

Abstract
Ozone (O3) is a serious public health concern. Recent findings indicate that the damaging health effects of O3 extend to multiple systemic organ systems. Herein, we hypothesize that O3 inhalation will cause downstream alterations to the liver. To test this, male Sprague-Dawley rats were exposed to 0.5 ppm O3 for 8 h/day for 5 days. Plasma liver enzyme measurements showed that 5 day O3 exposure did not cause liver cell death. Proteomic and mass spectrometry analysis identified 10 proteins in the liver that were significantly altered in abundance following short-term O3 exposure and these included several stress responsive proteins. Glucose-regulated protein 78 and protein disulfide isomerase increased, whereas glutathione S-transferase M1 was significantly decreased by O3 inhalation. In contrast, no significant changes were detected for the stress response protein heme oxygenase-1 or cytochrome P450 2E1 and 2B in liver of O3 exposed rats compared to controls. In summary, these results show that an environmentally-relevant exposure to inhaled O3 can alter the expression of select proteins in the liver. We propose that O3 inhalation may represent an important unrecognized factor that can modulate hepatic metabolic functions.
Keywords: Ozone, Liver, Proteome, Cytochrome P450, Stress response proteins, Endoplasmic reticulum stress
Graphical abstract
Highlights
-
•
Rats were exposed to filtered air (FA) or 0.5 ppm ozone (O3) 8 h/day for 5 days.
-
•
Using this exposure protocol, O3 caused no detectable lung injury or liver cell death.
-
•
O3 altered the expression of some drug metabolism and stress proteins in liver.
Introduction
Approximately 50% of the U.S. population resides in areas where ambient ozone (O3) concentrations exceed the current 0.075 ppm 8 h time average set by the National Ambient Air Quality Standards (NAAQS) [1]. O3 is a primary component of photochemical smog and is a serious public health concern especially when air quality is poor. Persons particularly sensitive to O3 exposure include the elderly, young children, and those with pre-existing pulmonary diseases such as asthma and chronic obstructive pulmonary disease. It is established that exposure to high levels of O3 decreases lung function, increases pulmonary hyper-reactivity, causes airway epithelial cell damage and remodeling, and increases epithelial permeability [2–4].
In addition to adverse pulmonary effects, emerging evidence shows that O3 inhalation can cause tissue injury and altered metabolism in other systemic organ systems. For example, studies in nonhuman primates show that postnatal episodic O3 exposure only during infancy (30 days to 6 months of age) resulted in long-term effects to the systemic innate immune system that were detectable up to 1 yr of age [5]. Recent epidemiologic studies report a positive correlation of air pollution exposure with increased cardiovascular-related morbidity and mortality resulting in increased hospital admissions related to cardiac events [6–9]. Additional studies reported that successive days of high ambient O3 exposure correlated with increased blood pressure, blood lipids, and decreased glucose tolerance in humans [8]. Ballinger, Postlethwait, and colleagues showed that exposure to 0.5 ppm O3 induced vascular dysfunction and increased aortic mitochondrial DNA damage in healthy wild-type mice, and increased progression of atherosclerosis in mice genetically predisposed to cardiovascular disease [10]. Moreover, a single 5-day regimen of O3 also increased mtDNA damage in the abdominal aorta of infant nonhuman primates [10]; a model more closely mimicking human exposures. This work is important because not only does it provide direct evidence that O3 inhalation damages the cardiovascular system, but also shows that O3 has the potential to exacerbate cardiovascular disease including atherosclerosis [11].
O3 inhalation has also been shown to induce metabolic changes in other peripheral organ systems. For example, pentobarbital-induced sleeping times increased after O3 exposure, suggesting alterations in hepatic drug metabolism and clearance mechanisms [12,13]. Since this early work, other groups have addressed the effect of inhaled O3 on the liver. Last and colleagues, using a microarray approach, examined the effects of O3 (1.0 ppm) on the liver transcriptome and showed that O3 inhalation significantly decreased the mRNA levels of several xenobiotic, carbohydrate, and fatty acid metabolism genes [14]. Other groups have shown that acute exposures of O3 (1.0–2.0 ppm) for 3 h increased nitric oxide production in isolated hepatocytes, as well as increased rates of protein synthesis [15]. Herein, our goal was to determine whether inhalation of O3 at a concentration lower than used in previous studies, alters the hepatic proteome and specific xenobiotic metabolizing enzymes in an animal model without pre-existing disease.
Materials and methods
O3 exposure protocol
Male Sprague-Dawley rats were purchased from Harlan Laboratories (Barrier 217 VAF, Indianapolis, IN) and were provided standard rat chow and water ad libitum. Animals were housed two per cage under barrier conditions and maintained using a standard 12 h light-dark cycle. Rats were exposed to either filtered air (FA) or ozone (O3) at 0.5 ppm for 8 h/day for 5 days between 9:00 AM to 5:00 PM in the University of Alabama at Birmingham (UAB) Environmental Exposure Facility. Standard laboratory rat chow was removed from cages before the start of exposures to prevent rats from ingesting food that may contain oxidized nutrients (e.g., lipids, thiols, antioxidants) as a consequence of O3 reaction. Rat chow was also removed from cages of FA exposed rats. Fresh food was returned to cages after exposures and present throughout the entire dark period when rats are active and consume food. The exposure protocol had no effect on body weight (FA: 348±4 g and O3: 341±15 g, p=0.69). Prior to exposures, animals were acclimated to the chambers for at least 72 h before beginning FA and O3 exposures to minimize stress associated with a novel environment. Importantly, rats remained in their home cage with bedding and water, with cages (stainless steel wire mesh coverings) placed into the exposure chamber. O3 was generated from 100% O2 using a model OZ1PCS-V/SW O3 generator (Ozotech Inc, Yreka, CA), mixed with FA, and flowed into 0.8 m3 stainless steel chambers (~22 °C, 50% relative humidity; 30 volume changes/hr). O3 concentrations were continuously monitored using a Thermo-Environmental Model UV Photometric analyzer (Thermo-Environmental, Franklin, MA). Measurements obtained inside the cages demonstrated that O3 concentrations in the animals breathing zones were equivalent to the chamber bulk phase concentrations. FA controls were housed in separate chambers receiving the same clean air supply at equal air exchange rates throughout the duration of the exposure regimen. Animals were anesthetized with a 50 mg/kg body weight (i.p.) injection of sodium pentobarbital and euthanized via exsanguination. Tissues were harvested within 1 h of cessation of the exposure. All procedures were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Bronchoalveolar lavage and cell differential analysis
A cannula was inserted into the trachea via a midline tracheotomy, the chest cavity opened via a midline thoracotomy, and 9 mL of warmed (37 °C) phosphate buffered saline (pH 7.0; 310 mOsm) was gently instilled and withdrawn 3 times to yield bronchoalveolar lavage (BAL) [16]. The BAL was subjected to a standard cytospin procedure (StatSpin Cytofuge, Norwood, MA) to determine cell counts and types. Slides were stained with a modified Wright-Giemsa stain and 300 cells (100 cells per lane) were counted under a light microscope using characteristics unique to each cell type [17]. Total cell counts were averaged for each cell type and percent of total cells calculated. The remaining BAL was centrifuged to generate BAL fluid (BALF) and protein concentrations in the BALF were measured to determine whether altered epithelial permeability [18] was detectable after the 5th period of O3.
Plasma chemistries for liver enzymes and histology
Blood was collected via the abdominal aorta using a heparin-coated syringe and centrifuged at 4 °C at 2000g for 10 min to obtain plasma. Alanine and aspartate aminotransferase (ALT and AST, respectively) activities were measured in plasma using a spectrophotometric assay per manufacturer's directions (Pointe Scientific, Inc, Canton MI). Briefly, NAD+ oxidation was recorded under constant temperature conditions (37 °C) over a time period of 10 min at 340 nm. The rate was measured and ALT and AST levels reported as international units/liter (IU/L). Liver tissue was fixed in formalin and paraffin embedded. Sections were mounted on slides and stained with hematoxylin and eosin (H&E). Slides were scored for injury, steatosis, and inflammation by a pathologist blinded to the experimental groups.
Two dimension isoelectric focusing/SDS-PAGE (2D IEF/SDS-PAGE)
Livers were excised and homogenized in ice-cold 0.25 M sucrose, 5 mM Tris–HCl, and 1 mM EDTA, pH 7.4, containing protease inhibitors [phenylmethylsulfonyl fluoride (40 µg/mL), leupeptin (5 µg/mL), and pepstatin A (7 µg/mL)] [19]. Protease inhibitors were included to prevent sample degradation prior to proteomic analyses. For proteomic studies there were six FA (control) and six O3 exposed rats per group. Post-nuclear supernatant fraction was prepared by centrifugation of liver homogenates at 568g for 10 min at 4°C. Protein concentrations were determined using the Bradford protein assay and bovine serum albumin (BSA) as a standard [20]. Proteomic analyses were performed by methods as previously described [21]. Post-nuclear supernatant (100 µg) from liver homogenates was added to IEF gel strip rehydration buffer containing 7 M urea, 2 M thiourea, 2% (w/v) CHAPS, 0.5% (w/v) n-dodecyl-ß-D-maltoside, 0.002% (w/v) bromophenol blue, ampholine electrophoresis reagent (Sigma, St. Louis, MO, range pH 3–10), 0.04 M DTT and 2 mM tributylphosphine. Following protein extraction, samples were applied to IEF gel strips (Invitrogen ZOOM Strips, pH 3–10, Carlsbad, CA) and rehydration of IEF strips was done overnight. For SDS-PAGE, IEF gel strips were placed horizontally on top of a 10% resolving gel with 4% stacking gel, and sealed into place using warm agarose (1%, w/v) and gels were run at 100 V for . After electrophoresis, gels were stained with Sypro Ruby™ (Invitrogen, Carlsbad, CA) for total protein. Protein stained gels were imaged using a Bio-Rad ChemiDoc XRS imaging system (Bio-Rad Laboratories, Inc, Hercules, CA).
2D gel image analyses
Methods for gel analyses are described in our previous work [21]. Differences in protein density were performed using PDQuest Image Analysis software (Bio-Rad, Hercules, CA). Individual protein spots on all twelve 2D gels (n=6 for FA and n=6 for O3) were identified using the program software and visually matched for accuracy. A master gel was created to serve as the reference gel for FA and O3 groups, which the PDQuest software program uses automatically to match the spots across all gels. Manual verification was then used to correct for incorrect spot matching to the reference gel or spots not initially detected by software. In order to correct for any differences in protein loading, the density for all spots in each gel was normalized to total density in verified protein spots for that particular gel. The protein densities were then entered into a spreadsheet and statistically significant differences were determined using an unpaired t-test (two-tail).
Protein identification with matrix assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry
Protein identification was performed as previously described [21]. Protein spots were cut from gels and processed using standard methods in the UAB mass spectrometry shared facility (http://www.uab.edu/proteomics). Briefly, protein samples were de-stained with three successive 30 min washes in 1:1 solution of 50 mM NH4HCO3 and acetonitrile. Samples were treated with 10 mM dithiothreitol in 50 mM NH4HCO3 for 60 min at 60 °C to reduce cysteine residues, which was followed by alkylation of free cysteines with 55 mM iodoacetamide in 50 mM NH4HCO3 for 60 min. This was followed by 16 h incubation at 37 °C with trypsin to digest proteins. The resulting solution was extracted by two successive 30 min washes in a 1:1 solution of 5% formic acid and acetonitrile, supernatants were collected, and dried using a Savant SpeedVac. Lyophilized samples were resuspended in 0.1% formic acid, desalted (C18 ZipTips, Millipore), and diluted 1:10 with a saturated solution of α-cyano-4-hydroxycinnamic acid matrix before being applied to MALDI-TOF plates. Samples were analyzed with a Voyager De-Pro mass spectrometer in the positive mode and resulting spectra were analyzed using Voyager Explorer software. Peptide masses were submitted to the Mascot database for protein identification (www.matrixscience.com). Mascot uses a probability based system of the Mowse algorithm to determine significant results [22]. Mowse is an algorithmic calculation used to assign statistical weight to each peptide match; thus, a higher Mowse score implies higher statistical likelihood of the match being correct. Herein, the Mascot results for proteins IDs ranged from 132–467. All scores were>40 indicating extensive homology and significant matches for this study (p≤0.05).
Western blotting
Based on gene expression results from other studies [12–14] we decided to examine the effect of inhaled O3 on select cytochrome P450 (CYP450) enzymes and the stress-inducible enzyme heme oxygenase 1 (HO-1) in the liver. Equal amounts of protein from post-nuclear supernatant fractions (40 or 50 µg) were loaded and separated on 12% SDS-PAGE gels then transferred for 1 h to nitrocellulose membranes. Membranes were blocked overnight in 5% (w/v) non-fat milk in Tris base saline solution with Tween-20 (TBS-T) while shaking at 5 °C. Levels of HO-1 protein were detected using a 1:5,000 dilution of primary antibody (Stressgen, Ann Arbor, MI). Levels of CYP2E1 and CYP2B were detected using 1:1,000 primary antibody dilutions (Chemicon International, Temecula, CA). After successive washes in TBS-T, blots were incubated in horseradish peroxidase (HRP) conjugated anti-rabbit secondary antibody (1:10,000) in 5% (w/v) non-fat milk in TBS-T for 1 h (Sigma, St. Louis, MO). Bands were visualized using luminol-based chemiluminescence reagents with a ChemiDoc XRS System (Bio-Rad, Hercules, CA) and quantification of band densities were achieved using Quantity One software (Bio-Rad, Hercules, CA) [23]. To ensure equal protein loading of lanes, GAPDH was used as a loading control at a dilution of 1:1000 anti-GAPDH primary antibody. A representative example of a CYP2B blot reprobed for GAPDH is shown in Fig. 2D. Further, equal protein loading of gels was confirmed using total protein staining (e.g., SYPRO® Ruby protein gel stain: FA=34,875±4241 and O3=36,079±3337 arbitrary density units; n=6, p=0.59) for blots not reprobed with GAPDH.
Fig. 2.
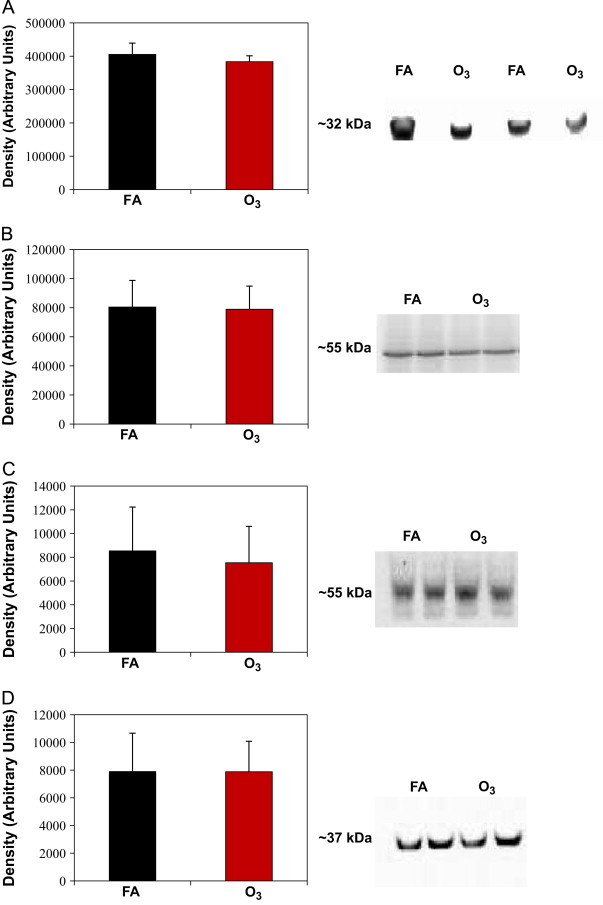
Protein levels of HO-1, CYP2E1, and CYP2B in liver of O3 and FA rats. Rats were exposed to filtered air (FA) or O3 (0.5 ppm) for 8 h/day for 5 days. After exposures, livers were removed and homogenates were analyzed via western blot technique for (A) HO-1, (B) CYP2E1, (C) CYP2B, and (D) GAPDH as a loading control. O3 exposure did not significantly alter protein expression for all 4 proteins measured (p>0.05). Full-size images of western blots were cropped to highlight protein bands of interest. Continuous gel images are shown with approximate molecular weights provided next to blot image. Data represent mean±SD for n=6 animals per group. HO-1, heme oxygenase-1; CYP2E1, cytochrome P450 2E1; CYP2B, cytochrome P450 2B; and GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Statistical analyses
Statistical significance for all measured parameters was assessed using an unpaired t-test and significance was determined with a p value≤0.05.
Results
Lung cell differential and BALF protein
BALF from FA and O3 exposed rats was assessed for epithelial permeability and inflammatory cell infiltration as described in Methods. No significant change in BALF protein concentration were observed between FA and O3 exposed groups; 63.0±27.9 and 72.9±46.2 μg/mL, respectively (p>0.05). Cell differential analysis also revealed that macrophage, lymphocyte, neutrophil, or basophil airspace populations in O3 exposed rats did not differ from FA controls at the end of the 5 days of O3 exposure protocol (Table 1).
Table 1.
Bronchoalveolar lavage cell differential analysis.
| Filtered air | Ozone | p value | |
|---|---|---|---|
| Volume recovered (mL) | 6.54±0.52 | 6.23±0.48 | 0.32 |
| % Yield | 73±7.00 | 69±7.00 | 0.32 |
| Cell counta | |||
| Macrophages | 99.2±0.76 | 97.5±1.95 | 0.10 |
| Neutrophils | 1.30±1.37 | 2.20±0.68 | 0.75 |
| Lymphocytes | 0.33±0.82 | 0.42±1.90 | 0.23 |
| Basophils | 0.00±0.00 | 0.17±0.40 | 0.34 |
Lavage fluid was recovered as described in Methods.
Cell counts were generated using the average of 3 independent counts of 100 total cells/count and are represented as a percentage of total counted cells.
Effect of O3 exposure on plasma liver enzymes and histology
Plasma ALT and AST enzyme activities were measured as indicators of hepatocellular damage due to O3 exposure. After 5 days of 0.5 ppm O3 exposure, AST enzyme activity levels for FA and O3 groups were 47.2±2.3 and 55.2±13.2 IU/L, respectively. Similarly, ALT enzymes activity levels for FA and O3 groups were 48.9±3.7 and 51.0±8.9 IU/L, respectively. No statistically significant changes were observed in liver enzyme activities in the O3 treated group compared to the FA group (p>0.05). Additionally H&E-stained liver sections scored for changes to liver morphology, inflammation, and injury showed no pathology and no differences between O3 and FA groups (data not shown). These results demonstrate that this inhaled O3 exposure regimen did not cause detectable toxicity to the liver.
Effect of ozone exposure on liver proteome
Representative 2D IEF/SDS-PAGE gel images from both FA and O3 treatment groups are shown in Fig. 1. A global 2D proteomic approach was performed to determine whether O3 inhalation altered liver protein abundance. Following separation of proteins in two dimensions (isoelectric point and molecular weight), proteins were analyzed for differences in abundance using PDQuest software tools as described in Methods. Using this mini-gel 2D system, we were able to detect approximately 275 individual proteins spots on each gel. O3 exposure resulted in a significant change in the expression of 23 proteins as compared to FA control. Of these 23 proteins, mass spectrometry analyses identified 10 proteins. A master map showing the location of the identified proteins and their assigned spot numbers is provided in Fig. 1A. A comparison of the protein densities of these 10 proteins is shown graphically in Fig. 1B. Unique protein identifications with statistical analysis (% change and p values) and mass spectrometry details are provided in Table 2. Each protein spot shown in the master map matched the expected molecular weight and isoelectric point of each identified protein (Table 2). Total protein density from 2D gels is presented in Fig. 1C and shows that changes in individual protein density were not due to differences in total protein loading of gels. The 10 identified proteins were categorized into 4 broad groups: cytoskeletal, energy metabolism, drug metabolism, and protein folding/ER stress (Table 3).
Fig. 1.
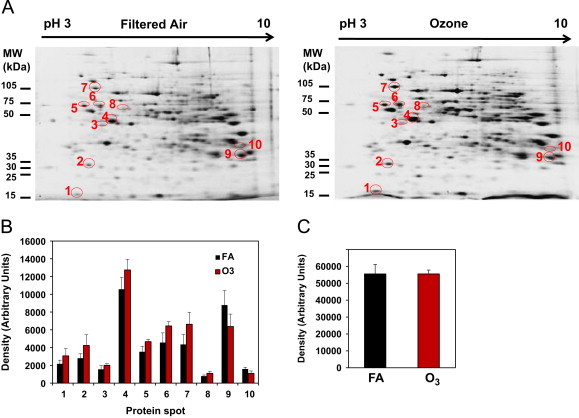
Master map of liver proteins differentially altered by inhaled O3. Rats were exposed to filtered air (FA) or O3 (0.5 ppm) for 8 h/day for 5 days. After exposures, livers were removed and the post-nuclear supernatant was analyzed for global protein expression using 2D gel proteomics and mass spectrometry techniques. (A) Shows the representative 2D gels of proteins from liver of FA and O3 exposed rats. Note that the circled protein “spots” were identified and are listed in Table 2 with respective p values. (B) Shows the change in abundance (increase or decrease) of liver proteins found to be altered by inhaled O3. The number below each pair of bar graphs corresponds to the protein spot shown in the master map (panel A) and match numbers used in Table 2. (C) Total protein density calculated from 2D gels generated from FA and O3 groups. This result shows equal protein loading across all gels (p>0.05). Data represent mean±SD of n=6 animals per group.
Table 2.
Hepatic proteins significantly altered in abundance as a result of 0.5 ppm O3 exposure: results from 2D IEF/SDS-PAGE and mass spectrometry.
| Spot # | Protein identification | Mass (kDa) | Isoelectric point (pI) | % Change | p value | MOWSE score | Peptides matched |
|---|---|---|---|---|---|---|---|
| 1 | Cytochrome b5 type A (microsomal) | 11.4 | 5.26 | 41.6 | 0.038 | 166 | 6 |
| 2 | Catechol-O-methyltransferase | 29.8 | 5.41 | 50.9 | 0.025 | 132 | 5 |
| 3 | Galactokinase 1 | 42.8 | 5.24 | 29.7 | 0.036 | 286 | 8 |
| 4 | Actin, beta like 2 | 42.2 | 5.30 | 20.6 | 0.014 | 277 | 13 |
| 5 | Protein disulfide-isomerase | 57.3 | 4.82 | 32.3 | 0.002 | 784 | 34 |
| 6 | Tubulin, alpha 1C | 50.6 | 4.96 | 41.1 | 0.003 | 336 | 16 |
| 7 | Glucose-regulated protein 78 | 72.5 | 5.07 | 52.0 | 0.002 | 467 | 15 |
| 8 | Glycerol kinase | 58.2 | 5.49 | 40.6 | 0.01 | 220 | 7 |
| 9 | Glutathione S-transferase mu 1 | 25.8 | 8.27 | −27.5 | 0.021 | 350 | 15 |
| 10 | D-β-hydroxybutyrate dehydrogenase, type I | 38.7 | 8.93 | −31.6 | 0.033 | 187 | 8 |
Proteins identified as significantly altered were matched to all Filtered Air (FA) and Ozone (O3) 2D IEF/SDS-PAGE gels. Spot # is the same number used to identify circled proteins in Fig. 1A and densities shown in Fig. 1B. Data represent p values determined using a Student's unpaired t-test (two-tail).
Table 3.
Description of cellular pathways and function of identified hepatic proteins altered O3 inhalation.
| Protein Identification | Accession # | Pathway | Function |
|---|---|---|---|
| Cytochrome b5 | GenBank:AAB67609 | Drug metabolism | Cypb5 mediates the rate of P450 dependent mono-oxygenation reactions through the transfer of the second electron from NADPH. |
| Catechol-O-methyltransferase | GenBank:NP_036663 | Drug metabolism/L-dopa metabolism | COMT enzymatically O-methylates catechol containing compounds. Both over expression and loss of activity has been implicated in vascular diseases as well as neuronal disorders. |
| Galactokinase 1 | GenBank:NP_001008283 | Cell cycle, energy metabolism | First enzyme required for the conversion of galactose to glucose forming galactose-1-phosphate. |
| Actin, beta like 2 | GenBank:NP_001099879 | Cytoskeletal | Important for cell motility, structure, and integrity. |
| Protein disulfide-isomerase | GenBank:NP_037130 | Protein Folding/ER stress | PDI is responsible for inter and intra molecular disulfide bond formation within the endoplasmic reticulum. |
| Tubulin, alpha 1C | GenBank:NP_001011995 | Cytoskeletal | Alpha tubulin is one of two components of tubulin which is assembled to form microtubules. These are important for cell movement and maintenance of shape. |
| Glucose-regulated protein 78 | GenBank:NP_037215 | Protein Folding/ER stress | Located within the ER; GRP78/BiP binds and inactivates the proteins responsible for UPR activation. Also directly binds misfolded proteins to assist in proper folding or clearance. |
| Glycerol kinase | GenBank:NP_077357 | Energy metabolism | Catalyzes formation of glycerol 3-phosphate. Overexpression in H4IIE cells resulted in increased fat storage and alters activity of PPAR-alpha and other transcription factors. |
| Glutathione S-transferase mu 1 | GenBank:AAA41286 | Phase II drug metabolism | Facilitates conjugation of reduced glutathione to electrophilic compounds for increased excretion. |
| D-β-hydroxybutyrate dehydrogenase | GenBank:AAB59684 | Energy/lipid metabolism | Oxidoreductase, mitochondrial. Involved in fatty acid catabolism. BDH1 facilitates formation of the ketone body 3-hydroxybutyrate. |
Protein folding/ER stress proteins
Two proteins involved in ER stress were increased by O3 exposure. Protein disulfide isomerase (PDI) increased by 32% and glucose-regulated protein 78 (GRP78) increased by 52% in liver of O3 exposed rats compared to protein levels measured in FA controls. PDI assists in the proper folding and disulfide bond formation of proteins within the ER [24]. GRP78, a chaperone protein located in the ER, plays an important role in the regulation of the unfolded protein response activated during ER stress [25].
Drug metabolism proteins
Proteins that demonstrated significant expression changes due to O3 exposure compared to FA control samples included microsomal cytochrome b5, catechol-O-methyltransferase (COMT), and glutathione-S-transferase mu 1 (GSTM 1). Cytochrome b5 increased in expression by 43% in liver of O3 exposed rats compared to FA controls. This protein is important for CYP450 enzymatic reactions involving fatty acid desaturation, metabolism of xenobiotics, and cholesterol synthesis and breakdown [26]. COMT protein, a highly abundant liver protein, increased by 50% in liver of O3 exposed rats. COMT catalyzes O-methylation of catechol containing molecules [27] and has been implicated in pathologies such as vascular disease and Parkinson's disease due to the generation of reactive catecholamine semiquinone species [28]. GSTM 1 decreased in expression by 27% in liver of O3 exposed rats compared to FA controls. GSTM 1 is a phase II xenobiotic metabolism enzyme responsible for the conjugation of glutathione groups to endogenous and exogenous compounds for excretion [29]. This enzyme is largely considered a hepatoprotective enzyme due to its ability to conjugate reactive metabolites produced as byproducts from oxidative stress mechanisms [29].
Energy metabolism and cytoskeletal proteins
Three proteins involved in energy metabolism, galactokinase, glycerol kinase, and β-hydroxybutyrate dehydrogenase 1 (BDH1) were increased by O3 exposure. Both galactokinase and glycerol kinase increased in expression by 41% and 30%, respectively, in response to O3, whereas BDH significantly decreased in expression by 32% in liver of O3 exposed rats compared to FA controls. BDH1 is a mitochondrial protein that catalyzes the first step of ketone body metabolism, converting β-hydroxybutyrate to acetoacetate [30]. Cytoskeletal proteins that significantly increased in response to O3 exposure were β-actin and α-tubulin, which are involved in cell cycle, division, and motility [31].
Effect of O3 exposure on CYP450 and HO-1 protein levels
Previous studies showed that high O3 exposure levels (1–3 ppm) prolonged pentobarbital-induced sleeping times in rodents, suggesting a decrease in xenobiotic metabolism [13,32]. Herein, protein levels of CYP2E1 and 2B were measured to determine if lower, more environmentally relevant exposures of O3 (0.5 ppm) altered key drug metabolizing enzymes. After 5 days of 0.5 ppm O3 exposure, protein expression levels of CYP2E1 or CYP2B were not significantly changed as compared to FA controls (Fig. 2B and C). Additionally, the stress inducible form of heme oxygenase, HO-1, was measured in this study. Analysis showed no significant difference in HO-1 protein levels between FA and O3 groups at the end of 5 exposure periods (Fig. 2A).
Discussion
A new disease paradigm has emerged in the field of environmental health sciences suggesting that inhalation of environmental pollutants, like O3, can exacerbate diseases that extend beyond the pulmonary system; the initial site of exposure. Importantly, O3 inhalation may initiate a cascade of events leading to tissue perturbations in other organ systems, including cardiovascular, hematopoietic, and hepatic systems. In support of this new concept, particulate matter (PM) and other gaseous pollutants generated from industry and diesel exhaust exacerbate not only pulmonary injury, but also cardiovascular disease [6]. Epidemiological studies also report increased cardiovascular risk from high ambient O3 days with significant changes in blood lipids, blood pressure, and factors important for glucose metabolism [8]. While human exposures to inhaled pollutants typically involve heterogeneous mixtures of PM and O3, animal studies show that O3 alone can damage cardiovascular tissues and cause hepatic changes [10,14]. Importantly, in nonhuman primates exposure to O3 also induces cardiovascular injury [10] and induces long-term alterations in immune function [5].
The molecular events responsible for O3 induced systemic effects are poorly defined. The reaction of O3 within the lung is complex and results in the production of a diverse array of biologically active products. O3 reacts with mono-unsaturated fatty acids to produce multiple aldehyde and hydroxyhydroperoxide species, as well as small amounts of the Criegee ozonide [33,34]. Studies by Murphy, White, and colleagues demonstrated that the reaction of O3 with calf lung surfactant resulted in the formation of biologically active oxidized phospholipid species that reduced cell viability by both necrotic and apoptotic mechanisms [35]. Moreover, Friedman and colleagues have demonstrated that multiple lipid ozonation products activate cellular signaling pathways and induce inflammatory mediators [36].
As most air pollution episodes last several days, single event exposures in experimental laboratory studies are not environmentally relevant. Most exposures are also not continuous but occur for several hours per day for several consecutive days. For these reasons, we designed an O3 exposure model that encompassed these features; i.e., 8 h per day for 5 consecutive days. Further, the exposure regimen used in our current study better replicates a typical day where ambient O3 concentrations have increased beyond the current NAAQS standard of 0.075 ppm. The concentration of O3 used in the current study is 0.5 ppm, which is higher than the NAAQS standard. Notably, most experimental animal studies use O3 concentrations in the 0.8–3.0 ppm range to examine health impacts. Higher doses of O3 are used in experimental studies for a variety of reasons [11]. First, it is generally accepted that because rats are obligate nose breathers and that they have a more complex nasopharyngeal architecture that facilitates removal of inhaled reactive gases; concentrations above those encountered by humans during typical ambient exposures should be employed. Second, O3 exposures with animals are typically done during the day when rats are less active/asleep (i.e., the light period of a 12 h light:12 h dark cycle). Consequently, the combination of nose breathing and low physical activity function to reduce the dose delivered to the lower respiratory track of rats. These factors will ultimately result in a lower composite dose of O3 reaching the lungs in experimental animals as compared to typical human O3 exposures.
Following a 5 day O3 exposure protocol, we did not observe any significant changes to overall protein levels or inflammatory cells within BALF extracted from lungs (Table 1). Increased protein in BALF would indicate significant epithelial damage and loss of barrier integrity. It has been shown that repeated O3 exposures increase neutrophils, lymphocytes, and macrophages in the lungs. In rodents, neutrophilic inflammation generally begins to resolve by day 3 following a 1.0 ppm O3 exposure [37]. We also did not detect changes in plasma levels of ALT or AST enzyme activities; common measurements for hepatocyte necrosis. Taken together, these findings indicate that by the 5th day of O3 exposure, injury may have resolved sufficiently to reduce injury metrics below detectable levels. Similarly, we observed no change in CYP2E1 and 2B protein levels following O3 exposure, which supports recent results showing no change in gene expression for these two CYP450 enzymes following inhaled O3 exposure [14]. In addition to looking at CYP450s, we measured expression of another key stress responsive protein HO-1; the inducible member of the heme-oxygenase enzyme family. Herein, we saw no change in HO-1 protein after a 5 day O3 exposure regimen. The absence of any change in HO-1 expression could be anticipated considering HO-1 up-regulation typically occurs very early in response to stress. Further, it is important to note that we measured HO-1 levels only at one time point. If O3 exposure elicited an early hepatic stress response (e.g., HO-1 induction) at any time before 5 days it would have been missed in this study.
Despite the absence of overt lung and liver toxicity at Day 5, O3 exposure did alter the levels of other key proteins in the liver as evidenced by our proteomics analyses. Considering that only a few studies have investigated the effects of inhaled O3 on the liver [13–15] we took a global proteomic approach to determine whether O3 exposure alters expression of liver proteins. Moreover, we propose that investigating biological events and/or alterations that occur after pulmonary inflammation has resolved is a more relevant question for understanding human health impacts from inhaled pollutants. Specifically, it is more important to determine whether systemic alterations and/or pathological events induced by O3 are still obvious after pulmonary inflammation has resolved versus simply looking at downstream systemic sequelae that occur at the same time as the toxic insult.
Using 2D gel electrophoresis techniques and mass spectrometry we detected over 20, and successfully identified 10, proteins that were significantly altered by O3 inhalation. These proteins were grouped into 4 different categories: cytoskeletal, drug metabolism, energy metabolism, and ER stress-related proteins; with 8 proteins significantly increased and 2 decreased in response to inhaled O3 exposure. Two of the most intriguing changes observed from our mass spectrometry data are the increases in PDI and GRP78. Both proteins are located in the endoplasmic reticulum and are involved in the proper folding of proteins and maintenance of ER homeostasis. The ER is a complex intracellular membranous network acting as the site for proper folding and maturation of newly synthesized proteins. It is also sensitive to a variety of signals and tightly regulated in order to maintain proper protein and lipid homeostasis. One such signal is excess accumulation of misfolded proteins, which activates the unfolded protein response (UPR). GRP78 binds all three proteins responsible for the activation of UPR (IRE1α, ATF6, and PERK), rendering the signaling pathway inactive [24]. GRP78 is believed to be released from the previous three proteins responsible for UPR activation and is sequestered in the ER lumen where it binds misfolded proteins and subsequently the UPR is activated [38]. An upregulation of GRP78 suggests that a mild form of ER stress occurred by the Day 5 time point in order to bring the ER lumen back to homeostasis. Indeed, overexpression of GRP78 protects cells from known stressors such as a high fat diet; alleviating injury and promoting restoration of ER homeostasis [39,40]. Additionally, studies investigating the effect of PM exposure show induction of ER stress pathways in the liver of animal models with pre-existing liver disease [41].
Despite no detectable effects on CYP2E1 and 2B protein levels we did observe changes in other drug metabolism enzymes. We saw a 50% increase in COMT protein levels. COMT, a phase II drug metabolism enzyme, O-methylates catechol-containing compounds including dietary phytochemicals and metabolites of toxic aryl hydrocarbons [28]. Increases in COMT may be a protective adaptive response because accumulation of endogenous catecholamines can spontaneously or enzymatically be converted to semiquinone and quinone intermediates that are cytotoxic [28,42]. In contrast, we saw a decrease in liver GSTM 1 in response to O3 exposure. The GST super-family consists of phase II drug metabolism enzymes and GSTM 1 is one of seven GST M isoforms [29]. Substrates for GSTs include environmental chemicals, like acrolein and DDT, as well as, numerous drugs and endogenous reactive lipid species [43]. Conjugation of GSH with these compounds typically produces less reactive products that are subsequently excreted; thus, ameliorating cellular oxidative stress and toxicity. GST enzymes are typically increased in response to stress and considered a conserved adaptive response as it occurs throughout the animal kingdom [44]. Interestingly, we observed that hepatic GSTM 1 protein was significantly decreased by O3 inhalation. While several studies have reported that the GSTM 1 deficiency is associated with decreased lung function and airway inflammation following O3 exposure [45]; the impact of O3 and GSTM 1 deficiency on the liver is not known. Animal models null for GSTM 1 display altered hepatic drug clearance [46]. In the context of the current study, decreased GSTM 1 protein in the liver after O3 inhalation could impair adaptive stress responses and/or impair hepatic drug metabolism and clearance mechanisms. Importantly, this could manifest as a serious problem in individuals with GSTM 1 polymorphisms exposed to O3. Indeed, individuals with the GSTM 1 null polymorphism are at higher risk for hepatocellular carcinoma, alcoholic liver disease, and obesity associated fatty liver disease [47–50]. A proteomic analysis of alcohol-induced liver injury found that chronic, binge ethanol administration decreased GSTM 1 levels in liver [51]. Therefore, the failure of key adaptive stress responses may be critical in how liver responds and adapts to O3 [52]. Finally, β-actin and α-tubulin, were found to be altered by O3. Adduction of these proteins by reactive lipid species and acetaldehyde disrupts the cytoskeletal network in hepatocytes [31] with increases in transcript and protein observed in an animal model of pancreatic injury [53]. It was postulated that these increases play a protective role by increasing the amount of cytoskeletal protein needed to replace damaged or modified β-actin and α-tubulin in the cell. Taken together, these results suggest that inhaled O3 alters protein expression patterns in an organ (liver) distal from initial site of exposure.
Conclusion
In summary, we report that an environmentally-relevant inhalation exposure to O3 alters the liver proteome. We observed significant changes in the expression of key stress response and drug metabolism proteins in the liver following O3 inhalation. While it is well established that O3 can exacerbate pulmonary diseases, an emerging literature indicates that inhaled O3 may have significant effects on other organ systems, including the liver. Thus, it will be important to better understand how these systemic effects could impact health. Importantly, the systemic effects observed with O3 alone may be exacerbated when coupled to pre-existing diseases and pathologies. For example, animals predisposed to heart disease display greater vascular injury when exposed to O3 [10]. In addition, rodents administered acetaminophen, a known hepatotoxicant, have increased liver injury following O3 exposure [54]. Also, genetic predisposition to obesity in multiple animal models modulates airway hypersensitivity and inflammatory cytokines as compared to unexposed obese controls [55–57]. Together, the results from the current study provides new insight into the growing field of environmental-related disease and health impacts from inhaled pollutants and suggest that the liver is a systemic site impacted by O3.
Funding
This work was supported by the National Institute of Health grants to SMB (R01 AA15172 and R01 AA18841) and EMP (P01 ES111617 and R01 HL54696).
Acknowledgments
The authors would like to thank Dr. S. Barnes and Mr. L. Wilson of the UAB Mass Spectrometry Shared Facility. Mass spectrometers in the Shared Facility came from funds provided by NCRR Grants S10RR11329 and S10RR13795 plus UAB HSF General Endowment Fund. Operational funds came from the UAB Comprehensive Cancer Center Core Grant (P30CA13148), Purdue-UAB Botanicals Center for Age-Related Disease (P50AT00477), UAB Center for Nutrient-Gene Interaction in Cancer Prevention (U54CA100949), UAB Skin Disease Research Center (P30AR050948), and UAB Polycystic Kidney Disease Center (P30DK74038). The Exposure Facility received support from the UAB Center for Free Radical Biology.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Whitney S. Theis, Email: wtheis01@uab.edu.
Kelly K. Andringa, Email: andringa@uab.edu.
Telisha Millender-Swain, Email: tmswain@uab.edu.
Dale A. Dickinson, Email: dadickin@uab.edu.
Edward M. Postlethwait, Email: epost@uab.edu.
Shannon M. Bailey, Email: sbailey@uab.edu.
References
- 1.Environmental Protection Agency, 2008. Office of the Federal Register, National Archives and Records Administration, Federal Register vol. 73 (60) (March 27, 2008), pp. 16179–16514.
- 2.Bhalla D.K., Crocker T.T. Pulmonary epithelial permeability in rats exposed to O3. J. Toxicol. Environ. Health. 1987;21(1–2):73–87. doi: 10.1080/15287398709531003. [DOI] [PubMed] [Google Scholar]
- 3.Dye J.A., Madden M.C., Richards J.H., Lehmann J.R., Devlin R.B., Costa D.L. Ozone effects on airway responsiveness, lung injury, and inflammation. Comparative rat strain and in vivo/in vitro investigations. Inhal. Toxicol. 1999;11(11):1015–1040. doi: 10.1080/089583799196664. [DOI] [PubMed] [Google Scholar]
- 4.Lippmann M. Effects of ozone on respiratory function and structure. Annu. Rev Public Health. 1989;10:49–67. doi: 10.1146/annurev.pu.10.050189.000405. [DOI] [PubMed] [Google Scholar]
- 5.Maniar-Hew K., Postlethwait E.M., Fanucchi M.V. Postnatal episodic ozone results in persistent attenuation of pulmonary and peripheral blood responses to LPS challenge. Am. J. Physiol. Lung Cell Mol. Physiol. 2011;300(3):L462–471. doi: 10.1152/ajplung.00254.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brook R.D., Franklin B., Cascio W. Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the american heart association. Circulation. 2004;109(21):2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- 7.Chuang K.J., Chan C.C., Su T.C., Lee C.T., Tang C.S. The effect of urban air pollution on inflammation, oxidative stress, coagulation, and autonomic dysfunction in young adults. Am. J. Respir Crit. Care Med. 2007;176(4):370–376. doi: 10.1164/rccm.200611-1627OC. [DOI] [PubMed] [Google Scholar]
- 8.Chuang K.J., Yan Y.H., Cheng T.J. Effect of air pollution on blood pressure, blood lipids, and blood sugar: a population-based approach. J. Occup. Environ. Med. 2010;52(3):258–262. doi: 10.1097/JOM.0b013e3181ceff7a. [DOI] [PubMed] [Google Scholar]
- 9.Park S.K., O'Neill M.S., Vokonas P.S., Sparrow D., Schwartz J. Effects of air pollution on heart rate variability: the VA normative aging study. Environ. Health Perspect. 2005;113(3):304–309. doi: 10.1289/ehp.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chuang G.C., Yang Z., Westbrook D.G. Pulmonary ozone exposure induces vascular dysfunction, mitochondrial damage, and atherogenesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297(2):L209–216. doi: 10.1152/ajplung.00102.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole M.P., Freeman B.A. Promotion of cardiovascular disease by exposure to the air pollutant ozone. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297(2):L205–208. doi: 10.1152/ajplung.00187.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardner D.E. Introductory remarks: session on genetic factors affecting pollutant toxicity. Environ. Health Perspect. 1979;29:45–48. doi: 10.1289/ehp.792945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham J.A., Menzel D.B., Miller F.J., Illing J.W., Gardner D.E. Influence of ozone on pentobarbital-induced sleeping time in mice, rats, and hamsters. Toxicol. Appl. Pharmacol. 1981;61(1):64–73. doi: 10.1016/0041-008x(81)90008-9. [DOI] [PubMed] [Google Scholar]
- 14.Last J.A., Gohil K., Mathrani V.C., Kenyon N.J. Systemic responses to inhaled ozone in mice: cachexia and down-regulation of liver xenobiotic metabolizing genes. Toxicol. Appl. Pharmacol. 2005;208(2):117–126. doi: 10.1016/j.taap.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Laskin D.L., Pendino K.J., Punjabi C.J., Rodriguez del Valle M., Laskin J.D. Pulmonary and hepatic effects of inhaled ozone in rats. Environ. Health Perspect. 1994;102(suppl. 10):61–64. doi: 10.1289/ehp.94102s1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballinger C.A., Cueto R., Squadrito G. Antioxidant-mediated augmentation of ozone-induced membrane oxidation. Free Radic. Biol. Med. 2005;38(4):515–526. doi: 10.1016/j.freeradbiomed.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Laviolette M., Carreau M., Coulombe R. Bronchoalveolar lavage cell differential on microscope glass cover. A simple and accurate technique. Am. Rev. Respir Dis. 1988;138(2):451–457. doi: 10.1164/ajrccm/138.2.451. [DOI] [PubMed] [Google Scholar]
- 18.Lu F.L., Johnston R.A., Flynt L. Increased pulmonary responses to acute ozone exposure in obese db/db mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290(5):L856–865. doi: 10.1152/ajplung.00386.2005. [DOI] [PubMed] [Google Scholar]
- 19.Mantena S.K., Vaughn D.P., Andringa K.K. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009;417(1):183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Andringa K.K., King A.L., Eccleston H.B. Analysis of the liver mitochondrial proteome in response to ethanol and S-adenosylmethionine treatments: novel molecular targets of disease and hepatoprotection. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;298(5):G732–745. doi: 10.1152/ajpgi.00332.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pappin D.J., Hojrup P., Bleasby A.J. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993;3(6):327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 23.King A.L., Swain T.M., Dickinson D.A., Lesort M.J., Bailey S.M. Chronic ethanol consumption enhances sensitivity to Ca(2±)-mediated opening of the mitochondrial permeability transition pore and increases cyclophilin D in liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;299(4):G954–966. doi: 10.1152/ajpgi.00246.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K., Kaufman R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454(7203):455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentile C.L., Frye M., Pagliassotti M.J. Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxid. Redox Signal. 2011;15(2):505–521. doi: 10.1089/ars.2010.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn R.D., McLaughlin L.A., Hughes C., Song C., Henderson C.J., Roland Wolf C. Cytochrome b5 null mouse: a new model for studying inherited skin disorders and the role of unsaturated fatty acids in normal homeostasis. Transgenic Res. 2011;20(3):491–502. doi: 10.1007/s11248-010-9426-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Axelrod J., Senoh S., Witkop B. O-Methylation of catechol amines in vivo. J. Biol. Chem. 1958;233(3):697–701. [PubMed] [Google Scholar]
- 28.Zhu B.T. Catechol-O-Methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr. Drug Metab. 2002;3(3):321–349. doi: 10.2174/1389200023337586. [DOI] [PubMed] [Google Scholar]
- 29.Hayes J.D., Flanagan J.U., Jowsey I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W.W., Churchill S., Lindahl R., Churchill P. Regulation of D-beta-hydroxybutyrate dehydrogenase in rat hepatoma cell lines. Cancer Res. 1989;49(9):2433–2437. [PubMed] [Google Scholar]
- 31.Shepard B.D., Tuma P.L. Alcohol-induced alterations of the hepatocyte cytoskeleton. World J. Gastroenterol. 2010;16(11):1358–1365. doi: 10.3748/wjg.v16.i11.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graham J.A., Menzel D.B., Mole M.L., Miller F.J., Gardner D.E. Influence of ozone on pentobarbital pharmacokinetics in mice. Toxicol. Lett. 1985;24(2–3):163–170. doi: 10.1016/0378-4274(85)90053-0. [DOI] [PubMed] [Google Scholar]
- 33.Kafoury R.M., Pryor W.A., Squadrito G.L., Salgo M.G., Zou X., Friedman M. Lipid ozonation products activate phospholipases A2, C, and D. Toxicol. Appl. Pharmacol. 1998;150(2):338–349. doi: 10.1006/taap.1998.8418. [DOI] [PubMed] [Google Scholar]
- 34.Pryor W.A., Squadrito G.L., Friedman M. The cascade mechanism to explain ozone toxicity: the role of lipid ozonation products. Free Radic. Biol. Med. 1995;19(6):935–941. doi: 10.1016/0891-5849(95)02033-7. [DOI] [PubMed] [Google Scholar]
- 35.Uhlson C., Harrison K., Allen C.B., Ahmad S., White C.W., Murphy R.C. Oxidized phospholipids derived from ozone-treated lung surfactant extract reduce macrophage and epithelial cell viability. Chem. Res. Toxicol. 2002;15(7):896–906. doi: 10.1021/tx010183i. [DOI] [PubMed] [Google Scholar]
- 36.Kafoury R.M., Hernandez J.M., Lasky J.A., Toscano W.A., Jr., Friedman M. Activation of transcription factor IL-6 (NF-IL-6) and nuclear factor-kappaB (NF-kappaB) by lipid ozonation products is crucial to interleukin-8 gene expression in human airway epithelial cells. Environ. Toxicol. 2007;22(2):159–168. doi: 10.1002/tox.20246. [DOI] [PubMed] [Google Scholar]
- 37.Schelegle E.S., Walby W.F., Alfaro M.F. Repeated episodes of ozone inhalation attenuates airway injury/repair and release of substance P, but not adaptation. Toxicol. Appl. Pharmacol. 2003;186(3):127–142. doi: 10.1016/s0041-008x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- 38.Rutkowski D.T., Kaufman R.J. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14(1):20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Ye R., Jung D.Y., Jun J.Y. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. 2010;59(1):6–16. doi: 10.2337/db09-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kammoun H.L., Chabanon H., Hainault I. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 2009;119(5):1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laing S., Wang G., Briazova T. Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am. J. Physiol. Cell Physiol. 2010;299(4):C736–749. doi: 10.1152/ajpcell.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stokes A.H., Hastings T.G., Vrana K.E. Cytotoxic and genotoxic potential of dopamine. J. Neurosci. Res. 1999;55(6):659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 43.Hayes J.D., McLellan L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999;31(4):273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 44.Leiers B., Kampkötter A., Grevelding C.G., Link C.D., Johnson T.E., Henkle-Dührsen K. A stress-responsive glutathione S-transferase confers resistance to oxidative stress in Caenorhabditis elegans. Free Radic. Biol. Med. 2003;34(11):1405–1415. doi: 10.1016/s0891-5849(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 45.Alexis N.E., Zhou H., Lay J.C. The glutathione-S-transferase Mu 1 null genotype modulates ozone-induced airway inflammation in human subjects. J. Allergy Clin. Immunol. 2009;124(6):1222–1228. doi: 10.1016/j.jaci.2009.07.036. (e1225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujimoto K., Arakawa S., Shibaya Y. Characterization of phenotypes in Gstm1-null mice by cytosolic and in vivo metabolic studies using 1,2-dichloro-4-nitrobenzene. Drug Metab. Dispos. 2006;34(9):1495–1501. doi: 10.1124/dmd.106.010009. [DOI] [PubMed] [Google Scholar]
- 47.Hori M., Oniki K., Nakagawa T. Association between combinations of glutathione-S-transferase M1, T1 and P1 genotypes and non-alcoholic fatty liver disease. Liver Int. 2009;29(2):164–168. doi: 10.1111/j.1478-3231.2008.01794.x. [DOI] [PubMed] [Google Scholar]
- 48.Marcos M., Pastor I., Chamorro A.J., Ciria-Abad S., Gonzalez-Sarmiento R., Laso F.J. Meta-analysis: glutathione-S-transferase allelic variants are associated with alcoholic liver disease. Aliment. Pharmacol. Ther. 2011;34(10):1159–1172. doi: 10.1111/j.1365-2036.2011.04862.x. [DOI] [PubMed] [Google Scholar]
- 49.Song K., Yi J., Shen X., Cai Y. Genetic polymorphisms of glutathione S-transferase genes GSTM1, GSTT1 and risk of hepatocellular carcinoma. PloS One. 2012;7(11):e48924. doi: 10.1371/journal.pone.0048924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang B., Huang G., Wang D. Null genotypes of GSTM1 and GSTT1 contribute to hepatocellular carcinoma risk: evidence from an updated meta-analysis. J. Hepatol. 2010;53(3):508–518. doi: 10.1016/j.jhep.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 51.Aroor A.R., Lowery R.J., Restrepo R.J., Mooney B.P., Shukla S.D.A. Proteomic analysis of liver after ethanol binge in chronically ethanol treated rats. Proteome Sci. 2012;10(1):29. doi: 10.1186/1477-5956-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergamaschi E., De Palma G., Mozzoni P. Polymorphism of quinone-metabolizing enzymes and susceptibility to ozone-induced acute effects. Am. J. Respir Crit. Care Med. 2001;163(6):1426–1431. doi: 10.1164/ajrccm.163.6.2006056. [DOI] [PubMed] [Google Scholar]
- 53.Zhong B., Omary M.B. Actin overexpression parallels severity of pancreatic injury. Exp. Cell Res. 2004;299(2):404–414. doi: 10.1016/j.yexcr.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 54.Aibo D.I., Birmingham N.P., Lewandowski R. Acute exposure to ozone exacerbates acetaminophen-induced liver injury in mice. Toxicol. Sci. 2010;115(1):267–285. doi: 10.1093/toxsci/kfq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shore S.A., Rivera-Sanchez Y.M., Schwartzman I.N., Johnston R.A. Responses to ozone are increased in obese mice. J. Appl. Physiol. 2003;95(3):938–945. doi: 10.1152/japplphysiol.00336.2003. [DOI] [PubMed] [Google Scholar]
- 56.Shore S.A., Lang J.E., Kasahara D.I. Pulmonary responses to subacute ozone exposure in obese vs. lean mice. J. Appl. Physiol. 2009;107(5):1445–1452. doi: 10.1152/japplphysiol.00456.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams A.S., Mathews J.A., Kasahara D.I. Augmented pulmonary responses to acute ozone exposure in obese mice: roles of TNFR2 and IL-13. Environ. Health Perspect. 2013;121(5):551–557. doi: 10.1289/ehp.1205880. [DOI] [PMC free article] [PubMed] [Google Scholar]