39–42]. We therefore hypothesized that the increased peroxynitrite stress induced by EFV/INH co-exposure was causally involved in triggering lethal cell injury via opening of the mPT pore. To monitor in situ the opening of the mPT pore, which involves both the outer and inner mitochondrial membrane, we preloaded hepatocytes by cold/warm incubation with calcein-AM/Co2+ (for rationale see Materials and methods) prior to exposure to the drugs. As expected, we found that untreated control hepatocytes exhibited a punctate pattern of calcein fluorescence that co-localized with TMRM fluorescence, indicating that calcein was sequestered in the mitochondria (not shown). Solvent controls retained the mitochondrial fluorescence for at least 2 h, indicative of an intact inner mitochondrial membrane (Fig. 5A). In contrast, the addition of combined EFV/INH induced a rapid loss of the mitochondrial fluorescence, suggesting opening of the mPT pore as evidenced by the quenching of calcein fluorescence by Co2+. INH alone had no apparent effect on mitochondrial calcein release (not shown). The loss of the mitochondrial fluorescence was not inhibitable by cyclosporin A (CsA, 1 μM), suggesting that a CsA-insensitive mode of mPT [43] was activated. To confirm that this event resulted in oncotic necrosis, which is the default mode of cell injury caused by the mPT [44], we co-exposed the hepatocytes to propidium iodide (PI), a fluorescent nuclear DNA marker, which has access to cells with damaged plasma membrane only, but not to apoptotic nuclei with intact cell membrane [45]. We found that EFV/INH co-treatment, but not treatment with solvent alone or INH alone, caused a large number of nuclei to become PI-positive (Fig. 5A). We also incubated cells with the fluorescent DNA stain Hoechst 33342, but did not find any evidence of fragmented or condensed nuclei, which would be indicative of apoptosis (not shown). Taken together, these data indicate that the combination of EFV/INH is a potent inducer of the CsA-insensitive mode of the mPT that results in hepatocellular necrosis.
Fig. 5.
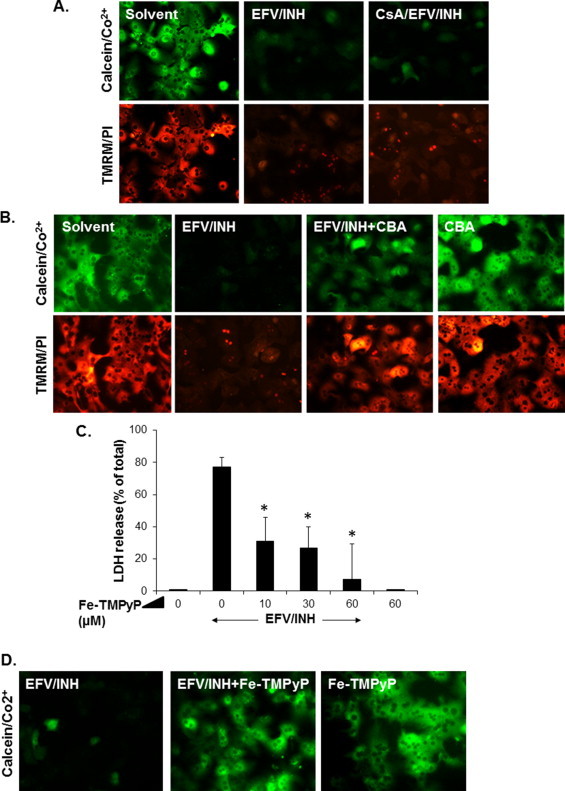
EFV/INH-induced opening of the mitochondrial permeability transition (mPT) pore and its modulation by peroxynitrite scavengers. Cultured mouse hepatocytes were cold/warm-loaded with calcein-AM/Co2+, TMRM, and PI, as described in Materials and methods, and exposed to combined INH (1000 μM) and EFV (30 μM) for 1 h. (A) The fluorescence micrographs show hepatocellular loss of mitochondrial labeling with calcein after drug treatment, indicative of mPT induction, loss of TMRM staining (100 nM, 20 min preloading), reflecting a decrease in the Δψm, and an increase in the number of PI (1 μM)-positive nuclei as it occurs in oncotic necrosis. Some cells were pretreated with cyclosporin A (CsA, 1 μM, for 20 min). (B) Preincubation (20 min) with the peroxynitrite scavenger, CBA (100 μM) afforded partial protection against EFV/INH-induced mPT, membrane depolarization, and cell injury. (C) Concentration-dependent protective effect of the peroxynitrite decomposition catalyst, Fe-TMPyP on EFV/INH-induced hepatocellular injury. LDH leakage was determined after 24 h. (D) Protective effects of Fe-TMPyP (30 μM, 1 h) against EFV/INH-induced mPT induction. All fluorescence micrographs are typical of three independent cell preparations.
To determine whether the increase in peroxynitrite levels might be causally involved in cell injury, as opposed to being a nonspecific consequence of the injury, we explored the effects of two unrelated peroxynitrite-reactive agents on EFV/INH-induced cell injury. First, in the presence of the peroxynitrite scavenger CBA itself, we found that the cells were protected against the treatment-related collapse of the Δψm as well as the development of PI-positive (necrotic) cells (Fig. 5B). Second, Fe-TMPyP similarly attenuated EFV/INH-induced LDH release in a concentration-dependent manner (Fig. 5C) and prevented the opening of the mPT pore (Fig. 5D). Taken together, these data indicate that ONOO− formation occurred prior to cell death and that it contributes to the treatment-related cell injury.
Methylene blue, an alternative electron carrier, bypasses complexes I and II and rescues hepatocytes from EFV/INH-induced lethal injury
If the mechanisms of EFV/INH-induced hepatocyte demise indeed involved a joint inhibitory effect on complexes I and II, then circumvention of this proximal ETC block with an alternative electron carrier that feeds electrons into the ETC at a more distal site should protect against cell injury. One of these alternative electron carriers is methylene blue (MB), a redox-active agent that has been shown to directly accept electrons from NADH and reduce cytochrome c without the involvement of ubiquinone [46]. We found that, in the presence of mitochondria and EFV (30 μM), MB greatly enhanced the consumption of NADH in a concentration-dependent manner (Fig. 6A), even exceeding the rate caused by normal complex I activity by several-fold. This confirms the ability of MB to oxidize NADH even under conditions of chemical inhibition of complex I. Next, we investigated whether MB was able to protect cultured hepatocytes against cell injury induced by exposure to combined INH (1000 μM)/EFV (30 μM). We found that MB (>30 μM) protected against the opening of the mPT pore and the subsequent hepatocellular necrosis, as demonstrated by the retention of calcein in the mitochondrial matrix in the presence of otherwise cytotoxic concentrations of EFV/INH (Fig. 6B). Furthermore, MB was able to almost completely prevent LDH release and loss of intracellular ATP (Fig. 6C and D). Control experiments confirmed that MB did not interfere with the LDH assay itself (data not shown). Taken together, these data strongly suggest that the toxicity caused by EFV/INH co-exposure is the consequence of a severe inhibition of ETC function, resulting in peroxynitrite stress, and that this impaired pathway can be bypassed by MB, resulting in full protection against cell injury.
Fig. 6.
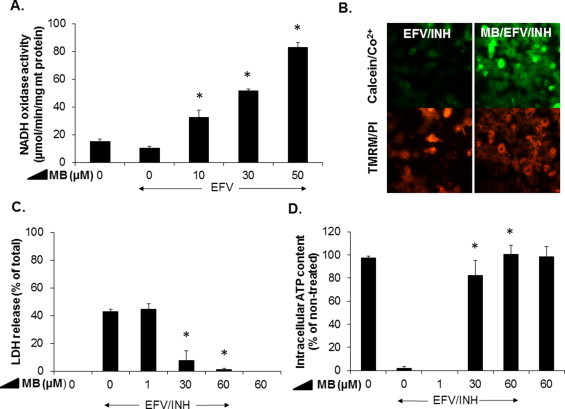
Protective effects of methylene blue (MB) against EFV/INH-induced hepatocellular injury. (A) Concentration-dependent effects of MB on EFV (30 μM)-induced inhibition of complex I activity (NADH oxidation) in isolated mouse liver mitochondria. Data are mean ± SD of three independent mitochondrial preparations using duplicate determinations. *P < 0.05 versus EFV alone. (B) Protective effects of MB (30 μM) against EFV (30 μM)/INH (1000 μM)-induced dissipation of the mitochondrial Δψm. Hepatocytes were cold/warm-loaded with calcein/Co2+, washed, and incubated with EFV/INH in the presence or absence of MB (30 μM). Hepatocytes were also preloaded with TMRM (100 nM) and PI (1 µM) for 20 min prior to drug exposure. (C and D) Concentration-dependent effects of MB on EFV (30 μM)/INH (1000 μM)-induced cell injury and intracellular ATP levels. Release of LDH into the extracellular medium and ATP concentrations were determined after 24 h exposure. Data are mean ± SD of three independent hepatocyte preparations using triplicate wells. *P < 0.05 versus EFV/INH alone.
Discussion
The aim of this study was to explore whether co-exposure of murine hepatocytes to EFV and INH, a clinically relevant drug combination, renders hepatocytes more susceptible to mitochondria-mediated lethal cell injury than either of the drugs alone. Our working hypothesis was that a joint inhibition of respiratory complexes I (by EFV) and II (by hydrazine, a major INH metabolite) would lead to a decrease in energy production, as well as an increase in superoxide leakage from the ETC, because ubiquinone reduction is impaired at the Q-binding site in both complexes I and II. We found that EFV indeed inhibits complex I activity in mouse hepatic mitochondria, and that this mitochondrial effect, which alone does not cause cell injury, can activate and amplify the latent cell injury caused by co-exposure to otherwise nontoxic concentrations of INH.
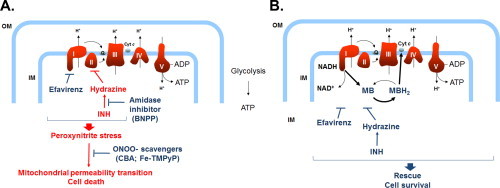
These conclusions were based on a number of experimental findings. First, the combined EFV/INH treatment, but not the individual drugs alone, resulted in a rapid loss of intracellular ATP, paralleled by a collapse of the Δψm and opening of the mPT pore, leading to hepatocellular necrosis. This suggests that, due to the combined inhibitory effects of EFV and INH, the energy flux along the ETC was severely impaired, leading to increased formation of superoxide and, hence, peroxynitrite (Fig. 7A).
Fig. 7.
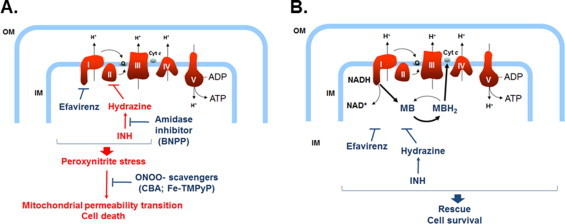
Schematic representation of the putative mechanisms involved in EFV/INH-mediated hepatic mitochondrial dysfunction and its prevention by MB. (A) Inhibition by EFV of complex I and hydrazine-mediated inhibition of complex II in combination results in peroxynitrite stress, opening of the mPT, and eventually oncotic necrosis. (B) MB acts as an electron acceptor from NADH at complex I, and MBH2 directly reduces cytochrome c, bypassing the upstream components of the ETC, thus protecting hepatocytes against EFV/INH-induced lethal cell injury.
Second, we show that bypassing complexes I and II with the redox-active, mitochondria-permeable electron carrier MB, normal energy fluxes could be maintained and cells could be protected against the toxicity caused by combined EFV/INH exposure (Fig. 7B). Methylene blue, a diaminophenothiazine, is reduced in the mitochondrial matrix by a number of NAD(P)H-dependent dehydrogenases, including complex I [46–48], resulting in the formation of MBH2 (leucomethylene blue). Because of the low redox potential (close to zero), MB can readily cycle between the oxidized and the reduced form [49,50] and function as electron donor to heme proteins including cytochrome c [46,49]. Although at high concentrations, MB can act as an uncoupling agent in isolated mitochondria [51], control experiments with TMRM had confirmed that, under the conditions used, MB did not alter the Δψm in mouse hepatocytes (data not shown). Methylene blue not only can be used as a biochemical tool to provide the proof-of-concept for a mitochondrial mechanism involved in the toxicity of a drug (here EFV/INH), but also could be used therapeutically to protect against drug-induced, mitochondria-mediated cell injury. In fact, MB is a clinically approved drug that has been used for many decades against methemoglobinemia and other disorders caused by imbalances in redox homeostasis. Furthermore, MB has been used in experimental models of drug-induced toxicity in which complex I dysfunction has been implicated, including doxorubicin-induced cardiotoxicity [52].
Third, the combined EFV/INH treatment resulted in the induction of the mPT as evidenced by in situ monitoring of calcein loss from mitochondria. Although many molecular details of the nature of the mPT pore are currently still unresolved, it has been demonstrated that besides the canonical “regulated” mode of mPT, which is induced by low-level chemical stress and which is CsA-sensitive, there is also an “unregulated” mode of mPT, which is caused by high-level chemical stress and which is CsA-insensitive [43]. We have previously shown in a mouse model of acetaminophen hepatotoxicity that the CsA-insensitive mode of mPT was activated by peroxynitrite [53]. Here, we show that peroxynitrite stress is critical in precipitating EFV/INH-induced cell death, as both the peroxynitrite scavenger CBA and the peroxynitrite decomposition catalyst Fe-TMPyP effectively protected against the mPT and the resulting cell death.
Fourth, we have provided evidence that bioactivation of INH was a key contributing factor to cell injury. This conclusion was supported by the findings that inhibition of acyl amidase-mediated hydrolytic cleavage of INH to hydrazine in the presence of BNPP was sufficient to protect cells against the toxicity caused by EFV/INH co-exposure. We also considered the possibility that the toxicity was dependent on a CYP-mediated oxidative metabolite generated from either INH or EFV. Indeed, previous studies had revealed that mitochondrial toxicity induced by EFV was inhibited by the pan-CYP inhibitor, 1-ABT, in primary human hepatocytes [54], pointing to a possible role of its major, CYP2B6-catalyzed metabolite, 8-hydroxy-EFV. However, here, 1-ABT had no apparent protective effects on combined EFV/INH toxicity. Also, INH itself is an irreversible CYP inhibitor, acting through its terminal hydrazine nitrogen-derived nitrene that tightly coordinates to the heme iron [55]. Because INH greatly amplified the toxicity in combination with EFV, rather than inhibiting it, it is reasonable to assume that the cell injury was not caused by an oxidative metabolite of EFV, but rather triggered by hydrazine cleaved off the parent INH.
How exactly EFV primes hepatocytes to the cell-killing activity induced by INH and/or hydrazine remains still unclear. EFV causes multiple toxic responses in cells, including endoplasmic reticulum stress [18], but the primary target of EFV is the mitochondrion [19], as convincingly demonstrated with Rhoo cells (depleted of mitochondrial DNA), in which the EFV-induced cellular effects were greatly attenuated. In Hep3B cells, EFV caused activation (phosphorylation) of AMP-activated protein kinase, the master switch of cellular energetics, causing a decrease in ATP-consuming processes and an increase in fatty acid uptake and β-oxidation [17], as well as increases in mitochondrial mass and induction of autophagy [20]. Together, these events likely reflect an adaptive response to the mitochondrial stress inflicted by EFV. However, the results of the present study suggest that the inhibition of complex I by EFV may be a major mechanism that leads to enhanced oxidant stress, lowering the threshold for inducing lethal cell injury if a second mitochondrial insult (e.g., by INH) is superimposed.
Translation of these concentration-dependent effects to the clinical situation is difficult, and a simple comparison ofin vitro concentrations (used for this study with cultured hepatocytes) with therapeutic plasma levels may be misleading. Clinical peak plasma levels of EFV are in the low micromolar range, but CYP2B6 polymorphisms (the major CYP form involved in EFV clearance) can lead to much higher plasma concentrations (e.g., >20 μM in CYP2B6 *6/*6 patients) [56]. However, EFV is highly (99.5%) plasma protein-bound, mostly albumin, thus greatly reducing the portion of “free”, pharmacologically active drug. In a mouse study, an EFV dose of 20 mg/kg resulted in a plasma Cmax ofapprox. 6 μM; however, the concentrations in the liver were much higher (approx. 150 μM) [57]. Therefore, the EFV concentrations used in this study (30 μM) seem reasonable. As to INH, the concentrations to which the mouse hepatocytes were exposed (1 mM), were clearly higher (approx. 10-fold) than therapeutic peak plasma levels in patients; however, they could be similar to portal concentrations. A recent study has quantitated the hepatic concentrations of INH and hydrazine after a single oral dose of 50 mg INH/kg body weight to mice; after 30 min, the hepatic levels were >200 μM for INH and >70 μM for hydrazine [34]. Clinical levels of hydrazine after INH have been reported to be in the low microM range [58,59], but hydrazine can accumulate following repeated doses of INH in slow acetylators. In view of our findings that, in combination with EFV, hydrazine at low microM concentrations can have dramatic effects on mitochondrial dysfunction and cell injury, the findings could be relevant for the in vivo situation.
In conclusion, we show that co-exposure of primary mouse hepatocytes to INH and EFV, two drugs often used in combination treatment clinically, can severely damage mitochondria and induce hepatocellular injury at otherwise nontoxic concentrations of the individual drugs. The findings are compatible with the concept that clinically silent genetic or pharmacologic abnormalities of mitochondrial function (e.g., EFV-induced complex I inhibition) can trigger and activate the hepatotoxic potential of drugs that have been implicated in causing idiosyncratic liver injury (e.g., INH-induced complex II inhibition and induction of peroxynitrite stress via its metabolite, hydrazine). Although this acute cell model does not exactly reflect the clinical situation, where the precipitation of overt liver injury is normally delayed, it provides a mechanistic model for cumulative prooxidative injury of liver mitochondria. Under normal conditions, due to the enormous energy reserve capacity of the liver, and the threshold effects for hepatocellular injury, the injury will not immediately manifest, but gradual oxidative injury can push the liver mitochondria to the point of no return.
Acknowledgments
This work was supported by a grant from Connecticut Innovations (11SCDIS02, to U.A.B.) and by the Boehringer Ingelheim Endowed Chair in Mechanistic Toxicology at UCONN.
References
- 1.Chan K., Truong D., Shangari N., O’Brien P.J. Drug-induced mitochondrial toxicity. Expert Opinion on Drug Metabolism and Toxicology. 2005;1:655–669. doi: 10.1517/17425255.1.4.655. 16863431 [DOI] [PubMed] [Google Scholar]
- 2.Dykens J.A., Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discovery Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. 17826691 [DOI] [PubMed] [Google Scholar]
- 3.Pereira C.V., Moreira A.C., Pereira S.P., Machado N.G., Carvalho F.S., Sardão V.A., Oliveira P.J. Investigating drug-induced mitochondrial toxicity: a biosensor to increase drug safety? Current Drug Safety. 2009;4:34–54. doi: 10.2174/157488609787354440. 19149524 [DOI] [PubMed] [Google Scholar]
- 4.Pessayre D., Fromenty B., Berson A., Robin M.A., Lettéron P., Moreau R., Mansouri A. Central role of mitochondria in drug-induced liver injury. Drug Metabolism Reviews. 2012;44:34–87. doi: 10.3109/03602532.2011.604086. 21892896 [DOI] [PubMed] [Google Scholar]
- 5.Nadanaciva S., Bernal A., Aggeler R., Capaldi R.A., Will Y. Target identification of drug induced mitochondrial toxicity using immunocapture based OXPHOS activity assays. Toxicology in Vitro. 2007;21:902–911. doi: 10.1016/j.tiv.2007.01.011. 17346924 [DOI] [PubMed] [Google Scholar]
- 6.Davey G.P., Peuchen S., Clark J.B. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. Journal of Biological Chemistry. 1998;273:12753–12757. doi: 10.1074/jbc.273.21.12753. 9582300 [DOI] [PubMed] [Google Scholar]
- 7.Inoue K., Nakada K., Ogura A., Isobe K., Goto Y., Nonaka I., Hayashi J.I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nature Genetics. 2000;26:176–181. doi: 10.1038/82826. 11017072 [DOI] [PubMed] [Google Scholar]
- 8.Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J.P., Letellier T. Mitochondrial threshold effects. Biochemical Journal. 2003;370:751–762. doi: 10.1042/BJ20021594. 12467494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schon E.A., Hirano M., DiMauro S. Drug effects in patients with mitochondrial diseases. In: Dykens J.A., Will Y., editors. Drug-Induced Mitochondrial Dysfunction. Wiley; Hoboken, NJ: 2008. pp. 311–324. [Google Scholar]
- 10.Krähenbühl S., Brandner S., Kleinle S., Liechti S., Straumann D. Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver. 2000;20:346–348. doi: 10.1034/j.1600-0676.2000.020004346.x. 10959815 [DOI] [PubMed] [Google Scholar]
- 11.Pelicano H., Feng L., Zhou Y., Carew J.S., Hileman E.O., Plunkett W., Keating M.J., Huang P. Inhibition of mitochondria respiration. A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. Journal of Biological Chemistry. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. 12853461 [DOI] [PubMed] [Google Scholar]
- 12.Perier C., Tieu K., Guégan C., Caspersen C., Jackson-Lewis V., Carelli V., Martinuzzi A., Hirano M., Przedborski S., Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. 16365298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boelsterli U.A., Lim P.L.K. Mitochondrial abnormalities—a link to idiosyncratic drug hepatotoxicity? Toxicology and Applied Pharmacology. 2007;220:92–107. doi: 10.1016/j.taap.2006.12.013. 17275868 [DOI] [PubMed] [Google Scholar]
- 14.Lee K.K., Fujimoto K., Zhang C., Schwall C.T., Alder N.N., Pinkert C.A., Krueger W., Rasmussen T., Boelsterli U.A. Isoniazid-induced cell death is precipitated by underlying mitochondrial complex I dysfunction in mouse hepatocytes. Free Radical Biology and Medicine. 2013;65:584–594. doi: 10.1016/j.freeradbiomed.2013.07.038. 23911619 [DOI] [PubMed] [Google Scholar]
- 15.Williams B.G., Dye C. Antiretroviral drugs for tuberculosis control in the era of HIV/AIDS. Science (New York, N.Y.) 2003;301:1535–1537. doi: 10.1126/science.1086845. 12920302 [DOI] [PubMed] [Google Scholar]
- 16.Bertrand J., Verstuyft C., Chou M., Borand L., Chea P., Nay K.H., Blanc F.X., Mentré F., Taburet A.M. Dependence of efavirenz- and rifampicin-isoniazid-based antituberculosis treatment drug-drug interaction on CYP2B6 and NAT2 genetic polymorphisms: ANRS 12154 study in Cambodia. Journal of Infectious Diseases. 2014;209:399–408. doi: 10.1093/infdis/jit466. 23990572 [DOI] [PubMed] [Google Scholar]
- 17.Blas-García A., Apostolova N., Ballesteros D., Monleón D., Morales J.M., Rocha M., Victor V.M., Esplugues J.V. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology. 2010;52:115–125. doi: 10.1002/hep.23647. 20564379 [DOI] [PubMed] [Google Scholar]
- 18.Gomez-Sucerquia L.J., Blas-Garcia A., Marti-Cabrera M., Esplugues J.V., Apostolova N. Profile of stress and toxicity gene expression in human hepatic cells treated with efavirenz. Antiviral Research. 2012;94:232–241. doi: 10.1016/j.antiviral.2012.04.003. 22554935 [DOI] [PubMed] [Google Scholar]
- 19.Apostolova N., Gomez-Sucerquia L.J., Alegre F., Funes H.A., Victor V.M., Barrachina M.D., Blas-Garcia A., Esplugues J.V. ER stress in human hepatic cells treated with efavirenz: mitochondria again. Journal of Hepatology. 2013;59:780–789. doi: 10.1016/j.jhep.2013.06.005. 23792026 [DOI] [PubMed] [Google Scholar]
- 20.Apostolova N., Gomez-Sucerquia L.J., Gortat A., Blas-Garcia A., Esplugues J.V. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology. 2011;54:1009–1019. doi: 10.1002/hep.24459. 21626526 [DOI] [PubMed] [Google Scholar]
- 21.Ramappa V., Aithal G.P. Hepatotoxicity related to anti-tuberculosis drugs: mechanisms and management. Journal of Clinical and Experimental Hepatology. 2013;3:37–49. doi: 10.1016/j.jceh.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma S., Kaplowitz N. Hepatotoxicity of antitubercular drugs. In: Kaplowitz N., DeLeve L., editors. Drug-Induced Liver Disease. Elsevier; Amsterdam: 2013. pp. 483–504. [Google Scholar]
- 23.Hoffmann C.J., Charalambous S., Thio C.L., Martin D.J., Pemba L., Fielding K.L., Churchyard G.J., Chaisson R.E., Grant A.D. Hepatotoxicity in an African antiretroviral therapy cohort: the effect of tuberculosis and hepatitis B. AIDS. 2013;21:1301–1308. doi: 10.1097/QAD.0b013e32814e6b08. [DOI] [PubMed] [Google Scholar]
- 24.Kirby D.M., Thorburn D.R., Turnbull D.M., Taylor R.W. Biochemical assays of respiratory chain complex activities. In: Pon L., Schon E., editors. Methods in Cell Biology: Mitochondria. Elsevier Science; Burlington: 2011. pp. 93–119. [DOI] [PubMed] [Google Scholar]
- 25.Lemasters J.J., Trollinger D.R., Qian T., Cascio W.E., Ohata H. Confocal imaging of Ca2+, pH, electrical potential, and membrane permeability in single living cells. Methods in Enzymology. 1999;302:341–358. doi: 10.1016/s0076-6879(99)02031-5. 12876784 [DOI] [PubMed] [Google Scholar]
- 26.Perry S.W., Norman J.P., Barbieri J., Brown E.B., Gelbard H.A. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques. 2011;50:98–115. doi: 10.2144/000113610. 21486251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zielonka J., Sikora A., Hardy M., Joseph J., Dranka B.P., Kalyanaraman B. Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chemical Research in Toxicology. 2012;25:1793–1799. doi: 10.1021/tx300164j. 22731669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Q., Harper T.W., Dierks E.A., Zhang L., Chang S., Rodrigues A.D., Marathe P. 1-aminobenzotriazole, a known cytochrome P450 inhibitor, is a substrate and inhibitor of N-acetyltransferase. Drug Metabolism and Disposition. 2011;39:1674–1679. doi: 10.1124/dmd.111.039834. 21677062 [DOI] [PubMed] [Google Scholar]
- 29.Preece N.E., Ghatineh S., Timbrell J.A. Course of ATP depletion in hydrazine hepatotoxicity. Archives of Toxicology. 1990;64:49–53. doi: 10.1007/BF01973376. 2306194 [DOI] [PubMed] [Google Scholar]
- 30.Ghatineh S., Morgan W., Preece N.E., Timbrell J.A. A biochemical and NMR spectroscopic study of hydrazine in the isolated rat hepatocyte. Archives of Toxicology. 1992;66:660–668. doi: 10.1007/BF01981506. 1336360 [DOI] [PubMed] [Google Scholar]
- 31.Hussain S.M., Frazier J.M. Cellular toxicity of hydrazine in primary rat hepatocytes. Toxicological Sciences. 2002;69:424–432. doi: 10.1093/toxsci/69.2.424. 12377991 [DOI] [PubMed] [Google Scholar]
- 32.Boelsterli, U.A., Lee, K.K., Mechanisms of isoniazid-induced liver injury—emerging role of mitochondrial stress. Journal of Gastroenterology and Hepatology; 2014: doi:10.11/jgh.12516 [DOI] [PubMed]
- 33.Pereira S.A., Wanke R., Marques M.M., Monteiro E.C., Antunes A.M. Insights into the role of bioactivation mechanisms in the toxic events elicited by non-nucleoside reverse transcriptase inhibitors. Advances Molecular Toxicology. 2012;6:1–39. [Google Scholar]
- 34.Liu K., Li F., Lu J., Gao Z., Klaassen C.D., Ma X. Role of CYP3A in isoniazid metabolism in vivo. Drug Metabolism and Pharmacokinetics. 2013 doi: 10.2133/dmpk.dmpk-13-nt-089. 24172716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Birch-Machin M.A., Turnbull D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods in Cell Biology. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. 11381612 [DOI] [PubMed] [Google Scholar]
- 36.Radi R., Cassina A., Hodara R., Quijano C., Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radical Biology and Medicine. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. 12446202 [DOI] [PubMed] [Google Scholar]
- 37.Crow J.P. Peroxynitrite scavenging by metalloporphyrins and thiolates. Free Radical Biology and Medicine. 2000;28:1487–1494. doi: 10.1016/s0891-5849(00)00249-5. 10927173 [DOI] [PubMed] [Google Scholar]
- 38.Mukhopadhyay P., Rajesh M., Bátkai S., Kashiwaya Y., Haskó G., Liaudet L., Szabó C., Pacher P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. American Journal of Physiology—Heart and Circulatory Physiology. 2009;296:H1466–H1483. doi: 10.1152/ajpheart.00795.2008. 19286953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radi R., Beckman J.S., Bush K.M., Freeman B.A. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. Journal of Biological Chemistry. 1991;266:4244–4250. 1847917 [PubMed] [Google Scholar]
- 40.Aulak K.S., Miyagi M., Yan L., West K.A., Massillon D., Crabb J.W., Stuehr D.J. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. 11593016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin L.J., Gertz B., Pan Y., Price A.C., Molkentin J.D., Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Experimental Neurology. 2009;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. 19272377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dranka B.P., Hill B.G., Darley-Usmar V.M. Mitochondrial reserve capacity in endothelial cells: the impact of nitric oxide and reactive oxygen species. Free Radical Biology and Medicine. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. 20093177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kon K., Kim J.S., Jaeschke H., Lemasters J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. 15486922 [DOI] [PubMed] [Google Scholar]
- 44.Armstrong J.S. The role of the mitochondrial permeability transition in cell death. Mitochondrion. 2006;6:225–234. doi: 10.1016/j.mito.2006.07.006. 16935572 [DOI] [PubMed] [Google Scholar]
- 45.Wrobel K., Claudio E., Segade F., Ramos S., Lazo P.S. Measurement of cytotoxicity by propidium iodide staining of target cell DNA. Application to the quantification of murine TNF-α. Journal of Immunological Methods. 1996;189:243–249. doi: 10.1016/0022-1759(95)00253-7. [DOI] [PubMed] [Google Scholar]
- 46.Atamna H., Nguyen A., Schultz C., Boyle K., Newberry J., Kato H., Ames B.N. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB Journal. 2008;22:703–712. doi: 10.1096/fj.07-9610com. 17928358 [DOI] [PubMed] [Google Scholar]
- 47.Atamna H., Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. Journal of Alzheimer’s Disease. 2010;20(Suppl. 2):S439–S452. doi: 10.3233/JAD-2010-100414. 20463399 [DOI] [PubMed] [Google Scholar]
- 48.Atamna H., Mackey J., Dhahbi J.M. Mitochondrial pharmacology: Electron transport chain bypass as strategies to treat mitochondrial dysfunction. BioFactors. 2012;38:158–166. doi: 10.1002/biof.197. 22419586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCord J.M., Fridovich I. The utility of superoxide dismutase in studying free radical reactions. II. The mechanism of the mediation of cytochrome c reduction by a variety of electron carriers. Journal of Biological Chemistry. 1970;245:1374–1377. 5462997 [PubMed] [Google Scholar]
- 50.Kelner M.J., Bagnell R., Hale B., Alexander N.M. Methylene blue competes with paraquat for reduction by flavo-enzymes resulting in decreased superoxide production in the presence of heme proteins. Archives of Biochemistry and Biophysics. 1988;262:422–426. doi: 10.1016/0003-9861(88)90393-1. 2835006 [DOI] [PubMed] [Google Scholar]
- 51.Visarius T.M., Stucki J.W., Lauterburg B.H. Stimulation of respiration by methylene blue in rat liver mitochondria. FEBS Letters. 1997;412:157–160. doi: 10.1016/s0014-5793(97)00767-9. 9257711 [DOI] [PubMed] [Google Scholar]
- 52.Hrushesky W.J.M., Olshefski R., Wood P., Meshnick S., Eaton J.W. Modifying intracellular redox balance: an approach to improving therapeutic index. Lancet. 1985;1:565–567. doi: 10.1016/s0140-6736(85)91218-8. 2857911 [DOI] [PubMed] [Google Scholar]
- 53.LoGuidice A., Boelsterli U.A. Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin D-regulated permeability transition. Hepatology. 2011;54:969–978. doi: 10.1002/hep.24464. 21626531 [DOI] [PubMed] [Google Scholar]
- 54.Bumpus N.N. Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicology and Applied Pharmacology. 2011;257:227–234. doi: 10.1016/j.taap.2011.09.008. 21958719 [DOI] [PubMed] [Google Scholar]
- 55.Kamel A., Harriman S. Inhibition of cytochrome P450 enzymes and biochemical aspects of mechanism-based inactivation (MBI) Drug Discovery Today: Technologies. 2013;10:e177–e189. doi: 10.1016/j.ddtec.2012.09.011. 24050247 [DOI] [PubMed] [Google Scholar]
- 56.Sukasem C., Chamnanphon M., Koomdee N., Puangpetch A., Santon S., Jantararoungtong T., Prommas S., Chantratita W., Manosuthi W. High plasma efavirenz concentrations and CYP2B6 polymorphisms in Thai HIV-1 infections. Drug Metabolism and Pharmacokinetics. 2013;28:391–397. doi: 10.2133/dmpk.dmpk-12-rg-120. 23399569 [DOI] [PubMed] [Google Scholar]
- 57.Destache C.J., Belgum T., Goede M., Shibata A., Belshan M.A. Antiretroviral release from poly(dl-lactide-co-glycolide) nanoparticles in mice. Journal of Antimicrobial Chemotherapy. 2010;65:2183–2187. doi: 10.1093/jac/dkq318. 20729545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blair I.A., Mansilla Tinoco R., Brodie M.J., Clare R.A., Dollery C.T., Timbrell J.A., Beever I.A. Plasma hydrazine concentrations in man after isoniazid and hydralazine administration. Human Toxicology. 1985;4:195–202. doi: 10.1177/096032718500400210. 4007883 [DOI] [PubMed] [Google Scholar]
- 59.Gent W.L., Seifart H.I., Parkin D.P., Donald P.R., Lamprecht J.H. Factors in hydrazine formation from isoniazid by paediatric and adult tuberculosis patients. European Journal of Clinical Pharmacology. 1992;43:131–136. doi: 10.1007/BF01740658. 1425868 [DOI] [PubMed] [Google Scholar]