

Abstract
The zinc tetrathiolate (ZnS4) cluster is an important structural feature of endothelial nitric oxide synthase (eNOS). The cluster is located on the dimeric interface and four cysteine residues (C94 and C99 from two adjacent subunits) form a cluster with a Zn ion in the center of a tetrahedral configuration. Due to its high sensitivity to oxidants this cluster is responsible for eNOS dimer destabilization during periods of redox stress. In this work we utilized site directed mutagenesis to replace the redox sensitive cysteine residues in the ZnS4 cluster with redox stable tetra-arginines. Our data indicate that this C94R/C99R eNOS mutant is active. In addition, this mutant protein is insensitive to dimer disruption and inhibition when challenged with hydrogen peroxide (H2O2). Further, the overexpression of the C94R/C99R mutant preserved the angiogenic response in endothelial cells challenged with H2O2. The over-expression of the C94R/C99R mutant preserved the ability of endothelial cells to migrate towards vascular endothelial growth factor (VEGF) and preserved the endothelial monolayer in a scratch wound assay. We propose that this dimer stable eNOS mutant could be utilized in the treatment of diseases in which there is eNOS dysfunction due to high levels of oxidative stress.
Keywords: Endothelial nitric oxide synthase, Protein engineering, Redox stability, Zinc tetrathiolate cluster
Graphical abstract
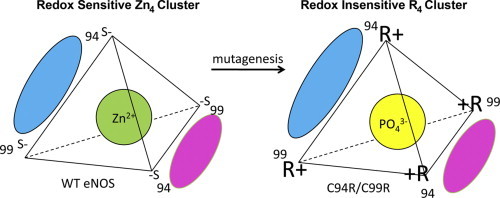
Highlights
-
•
The ZnS4 cluster is an important structural feature of eNOS.
-
•
This cluster is responsible for eNOS dimer destabilization during redox stress.
-
•
Site directed mutagenesis replaced ZnS4 clusters with redox stable tetra-arginines.
-
•
This eNOS mutant is insensitive to dimer disruption during redox stress.
-
•
This eNOS mutant continues to produce NO during redox stress.
Introduction
Metallo-enzymes can coordinate zinc ions (Zn) through cysteine or histidine residues. The Zn ion can coordinate four ligands in a tetrahedral structure. In this work we focused on the ZnS4 cluster in endothelial nitric oxide synthase (eNOS). Endothelial NOS, like all NOS isoforms, is a homodimeric enzyme with the ZnS4 cluster at the dimeric interface. The cluster is formed by four sulfur atoms from two cysteine residues C94 and C99 from each monomer (Fig. 1). It is well established that the dimeric configuration is required for nitric oxide (NO) generation by NOS [1,2]. Thus, the ZnS4 cluster is an important contributor to the proper folding of eNOS enzyme. However, the four sulfur atoms in the tetrahedral configuration are very sensitive to oxidation, leading to eNOS dimer disruption and attenuated NO production [3,4]. The distance between sulfur atoms in the ZnS4 cluster is equal to the distance between sulfur atoms in (S—S) disulfide bond. Therefore, the formation of an intermediate with a two-center three-electron bond between two sulfur atoms is very favorable (Fig. 1) [5]. Further oxidation of the three-electron (S–S) intermediate requires significantly less energy than oxidation of free cysteine, therefore, oxidation of cysteine residues in the ZnS4 cluster can occur even under conditions of mild oxidative stress.
Fig. 1.
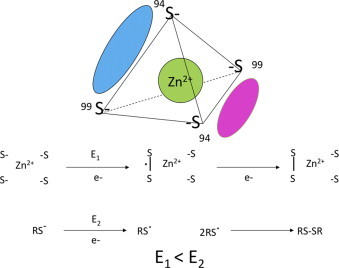
The structure of Zn-tetrathiolate cluster. The Zn4 cluster is composed of two cysteine residues (C94 and C99, human nomenclature) from each subunit of eNOS (cyan and magenta). Oxidation of cysteine residues within the Zn4 cluster requires less energy due to formation of three electron S–S bond in the tetrahedral coordination.
As eNOS dependent vasodilation is an important mechanism regulating vascular tone, the disruption of eNOS activity under conditions of oxidative stress, can induce pathological changes in blood vessels that can lead to a number of diseases including atherosclerosis, diabetes mellitus and hypertension [6–8]. Thus, maintaining NO production is a primary goal in the treatment of cardiovascular disorders. The purpose of this study was to design an eNOS enzyme that is insensitive to oxidative stress. It has been previously reported that arginine rich structures can be stabilized by the formation of strong electrostatic interactions between arginine residues and negative ions such as phosphate or chloride [9]. We report here that the replacement of the ZnS4 cluster with a tetra-arginine cluster results in a catalytically competent enzyme. This engineered eNOS is resistant to oxidative dimer disruption and is able to produce NO in an environment of enhanced oxidative stress.
Materials and methods
Molecular dynamic simulations
Molecular dynamic (MD) simulations were performed using Yasara. The Amber 99 all-hydrogen force field was used in the runs (22). For comparison, two 100 nsec simulations at 330 K were carried out using an eNOS dimeric structure. Simulations were carried out within a simulation cube filled with water molecules. All the atoms except those in the 70–125 aa region were fixed. The simulated systems contained ~2000 atoms. Snapshots were saved every 2 ns. Final structures after MD were structurally aligned using the Mustang algorithm in Yasara and the conformational changes were identified.
eNOS protein purification
For eNOS purification, 50 ml of terrific broth was premixed with ampicillin (100 mg/ml) and chlorophenicol (50 mg/ml), and inoculated with E. coli BL21 cells transformed with a polyHis-pCWeNOS plasmid containing wild type human eNOS sequence (9) or the mutant C94R/C99R. The polyHis-pCWeNOS vector was a gift from P. R. Ortiz de Montellano (University of California, San Francisco). The C94R/C99R eNOS mutant was prepared from the wildtype plasmid by Retrogen and sequenced to verify identity. Bacteria were grown overnight at 37 °C (260 rpm) then used to inoculate 2.8 L Fernabach flasks (6×1.5 L) again containing terrific broth (52 g/L) as the culture medium and supplemented with ampicillin (100 mg/ml), riboflavin (15 mg), and aminolevulinic acid (0.5 g). Flasks were placed on an orbital shaker and were allowed to grow at 37 °C (200 rpm). The OD600 was checked periodically during the growth period until it reached 0.8–1.0 (4–5 h) then adenosine-5′-triphosphate (ATP, 200 µM final concentration) and isopropyl-beta-d-thiogalactopyranoside, dioxane free (IPTG, 1 mM final concentration, to induce the T7 promoter) was added and the cells incubated for 18–20 h at 25 °C (200 rpm). Bacteria were then harvested by centrifugation using a FiberLite F6 6×1000 rotor at 4 °C (3500 rpm/2700g) for 20 min. The pellet was immediately transferred into lysis buffer (40 mM Tris–HCl, 5% glycerol, 1 mg/ml lysozyme, 100 mM NaCl, 4 mM FAD, 4 mM FMN, 100 µM BH4, 5 mM l-arginine) and a protease inhibitor cocktail for use with histidine-tagged proteins (Sigma), ribonuclease A from bovine pancreas (Sigma), and deoxyribonuclease I from bovine pancreas (106 units, Sigma) was added. The pellet was gently rocked for 30 min at 4 °C, sonicated on ice, then subjected to ultracentrifugation at 4 °C (60,000 rpm/37,1000g) for 1 h and 45 min. The supernatant was loaded onto a Hisprep FF 16/10 column (charged with 0.1 M NiSO4) using binding buffer (40 mM Tris–HCl, 100 mM NaCl, 5% glycerol, 30 mM imidazole, 100 µM BH4, 100 µM l-arginine) at 0.1 ml/min flow. The column was washed with washing buffer (40 mM Tris–HCl, 300 mM NaCl, 5% glycerol, 30 mM imidazole, 100 µM BH4, 100 µM l-arginine) using a flow rate of 1.5 ml/min, and a base line was obtained resulting in the washing out of non-histidine-tagged proteins. Elution of histidine-tagged protein was accomplished using elution buffer (40 mM Tris–HCl, 300 mM NaCl, 5% glycerol, 400 mM imidazole, 100 µM BH4, 100 µM l-arginine) at 1.0 ml/min flow. Collected fractions were loaded for size-exclusion gel filtration on a HiLoad 26/60 Superdex 200 prep grade column using eNOS gel filtration buffer (60 mM Tris–HCl, 100 mM NaCl, 5% glycerol, 100 µM BH4, 100 µM l-arginine) at 0.2 ml/min flow. Fractions were collected in 5 ml amounts for analysis by Coomassie blue staining and Western blot. Desalting was then performed for fractions containing eNOS using a HiPrep 26/10 desalting column and eNOS gel filtration buffer at flow rate of 0.5 ml/min. All purification steps were performed at 4 °C, and the purified protein was stored at −80 °C. Protein homogeneity was confirmed using Coomassie blue staining and Western blot with anti-eNOS antibody with 1:1000 dilutions (Transduction Labs). Final protein concentration was then measured in each fraction.
Gel filtration chromatography
To examine the extent of dimerization in the wildtype and mutant eNOS proteins we utilized analytical gel filtration. One hundred microlitres of each protein, at a concentration of 0.5 mg/ml, was injected into a Tosoh TSKgel G3000SW×l gel filtration column. Using a flow rate of 0.5 ml/min, monomer and dimer fractions were eluted in 100 mM phosphate buffer (pH=7.0) using an HPLC system (GE) and analyzed by measuring the absorption at 260 nm.
Determination of NOx levels
To measure NO production we utilized a chemiluminescence method. Wildtype eNOS and the C94R/C99R-eNOS were mixed with the cofactors calmodulin (10 µM) and BH4 (40 µM) as well as the substrate l-arginine (100 µM) and Ca2+ (100 µM CaCl2) in reaction buffer (50 mM HEPES, pH 7.4). The reaction was initiated with the addition of NADPH (10 µM). After 30 min of incubation at 37 °C the reaction mixture was analyzed for NOx levels. In our experiments, potassium iodide (KI)/acetic acid reagent was prepared fresh daily by dissolving 0.05 g of KI in 7 ml of acetic acid. This reagent was added to a septum sealed purge vessel and bubbled with nitrogen gas. The gas stream was connected via a trap containing 1 N NaOH to a Sievers 280i Nitric Oxide Analyzer (GE). Samples were injected with a syringe through a silicone/Teflon septum. Results were analyzed by measuring the area under curve of the chemiluminescence signal using the Liquid software (GE). To carry out NO measurements in cells experiment, we utilized cell lysate and measured cellular NO content per mg of protein.
Measurement of superoxide levels
To detect superoxide generation, EPR measurements were performed using the spin trap, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine.HCl (CMH) as we have described [10,11].
Optical absorption spectroscopy
The spectra of the wildtype and mutant eNOS proteins were determined using a Shimadzu spectrophotometer using a micro-cuvette. The final protein concentration used was 1 mg/ml. Spectra were scanned at 25 °C in the region 350–600 nm and the average of three measurements was plotted.
Overexpression of eNOS in COS-7 cells and dimer/monomer analysis
COS-7 cells were transfected with wildtype (WT) and C94R/C99R eNOS 48 h before experiment. Cells were treated with increasing concentrations of H2O2 (0-, 100-, 200-, 300-, 400-µM). Cells were lysed after the experiment. Lysis buffer containing 1% Triton X-100, 20 mM Tris pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Pierce) was then added and insoluble proteins precipitated by centrifugation at 13,000 rpm for 10 min at 4 °C. Samples were mixed with ice cold PBS and ice cold 5× sample buffer, incubated on ice for 10 min, centrifuged and loaded into 4–20% denaturating polyacrylamide gels (Mini-PROTEAN® TGXTM, BioRad) and low temperature gel electrophoresis (LT-PAGE) was utilized to evaluate the eNOS dimer/monomer ratio. The samples were separated on ice using ice-cold TRIS/Glycine running buffer with 0.1% SDS on 50 V for 3 h. All gels were electrophoretically transferred to PVDF membranes. The membranes were blocked with 1% BSA in Tris-buffered saline (TBS) containing 0.1% Tween 20 for 1 h and incubated overnight at 4 °C with an appropriate dilution of primary antibody anti-eNOS (BD). The membranes were then washed with TBST (3×10 min), incubated with secondary antibodies coupled to horseradish peroxidase, washed again with TBST (3×10 min), and the protein bands visualized using the SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) on a Kodak 440CF image station. Band intensity was quantified using Kodak 1D image processing software. Protein loading was normalized by reprobing membranes with HSP90 (Sigma).
Endothelial cell culture
Primary cultures of ovine pulmonary arterial endothelial cells (PAEC) were isolated as described previously [12]. Cells were maintained in DMEM containing 1 g/L glucose and supplemented with 10% fetal calf serum (Hyclone, Logan, UT), antibiotics, and antimycotics (MediaTech, Herndon, VA) at 37 °C in a humidified atmosphere with 5% CO2–95% air. Cells were utilized between passages 3 and 10. COS-7 cells were cultured and maintained in the same constituent medium but with a higher glucose concentration (4.5 g/L glucose).
Scratch wound assay
PAEC were transiently transfected with expression plasmids for either WT- or C94R/C99R-eNOS. The cells were then wounded by scratching the monolayer with a 200 µl pipette tip and washed with medium to remove any debris. Cells were then exposed or not to H2O2 (400 µM) and then photographed using phase-contrast microscope (Olympus, Japan) with digital camera 0-, 4-, and 8-h later. The rate of wound healing was determined by measuring the change in distance between the wounded edges over time. The data were plotted as percent change compared to untreated controls.
HMEC-1 migration analysis
An immortalized human microvascular endothelial cell line (HMEC-1) was provided by Dr. Robert Debs and used with permission of its creators, Dr. Edwin Ades and Mr. Francisco J. Candal of the Centers for Disease Control and Dr. Thomas Lawley of Emory University. HMEC-1 cells (passage 8) were seeded in a 6-well plate at 2×105 per well and cultured with EBM-2 supplemented with Singlequots (Cambrex) and 5% FBS. 24 h later, 2 µg of DNA (1:1 ratio of WT eNOS with GFP, or C94R/C99R eNOS with GFP) were transfected with PEI solution (gift from R. Debs). After 24 h incubation, GFP+ transfected cells were sorted using a FACS Aria II YG and were cultured for another 24 h before being harvested for migration assays. After detachment, 6×103 cells were resuspended in basal cell media (without supplements, 0.5% BSA) and plated in the upper of two chambers divided by a membrane with 8 µm pores (Corning Transwell, Corning, NY). H2O2 (Sigma) was added to both the upper and lower chambers at 300 µM. No chemoattractants were used. After 3 h, the membranes were washed twice in PBS and fixed in 4% formaldehyde. After wiping cells off of the upper side of the membrane with a cotton swab (Q-tip), the membranes were detached, dipped briefly in Hoechst 33258 (Invitrogen; 1:2000 in PBS) and mounted on glass slides. Migrated cells were counted on the lower side of the membrane using fluorescence microscopy on 5 random 100× optical fields per membrane. Each experimental condition was performed in triplicate.
Statistical analysis
Statistical calculations were performed using the GraphPad Prism V. 4.01 software. The mean±SD or SEM was calculated for all the samples and significance determined using either the unpaired t-test or ANOVA. For ANOVA, Newman–Kuels post-hoc testing was also utilized. A value of P<0.05 was considered significant.
Results
Protein engineering of a tetra-arginine cluster based endothelial NOS
We hypothesized that substituting the cysteines that formed ZnS4 cluster with arginines would still form an active eNOS homodimer, but one that was redox insensitive. Molecular dynamic (MD) experiments were initially undertaken using the available crystal stucture of human eNOS (PDB ID 3NOS) to determine if mutations of the two tetrathiolate clusters generating cysteine residues to arginines (C94R/C99R) would grossly alter the eNOS structure. A phosphate ion () was introduced as a replacement for the Zn2+ ion. The resulting mutant protein was simulated in a water filled cube for 100 ns using the MD simulation module in Yasara. After minimization of free energy, the structure of eNOS was still predicted to be dimeric and three arginine residues were found to interact with the phosphate ion (Fig. 2). Four arginines failed to form a compact tetrahedral structure, however, the structure of catalytic center and substrate channel did not appear to be disturbed in the C94R/C99R mutant eNOS.
Fig. 2.
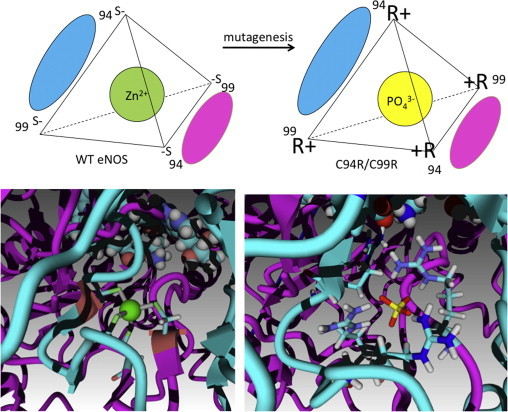
Ion cluster inversion in C94R/C99R eNOS. The upper panel demonstrates that wildtype eNOS contains a Zn4 cluster with four negatively charged sulfur atoms surrounding the Zn2+ cation in a tetrahedral configuration. In the C94R/C99R mutant the introduction of four arginine residue are proposed to stabilize the dimeric interface of eNOS via electrostatic interactions with a anion. The lower panel shows both the crystal structure of the Zn cluster (PDB ID 3NOS) in wildtype eNOS and computer simulated structure of the C94R/C99R mutant.
Protein purification and characterization of tetra-arginine C94R/C99R eNOS
To begin to analyze the properties of C94R/C99R mutant protein, a histidine tagged protein was expressed and purified using a bacterial expression system and Ni-NTA affinity chromatography. Gel filtration analysis demonstrated that the level of dimer in the purified C94R/C99R eNOS mutant protein was equivalent to that found in WT eNOS (Fig. 3A). This was in contrast to an eNOS mutant in which the ZnS4 cluster was disrupted by insertion of alanine residues [13] (C94A/C99A, Fig. 3A). Oxidation of the ZnS4 cluster by H2O2 resulted in the disruption of the WT eNOS dimeric structure (Fig. 3B) as previously published [3] while the C94R/C99R mutant eNOS was resistant to H2O2-mediated dimer disruption (Fig. 3B). Further, we found that H2O2 decreased NO generation in WT (Fig. 3C), but not in the C94R/C99R eNOS mutant (Fig. 3D). Interestingly, under basal conditions, the level of NO production was higher in the C94R/C99R mutant compared with that of WT eNOS (Fig. 3C and D). This increase in eNOS activity could be due to the fact that C94R/C99R eNOS mutant is not susceptible to the NO-mediated dimer disruption we have previously shown to occur in WT eNOS. However, further studies will be required to confirm this. Superoxide generation rate of C94R/C99R eNOS was similar to that of wild-type (Fig. 3E). Further we found that the heme spectra in both WT- and C94R/C99R-eNOS have a characteristic band at 396 nm that indicates active high spin heme state (Fig. 3F). Together these data suggest that the inversion of the ion cluster from cation centered ZnS4 cluster to an anion centered tetra-arginine cluster does not appear to affect the dimeric interface, catalytic and spectral characteristics of the recombinant protein. Moreover, the presence of the tetra-arginine cluster enhances the redox stability of the dimeric eNOS.
Fig. 3.
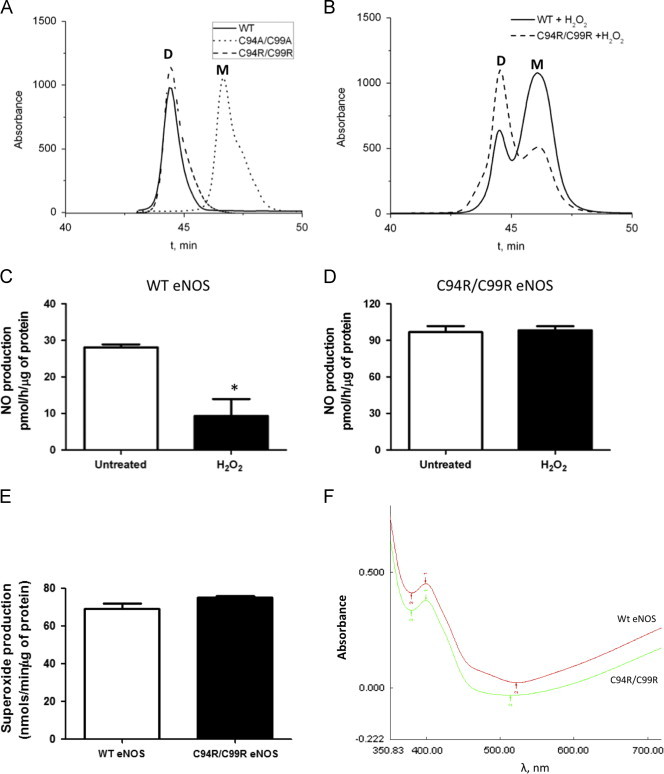
Characterization of C94R/C99R eNOS mutant. Gel filtration profiles (absorbance at λ260 nm) of WT (solid)-, C94R/C99R (dash)- and C94A/C99A (dot)-eNOS in the presence and absence of H2O2 (0.5 mM). WT- and C94R/C99R-eNOS form dimeric proteins in contrast to the C94A/C99A-eNOS mutant (A). H2O2 treatment disrupts the dimeric structure of WT eNOS, but the C94R/C99R mutant is resistant to monomerization (B). The disruption of the dimeric structure of WT eNOS correlates with a reduction in NO generation (C). NO generation is maintained in the C94A/C99A-eNOS mutant exposed to H2O2 (D). NO production in the C94R/C99R-eNOS under basal conditions is significantly higher that WT-eNOS (C and D) although superoxide generation is similar (E). Both WT- and C94R/C99R-eNOS enzymes exhibited high spin heme absorption peak at 396 nm that indicates catalytically active enzymes. (F). Data are mean±SEM, N=3, ⁎p<0.05 vs. untreated.
Redox stability of C94R/C99R eNOS in COS-7 cells
In order to evaluate the effect of the C94R/C99R mutation on dimeric assembly and susceptibility to oxidative mediated disruption in cells, plasmids encoding WT- and C94R/C99R-eNOS were transiently transfected into COS-7 cells. Our data demonstrated that COS-7 cells expressing the C94/C99R mutant had a higher dimer:monomer ratio than those expressing wildtype eNOS (Fig. 4A and B). Treatment with H2O2, which disrupts the ZnS4 cluster inducing eNOS monomerization [3], led to dose dependant decrease in eNOS dimer levels in WT- but not C94R/C99R-eNOS expressing cells (Fig. 4A and B). Thus, the replacement of redox sensitive tetrathiolate cluster with redox stable four arginines resulted in the stabilization of the enzyme to oxidative stress. We also measured NO generation for both WT- and the C94R/C99R mutant-eNOS. WT- and C999R-eNOS produced similar amount of NOx under basal conditions (Fig. 4C). H2O2 exposure significantly decreased the rate of NOx generation in WT eNOS expressing cells (Fig. 4C). However, NOx generation in the C94R/C99R eNOS expressing cells was unaffected by H2O2.
Fig. 4.
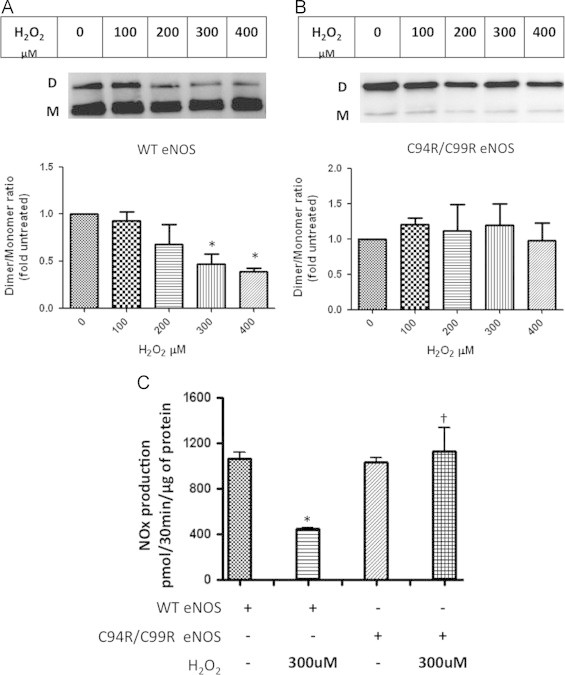
eNOS dimer stability under conditions of oxidative stress. COS-7 cells were transiently transfected with either WT- or C94R/C99R-eNOS. After 48 h, cells were exposed or not to increasing concentrations of H2O2 (0–400 µM, 30 min). Western blot analysis using LT-PAGE, was then used to determine the effect on the eNOS dimer: monomer ratio. H2O2 dose dependently decreased the dimer level in WT-eNOS expressing cells (A). However, the C94R/C99R mutant was resistant to dimer disruption even at 400 µM H2O2 (B). Basal levels of NOx were similar in WT- or C94R/C99R-eNOS expressing cells. H2O2 exposure (300 µM) significantly reduced NOx levels in WT eNOS expressing cells but not in the cells expressing the C94R/C99R eNOS mutant (C). Data are mean±SEM, N=3, ⁎p<0.05 vs. untreated; † vs. WT eNOS +H2O2.
C94R/C99R eNOS maintains endothelial cell angiogenesis under conditions of oxidative stress
To determine whether dimer-stable C94R/C99R eNOS protects eNOS-dependent endothelial cell function when exposed to oxidative stress, we transfected ovine PAEC with WT- and C94R/C99R-eNOS and utilized the scratch wound assay. Initially, we confirmed that eNOS protein levels (Fig. 5A) and NO production levels (Fig. 5B) were both increased. Then we assessed the effect on PAEC migration NO production in the presence of H2O2. Both NO production (Fig. 5B) and the migration rate (Fig. 5C) were significantly attenuated by H2O2 (400 µM) in WT-eNOS expressing cells. However, in PAEC transfected with the redox insensitive C94R/C99R eNOS both NO generation (Fig. 5B) and migration ability were maintained (Fig. 5C).
Fig. 5.
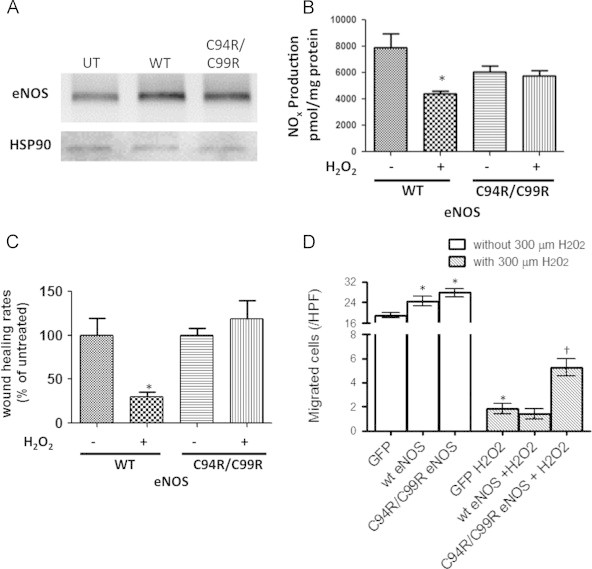
Effect of eNOS overexpression on endothelial cell migration. PAEC were transiently transfected with either WT- or C94R/C99R-eNOS. After 48 h, eNOS protein levels (A) and basal NO generation were determined. A representative Western blot image is shown that verifies over-expression of both proteins. The blots were reprobed with HSP90 to confirm equal loading. Basal NOx levels were also significantly increased in WT- and C94R/C99R-eNOS over-expressing cells (B). In the presence of H2O2 (400 µM, 1 h), NOx levels were significantly attenuated in WT- but not C94R/C99R-eNOS over-expressing cells (B). H2O2 (400 µM) significantly attenuated wound closure in WT- but not C94R/C99R-eNOS over-expressing cells (C). Similarly, in HMEC-1 cells, cell migration was significantly increased in WT- and C94R/C99R-eNOS over-expressing cells under basal conditions (D). H2O2 (300 µM) significantly attenuated HMEC-1 cell migration in both WT- and C94R/C99R-eNOS over-expressing cells (D). However, migration was attenuated less in C94R/C99R-eNOS over-expressing cells (D). Data are mean±SEM, N=3–49, ⁎p<0.05 vs. untreated; †p<0.05 vs. WT-eNOS.
Finally, we employed a well established, and commonly used, transwell assay for cell migration [14] that has been effective at detecting inhibitory effects on endothelial and endothelial-like cells [15]. HMEC-1 cells, a microvascular endothelial cell line, were transfected with either WT- or C94R/C99A-eNOS and compared to cells transfected with GFP. Our data indicate that under conditions where there were no chemoattractants in the lower chamber, cells both WT- or C94R/C99A-eNOS expressing cells exhibited significantly more random migration than cells transfected with GFP alone (Fig. 5D). However, in the presence of H2O2, although migration was decreased in all three groups, there was significantly less reduction in migration in cells transfected with C94R/C99A-eNOS (Fig. 5D).
Discussion
There is increasing histologic and physiologic evidence that endothelial injury and the resulting aberration in the balance of its regulatory mechanisms play a major role in the development of endothelial dysfunction and vascular remodeling. Endothelial dysfunction is a hallmark of many diseases of the vasculature, including hypertension (both pulmonary and systemic), atherosclerosis, and diabetes [16,17]. The most important consequence of endothelial dysfunction is the observed decrease in the ability of the endothelium to mediate vasodilation [18]. An increasing number of studies now implicate oxidative stress in the pathogenesis of many cardiovascular diseases [19–22]. Oxidative stress is now believed to be a major player in the development of endothelial dysfunction. Further, evidence from both our group and others, has shown a clear correlation between increase in oxidative stress and decrease in both eNOS dimer levels and NO signaling [3,23]. Published data demonstrate that both ROS, such as H2O2 [3] and RNS such as NO and peroxynitrite [23–25], can disrupt the eNOS dimer. The mechanism by which this dimer disruption occurs through the oxidation of the ZnS4 cluster located at the NOS dimer interface was reported previously [3]. Importantly, there is a great deal of interest in developing eNOS gene based therapies for the treatment of cardiovascular disease. For example, there is an ongoing clinical trial of a gene therapy for pulmonary hypertension, that is using endothelial progenitor cells, or more accurately, circulating angiogenic cells (CACs) engineered to over-express eNOS [26]. In addition, we, and others have shown that functional deficiencies in CAC migration toward VEGF or association with co-cultured endothelial tubes correlates with endothelial dysfunction and greater risk of cardiovascular events [15,26–31]. Further, CACs have been reported to provide therapeutic benefit when administered directly or i.v. to several models of tissue ischemia or injury [32–35]. The therapeutic effects of CACs can be augmented by transducing them to express vascular regulators, such as VEGF and eNOS [26,36,37]. However, a number of reports have shown that autologous cell therapy can be thwarted if the patient′s disease impairs the therapeutic functionality of the cells in question [30,38,39]. In addition, we have demonstrated that CACs isolated from older healthy and CAD donors produce less NO and more ROS than those from young healthy donors and that this is associated not only with impairment of migration toward VEGF, but also with reduced therapeutic capacity when injected into post-MI mouse hearts (MLS, unpublished data). Both negative effects can be improved by transduction of the CACs with an eNOS adenovirus. However, only partial restoration of cardiac function is achieved, even though resistance to NO is not apparent in the impaired CACs [28]. This underscores an important problem in the use of gene based therapies to treat cardiovascular diseases that involve insufficient levels of NO: therapeutic approaches to increase active eNOS protein may nonetheless have limited efficacy due to oxidative-mediated inhibition of the protein through the disruption of the ZnS4 cluster. Based on these factors, we postulated that there is a potential therapeutic niche for an eNOS protein that was insensitive to oxidative stress mediated dimer disruption and would be able to continue to produce NO under the oxidative stress conditions associated with endothelial dysfunction.
ZnS4 or zinc histidine–hiolate structural features are found in proteins, such as PKC family enzymes [40], zinc fingers of transcription factors [41], sirtuins [42] and metallothionein [43], and NOS [1]. In all cases these form conformational stabilizing structures. In addition, these structural elements share redox sensitivity due to availability of readily oxidazible cysteines [44–46]. However, the presence of a stabilizing tetrathiolate cluster at the dimer interface appears to be an exclusive feature of the NOS isoforms, as a similar structural element has, so far, not been found in other enzymatic systems. Thus, NOS is a unique enzyme that stabilizes its homodimeric structure by employing redox sensitive ZnS4 cluster. In this work we utilized protein engineering to eliminate this redox sensitivity. There have been several reports utilizing protein engineering to stabilize inter-subunit assembly and, thus, increase the stability of multimeric proteins. These include nucleoside diphosphate kinase [47], acid-sensing ion channel-1a (ASIC1a) [48], and seminal RNAse [49]. The formation of intersubunit bridges also plays an important role in the thermal stability of glucose-6-phosphate dehydrogenase [50]. As with NOS, stabilizing intersubunit disulfide bond formation occurs in many native multimeric proteins. Importantly, for our approach to eNOS, the introduction of an artificial intersubunit disulfide bridge is a well described tactic for the stabilization of a multimeric interface. Indeed, a similar strategy has been utilized to stabilize Fe-superoxide dismutase [51], thymidylate synthase [52] and triosephosphate isomerase [53]. From previous studies we know that there is a disruption of the NOS dimer when the proteins are oxidized by NO, H2O2 or peroxynitrite. This involves disulfide oxidation, but does not induce dimer stabilization via disulfides formation in the ZnS4 cluster. Based on this, we decided to utilize a charge based interactions rather than relying on covalent bonding. In our engineered protein we demonstrate that the replacement of the oxidant sensitive, ZnS4 cluster with a redox stable, tetra-arginine cluster, produces a fully functional enzyme that is resistant to dimer disruption by oxidative stress and that retains the ability to produce NO under conditions in which the wildtype enzyme is severely inhibited.
It is also worth noting that there is controversy in the literature regarding the relative roles of the ZnS4 cluster and BH4, a cofactor essential for the catalytic activity of all three NOS isoforms [54–57], in stabilizing the dimeric interface of NOS [23,24,58–60]. Both are redox sensitive, and the binding site of BH4 is located in close proximity to the ZnS4 cluster [1]. Thus, it has been difficult to differentiate the effects mediated by the ZnS4 cluster from those of BH4. In addition, prior studies have shown that cellular BH4 levels have important consequences for the structure of NOS. These include the ability of NOS to shift its heme iron to a high spin state [61], increase arginine binding [61], and at least in some NOS isoforms, potentially stabilize the active dimeric form of the enzyme [54]. However, based on our published data [3]it is possible that the antioxidant properties of BH4 is used to protect the ZnS4 cluster from oxidation during the catalytic cycling of NOS which in turn could explain the apparent dimer stabilizing ability of BH4. As we show here that the replacement of the ZnS4 cluster with redox stable tetra-arginines is sufficient to maintain eNOS in a dimeric form under conditions of oxidative stress, we conclude that the ZnS4 cluster plays the primary role in NOS dimerization.
In conclusion, our engineered C94R/C99R dimer stable mutant opens up the possibility of gene therapy for the treatment of conditions where eNOS dysfunction is associated with increased oxidative stress. This dimer stable enzyme should be able to maintain NO signaling under conditions in which the introduction of the wildtype enzyme would result in dimer disruption and reduced NO generation.
Acknowledgements
This research was supported in part by grants, HL60190 (to SMB), HL67841 (to SMB), HL084739 (to SMB), R21HD057406 (to SMB), HL086917 (to MLS) and P01HL101902 (to SMB) all from the National Institutes of Health, and by a grant from the Foundation Leducq (to SMB). RR and FVF were supported in part by National Institutes of Health Training Grant, 5T32-HL-06699 and OR by F32HL103136. We acknowledge Lejla Medzikovic for technical assistance on this project.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Raman C.S., Li H., Martasek P., Kral V., Masters B.S., Poulos T.L. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 2.Li H., Raman C.S., Glaser C.B., Blasko E., Young T.A., Parkinson J.F., Whitlow M., Poulos T.L. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J. Biol. Chem. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 3.Fonseca F.V., Ravi K., Wiseman D., Tummala M., Harmon C., Ryzhov V., Fineman J.R., Black S.M. Mass spectroscopy and molecular modeling predict endothelial nitric oxide synthase dimer collapse by hydrogen peroxide through zinc tetrathiolate metal-binding site disruption. DNA Cell Biol. 2010;29:149–160. doi: 10.1089/dna.2009.0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou M.H., Shi C., Cohen R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Invest. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu C., Yao S., Lin N. Studies on reactions of oxidizing sulfur-sulfur three-electron-bond complexes and reducing alpha-amino radicals derived from OH reaction with methionine in aqueous solution. Biochim. Biophys. Acta. 2001;1525:89–96. doi: 10.1016/s0304-4165(00)00174-4. [DOI] [PubMed] [Google Scholar]
- 6.Knowles J.W., Reddick R.L., Jennette J.C., Shesely E.G., Smithies O., Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J. Clin. Invest. 2000;105:451–458. doi: 10.1172/JCI8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai S., Khoo J., Mussa S., Alp N.J., Channon K.M. Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia. 2005;48:1933–1940. doi: 10.1007/s00125-005-1857-5. [DOI] [PubMed] [Google Scholar]
- 8.Escobales N., Crespo M.J. Oxidative-nitrosative stress in hypertension. Curr. Vasc. Pharmacol. 2005;3:231–246. doi: 10.2174/1570161054368643. [DOI] [PubMed] [Google Scholar]
- 9.Mrabet N.T., Van den Broeck A., Van den brande I., Stanssens P., Laroche Y., Lambeir A.M., Matthijssens G., Jenkins J., Chiadmi M., van Tilbeurgh H. Arginine residues as stabilizing elements in proteins. Biochemistry. 1992;31:2239–2253. doi: 10.1021/bi00123a005. [DOI] [PubMed] [Google Scholar]
- 10.Sud N., Sharma S., Wiseman D.A., Harmon C., Kumar S., Venema R.C., Fineman J.R., Black S.M. Nitric oxide and superoxide generation from endothelial NOS: modulation by HSP90. Am. J. Physiol. 2007;293:L1444–1453. doi: 10.1152/ajplung.00175.2007. [DOI] [PubMed] [Google Scholar]
- 11.Oishi P.E., Wiseman D.A., Sharma S., Kumar S., Hou Y., Datar S.A., Azakie A., Johengen M.J., Harmon C., Fratz S., Fineman J.R., Black S.M. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: a role for oxidative stress. Am. J. Physiol. 2008;295:L756–766. doi: 10.1152/ajplung.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wedgwood S., Black S.M. Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension. Antioxid. Redox Signal. 2003;5:759–769. doi: 10.1089/152308603770380061. [DOI] [PubMed] [Google Scholar]
- 13.Rafikov R., Fonseca F.V., Kumar S., Pardo D., Darragh C., Elms S., Fulton D., Black S.M. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 2011;210:271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falk W., Goodwin R.H., Jr., Leonard E.J. A 48-well micro chemotaxis assembly for rapid and accurate measurement of leukocyte migration. J. Immunol. Methods. 1980;33:239–247. doi: 10.1016/0022-1759(80)90211-2. [DOI] [PubMed] [Google Scholar]
- 15.Heiss C., Schanz A., Amabile N., Jahn S., Chen Q., Wong M.L., Rassaf T., Heinen Y., Cortese-Krott M., Grossman W., Yeghiazarians Y., Springer M.L. Nitric oxide synthase expression and functional response to nitric oxide are both important modulators of circulating angiogenic cell response to angiogenic stimuli. Arterioscler. Thromb. Vasc. Biol. 2010;30:2212–2218. doi: 10.1161/ATVBAHA.110.211581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griendling K.K., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ. Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 17.Griendling K.K., Sorescu D., Lassegue B., Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vascular Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 18.Vanhoutte P.M., Boulanger C.M. Endothelium-dependent responses in hypertension. Hypertens. Res. 1995;18:87–98. doi: 10.1291/hypres.18.87. [DOI] [PubMed] [Google Scholar]
- 19.Anatoliotakis N., Deftereos S., Bouras G., Giannopoulos G., Tsounis D., Angelidis C., Kaoukis A., Stefanadis C. Myeloperoxidase: expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top Med. Chem. 2013;13:115–138. doi: 10.2174/1568026611313020004. [DOI] [PubMed] [Google Scholar]
- 20.Vassalle C., Bianchi S., Bianchi F., Landi P., Battaglia D., Carpeggiani C. Oxidative stress as a predictor of cardiovascular events in coronary artery disease patients. Clin. Chem. Lab. Med. 2012;50:1463–1468. doi: 10.1515/cclm-2011-0919. [DOI] [PubMed] [Google Scholar]
- 21.Selvaraju V., Joshi M., Suresh S., Sanchez J.A., Maulik N., Maulik G. Diabetes, oxidative stress, molecular mechanism, and cardiovascular disease—an overview. Toxicol. Mech. Methods. 2012;22:330–335. doi: 10.3109/15376516.2012.666648. [DOI] [PubMed] [Google Scholar]
- 22.Strobel N.A., Fassett R.G., Marsh S.A., Coombes J.S. Oxidative stress biomarkers as predictors of cardiovascular disease. Int. J. Cardiol. 2011;147:191–201. doi: 10.1016/j.ijcard.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Chen W., Druhan L.J., Chen C.A., Hemann C., Chen Y.R., Berka V., Tsai A.L., Zweier J.L. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry. 2010;49:3129–3137. doi: 10.1021/bi9016632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravi K., Brennan L.A., Levic S., Ross P.A., Black S.M. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc. Natl. Acad. Sci. USA. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molnar J., Yu S., Mzhavia N., Pau C., Chereshnev I., Dansky H.M. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ. Res. 2005;96:1178–1184. doi: 10.1161/01.RES.0000168634.74330.ed. [DOI] [PubMed] [Google Scholar]
- 26.Ward M.R., Thompson K.A., Isaac K., Vecchiarelli J., Zhang Q., Stewart D.J., Kutryk M.J. Nitric oxide synthase gene transfer restores activity of circulating angiogenic cells from patients with coronary artery disease. Mol. Ther. 2011;19:1323–1330. doi: 10.1038/mt.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heiss C., Wong M.L., Block V.I., Lao D., Real W.M., Yeghiazarians Y., Lee R.J., Springer M.L. Pleiotrophin induces nitric oxide dependent migration of endothelial progenitor cells. J. Cell Physiol. 2008;215:366–373. doi: 10.1002/jcp.21313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heiss C., Jahn S., Taylor M., Real W.M., Angeli F.S., Wong M.L., Amabile N., Prasad M., Rassaf T., Ottaviani J.I., Mihardja S., Keen C.L., Springer M.L., Boyle A., Grossman W., Glantz S.A., Schroeter H., Yeghiazarians Y. Improvement of endothelial function with dietary flavanols is associated with mobilization of circulating angiogenic cells in patients with coronary artery disease. J. Am. Coll Cardiol. 2010;56:218–224. doi: 10.1016/j.jacc.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 29.Aschbacher K., Chen Q., Varga M., Haddad D.J., Yeghiazarians Y., Epel E., Wolkowitz O.M., Springer M.L. Higher fasting glucose levels are associated with reduced circulating angiogenic cell migratory capacity among healthy individuals. Am. J. Cardiovasc. Dis. 2012;2:12–19. [PMC free article] [PubMed] [Google Scholar]
- 30.Vasa M., Fichtlscherer S., Aicher A., Adler K., Urbich C., Martin H., Zeiher A.M., Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 31.Heiss C., Keymel S., Niesler U., Ziemann J., Kelm M., Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J. Am. Coll. Cardiol. 2005;45:1441–1448. doi: 10.1016/j.jacc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 32.Kalka C., Tehrani H., Laudenberg B., Vale P.R., Isner J.M., Asahara T., Symes J.F. VEGF gene transfer mobilizes endothelial progenitor cells in patients with inoperable coronary disease. Ann. Thorac. Surg. 2000;70:829–834. doi: 10.1016/s0003-4975(00)01633-7. [DOI] [PubMed] [Google Scholar]
- 33.Kawamoto A., Gwon H.C., Iwaguro H., Yamaguchi J.I., Uchida S., Masuda H., Silver M., Ma H., Kearney M., Isner J.M., Asahara T. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634–637. doi: 10.1161/01.cir.103.5.634. [DOI] [PubMed] [Google Scholar]
- 34.Kocher A.A., Schuster M.D., Szabolcs M.J., Takuma S., Burkhoff D., Wang J., Homma S., Edwards N.M., Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 35.Schachinger V., Assmus B., Britten M.B., Honold J., Lehmann R., Teupe C., Abolmaali N.D., Vogl T.J., Hofmann W.K., Martin H., Dimmeler S., Zeiher A.M. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction: final one-year results of the TOPCARE-AMI Trial. J. Am. Coll. Cardiol. 2004;44:1690–1699. doi: 10.1016/j.jacc.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Kong Y.G., Ren Z.Y., Su C.B., Wang R.Z., Xing B. [Expressive level of vascular endothelial growth factor in peripheral blood in patients with pituitary adenomas] Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2004;26:164–167. [PubMed] [Google Scholar]
- 37.Zhao Y.D., Courtman D.W., Deng Y., Kugathasan L., Zhang Q., Stewart D.J. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 2005;96:442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 38.Rauscher F.M., Goldschmidt-Clermont P.J., Davis B.H., Wang T., Gregg D., Ramaswami P., Pippen A.M., Annex B.H., Dong C., Taylor D.A. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108:457–463. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- 39.Heeschen C., Lehmann R., Honold J., Assmus B., Aicher A., Walter D.H., Martin H., Zeiher A.M., Dimmeler S. Profoundly reduced neovascularization capacity of bone marrow mononuclear cells derived from patients with chronic ischemic heart disease. Circulation. 2004;109:1615–1622. doi: 10.1161/01.CIR.0000124476.32871.E3. [DOI] [PubMed] [Google Scholar]
- 40.Knapp L.T., Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J. Biol. Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- 41.Vallee B.L., Coleman J.E., Auld D.S. Zinc fingers, zinc clusters, and zinc twists in DNA-binding protein domains. Proc. Natl. Acad. Sci. USA. 1991;88:999–1003. doi: 10.1073/pnas.88.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakrabarty S.P., Balaram H. Reversible binding of zinc in Plasmodium falciparum Sir2: structure and activity of the apoenzyme. Biochim. Biophys. Acta. 2010;1804:1743–1750. doi: 10.1016/j.bbapap.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Furey W.F., Robbins A.H., Clancy L.L., Winge D.R., Wang B.C., Stout C.D. Crystal structure of Cd,Zn metallothionein. Science. 1986;231:704–710. doi: 10.1126/science.3945804. [DOI] [PubMed] [Google Scholar]
- 44.Baldwin M.A., Benz C.C. Redox control of zinc finger proteins. Methods Enzymol. 2002;353:54–69. doi: 10.1016/s0076-6879(02)53036-6. [DOI] [PubMed] [Google Scholar]
- 45.Giles N.M., Watts A.B., Giles G.I., Fry F.H., Littlechild J.A., Jacob C. Metal and redox modulation of cysteine protein function. Chem. Biol. 2003;10:677–693. doi: 10.1016/s1074-5521(03)00174-1. [DOI] [PubMed] [Google Scholar]
- 46.Maret W. New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 2012;111:110–116. doi: 10.1016/j.jinorgbio.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 47.Boissier F., Georgescauld F., Moynie L., Dupuy J.W., Sarger C., Podar M., Lascu I., Giraud M.F., Dautant A. An intersubunit disulfide bridge stabilizes the tetrameric nucleoside diphosphate kinase of Aquifex aeolicus. Proteins. 2012;80:1658–1668. doi: 10.1002/prot.24062. [DOI] [PubMed] [Google Scholar]
- 48.Chu X.P., Close N., Saugstad J.A., Xiong Z.G. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J. Neurosci. 2006;26:5329–5339. doi: 10.1523/JNEUROSCI.0938-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sica F., Di Fiore A., Merlino A., Mazzarella L. Structure and stability of the non-covalent swapped dimer of bovine seminal ribonuclease: an enzyme tailored to evade ribonuclease protein inhibitor. J. Biol. Chem. 2004;279:36753–36760. doi: 10.1074/jbc.M405655200. [DOI] [PubMed] [Google Scholar]
- 50.Altamirano M.M., Mulliert G., Calcagno M. Sulfhydryl groups of glucosamine-6-phosphate isomerase deaminase from Escherichia coli. Arch. Biochem. Biophys. 1987;258:95–100. doi: 10.1016/0003-9861(87)90326-2. [DOI] [PubMed] [Google Scholar]
- 51.Bunting K.A., Cooper J.B., Tickle I.J., Young D.B. Engineering of an intersubunit disulfide bridge in the iron-superoxide dismutase of Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2002;397:69–76. doi: 10.1006/abbi.2001.2635. [DOI] [PubMed] [Google Scholar]
- 52.Gokhale R.S., Agarwalla S., Francis V.S., Santi D.V., Balaram P. Thermal stabilization of thymidylate synthase by engineering two disulfide bridges across the dimer interface. J. Mol. Biol. 1994;235:89–94. doi: 10.1016/s0022-2836(05)80018-x. [DOI] [PubMed] [Google Scholar]
- 53.Samanta M., Banerjee M., Murthy M.R., Balaram H., Balaram P. Probing the role of the fully conserved Cys126 in triosephosphate isomerase by site-specific mutagenesis--distal effects on dimer stability. FEBS J. 2011;278:1932–1943. doi: 10.1111/j.1742-4658.2011.08110.x. [DOI] [PubMed] [Google Scholar]
- 54.Gross S., Jones C., Hattori Y., Raman C. Academic Press; San Diego, CA: 2000. Tetrahydrobiopterin: an essential cofactor of nitric oixde synthase with an elusive role. Nitric oixde biology and pathobiology; pp. 167–187. [Google Scholar]
- 55.Kwon N.S., Nathan C.F., Stuehr D.J. Reduced biopterin as a cofactor in the generation of nitrogen oxides by murine macrophages. J. Biol. Chem. 1989;264:20496–20501. [PubMed] [Google Scholar]
- 56.Mayer B., Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells [erratum appears in Trends Biochem Sci 1998 Feb;23(2):87] Trends Bioch. Sci. 1997;22:477–481. doi: 10.1016/s0968-0004(97)01147-x. [DOI] [PubMed] [Google Scholar]
- 57.Tayeh M.A., Marletta M.A. Macrophage oxidation of l-arginine to nitric oxide, nitrite, and nitrate. Tetrahydrobiopterin is required as a cofactor. J. Biol. Chem. 1989;264:19654–19658. [PubMed] [Google Scholar]
- 58.Tummala M., Ryzhov V., Ravi K., Black S.M. Identification of the cysteine nitrosylation sites in human endothelial nitric oxide synthase. DNA Cell Biol. 2008;27:25–33. doi: 10.1089/dna.2007.0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai S., Khoo J., Channon K.M. Augmented BH4 by gene transfer restores nitric oxide synthase function in hyperglycemic human endothelial cells. Cardiovasc. Res. 2005;65:823–831. doi: 10.1016/j.cardiores.2004.10.040. [DOI] [PubMed] [Google Scholar]
- 60.Erwin P.A., Lin A.J., Golan D.E., Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 2005;280:19888–19894. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 61.Rodriguez-Crespo I., Gerber N.C., Ortiz de Montellano P.R. Endothelial nitric-oxide synthase. Expression in Escherichia coli, spectroscopic characterization, and role of tetrahydrobiopterin in dimer formation. J. Biol. Chem. 1996;271:11462–11467. doi: 10.1074/jbc.271.19.11462. [DOI] [PubMed] [Google Scholar]