

Abstract
Here, we review the role of oxidative protein modification as a signal for recognition and degradation of proteins. It was clearly demonstrated that the ATP- and ubiquitin-independent 20S proteasome is playing a key role in the selective removal of oxidized proteins. Furthermore, the current knowledge of the substrate susceptibility on the degradation of oxidized proteins and the role of the immunoproteasome will be highlighted.
Keywords: 20S Proteasome, Protein oxidation, Immunoproteasome, Protein aggregates
Introduction
In the first part of this series we described the principle proteasomal structure and its regulatory complexes [1]. Here we focus on the role of protein oxidation in the substrate recognition of the proteasome.
Cellular metabolism is accompanied by constant formation of free radicals and oxidants. Depending on the cell type, some 200 different reactive species can be found in humans [2], physiological/pathophysiological conditions, cellular substrate turnover, specialization and the functional proteome, age and environmental factors the cellular radical formation may vary in a broad range.
Reactive oxygen (ROS) and nitrogen species (RNS) have been considered as the major cause of aging, aging associated cellular dysfunctions, and a main factor in many pathologies. Though, meanwhile it became clear that ROS are also integral mediators of cellular adaption, summarized under the term redox signaling. Nevertheless, ROS are able to oxidatively damage/modify cell structures as proteins, lipids or nucleic acids. Due to the abundance of proteins, a bulk of ROS-induced oxidative damage is taken by them, interestingly predominantly by cytosolic proteins. Usually, the nuclear compartment shows only very low amounts of oxidatively modified proteins or aggregates of oxidized proteins, even after phases of severe oxidative stress [3–5].
However, since the permanent oxidative modification of proteins is an inevitable by-product of metabolism in every living cell, several different “counteracting” systems have evolved. A few of them are specialized in the repair of oxidatively damaged proteins, but their capacity is very limited: the only two known amino acids that can be repaired in an enzymatic way in mammalian cells are cysteine and methionine, while the bulk of oxidative modification is irreversible. Fig. 1 gives an overview over the most common reversible and irreversible protein-modifications. If proteins become oxidatively modified/damaged in an irreversible way, cells need effective systems for recognition and removing. For this purpose a cell provides different systems for degradation of proteins, as the lysosomal system, mitochondrial proteases (mainly the Lon protease [6,7]), different calcium-dependent proteases and the proteasomal system [8]. The multicatalytic 20S proteasome, an evolutionary very ancient system, has been described in detail in the first part of this series [1]. Proteasomes can be found in all three kingdoms of life: bacteria (in the archaea), plants, and animals.
Fig. 1.
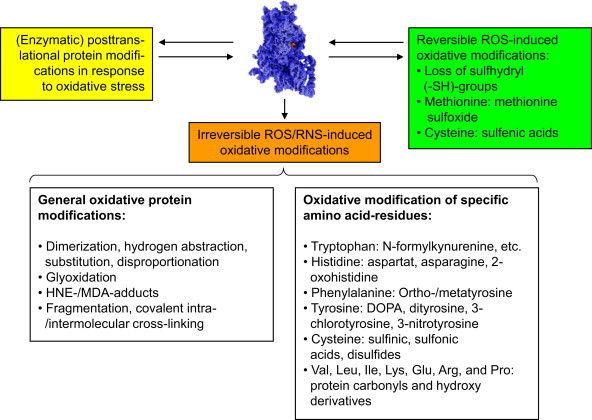
Typical ROS/RNS-mediated protein and side-chain modifications. This figure depicts some of the main reversible and irreversible protein modifications caused by ROS/RNS. The upper part shows different modifications some proteins can undergo in cells exposed to oxidative stress. Some of them are reversible oxidative modification (green box), that can be reversed by the cellular enzymatic machinery (see text below); another reversible pathway is the modification by cellular enzymes, that occurs in response to oxidative stress (yellow box). These modifications can be induced by ROS/RNS directly or by enzymatic reactions in response to ROS/RNS or a shifted redox-state of the cell, a common example for this is the so-called S-glutathionylation, mainly induced by oxidation of cysteine residues and reversed in an enzymatic way [23–25]. Another category is the formation of irreversible oxidative modifications by ROS/RNS that cannot be reversed by cellular enzymes (orange). Such proteins are usually recognized and degraded by specialized cellular enzymatic systems [8,9,26–30]. The lower part of the figure lists oxidative protein modification, classified by general principles or specific amino acid side-chains reactions. Reversible modifications are mainly found in cysteine and methionine residues, the only two amino acids that can be reduced/repaired by the cellular antioxidative enzymatic machinery. Methionine sulfoxide (MetSO) can be reduced by the methionine sulfoxide reductases Msr-A (specific for the S-stereoisomer) and Msr-B (specific for the R-stereoisomer of MetSO); both of them (Msr-A/B) use thioredoxin (Th-(SH)2) as reducing elements; after this, Th-(S-S) is reduced to Thr-(SH)2 again by the enzyme thioredoxin reductase in a NADPH-consuming way. The other amino acid residue very susceptible to ROS/RNS is cysteine. Its oxidation causes in proteins intra- or intermolecular cross-links (disulfides). Similar to MetSO, cysteine can be reduced by thioltransferases, that use either glutathione (GSH) or reduced thioredoxin (Th-(SH)2) in order to reduce a disulfide (–S–S–) into two separate –SH-groups (sulfhydryls). Of the different stages of cysteine oxidation, only the formation of the cysteinyl radical (protein-Cys-S•) and oxidation to sulfenic acid (protein-Cys-SOH) is reversible, while oxidation to sulfinic and sulfonic acid is irreversible, despite of a single known and highly specialized exception: sulfiredoxin is actually able to reduce sulfinic acid (protein-Cys-SO2H) in peroxiredoxins in an ATP-consuming reaction [31]. A loss of SH-groups may result in protein mis-/unfolding, inactivation (catalytic center), decreased antioxidative capacity, as well as the loss of specific functions. The variation of irreversible protein modifications outnumber the reversible ones by far and have in common that they cannot be repaired/reduced by the antioxidative machinery of the cell. Such general modifications (left description field of the lower part of this figure) can be induced by attacks of highly reactive radicals like hydroxyl, that are able to induce fragmentation of the protein, while attack on glycine seems to play a major role, as well as on proline, histidine and lysine [32]; furthermore, histidine is important in the formation of covalent cross-links [33]. Other events are de- and transamination (of glutamine and asparagine residues) that even can occur in a spontaneous way and does not have to be mediated/induced by ROS/RNS [34]. Furthermore, the formation of so-called advanced glycation end products (AGE's) [20,35,36] has been shown: Nε-carboxymethyllysine (CML) and Nε-carboxyethyllysine (CEL), as well as different glyoxal-lysine dimers (GOLDs) and methylglyoxal-lysine dimers (MOLDs) or pentosidine [37]. These AGEs are products of sugars and proteins, forming glycated proteins that may occur also from methylglyoxal, a potent glycating agent derived from trioses. Very prone to oxidative modifications are also the lipids in a cell. After ROS/RNS-mediated damage, amongst others highly reactive aldehydes are formed, that are able to react with proteins. The main reactive aldehydes are 4-hydroxy-2,3-nonenal (HNE, one of the most abundant products of lipid peroxidation, a bifunctional aldehyde, able to covalently cross-link proteins via reaction with either cysteine, lysine or histidine, followed by reaction with a lysine residue of another protein) [38], 4-hydroxyhexenal (HHE), malondialdehyde (MDA, forms Nε-malondialdehydelysine with lysine residues or the fluorescent adduct 1,4-dihydropyridine-3,5-dicarbaldehydes) [39]. The aldehydes glyoxal and acrolein react mainly with lysine, arginine, and histidine. The according end products of the mentioned reactions are referred to in the literature as “advanced lipid peroxidation end products” (ALEs)[37]. A typical step in the fragmentation of the protein backbone is the formation of an alkoxyl radical within the protein, that can decay either via the so-called diamide or α-amination pathways [40]. The irreversible oxidative modifications of specific residues show a large variety, but in biological systems several predominant modifications can be found some of them listed in the right description field of the lower part of this figure. In cells, the formation of 3-nitrotyrosine is mainly a hint to the presence of peroxynitrite (ONOO−), and thus the immunochemical detection of 3-nitrotyrosine became a quantitative and qualitative marker for ONOO−-mediated protein oxidation [41]. Dityrosines are mainly formed via the reaction of two tyrosyl radicals [42]. Those can be formed by the reaction of tyrosine side chains with hydroxyl radicals, hypochlorite or peroxynitrite [43]. Furthermore, hydroxyl radical mediated hydroxylation of phenylalanine, tyrosine and tryptophan plays a major role as well as comparable reactions of histidine, forming 2-oxohistidine [44]. Protein carbonyls [3,5] are the most abundant oxidative protein modification – their rate of formation is about 10-times higher than for any other oxidative protein modification. Protein carbonyls are mainly formed by oxidation of valine, leucine, isoleucine, lysine, glutamine, arginine, and proline side chains. Due to their high occurrence and establishment of easy to handle methods, protein carbonyls are the most often used quantitative marker of oxidative proteinmodification. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
A large body of evidence demonstrated that the 20S proteasome is the main proteolytic system removing oxidatively damaged proteins [1,9–12]. In contrast to the degradation of native proteins, the degradation of oxidatively damaged proteins does neither require the presence of ATP nor the polyubiquitination of the substrate, since an inhibition of the ubiquitinating system did only reveal any impact on the degradation of oxidized proteins as shown in Fig. 2. In fact, the 26S proteasome turned out to be very poor at degrading oxidatively damaged proteins [13]. Especially in phases of oxidative stress, the 26S proteasome disassembles (release of 19S) as well as the ubiquitinating system (especially E1- and E2-enzymes [14]) become deactivated already at peroxynitrite, hypochlorite or H2O2 concentrations that are about one magnitude lower than the concentrations needed to inactivate the uncapped 20S proteasome [15]. Oxidative damage/modification is not really a defined state or structure, but a slight transition from a natively folded and fully functional protein to a massively oxidized and covalently cross-linked state as depicted in Fig. 3. Thus, this transition is not defined stages by discreet stages of oxidation. Therefore, certain criteria as the exposure of hydrophobic structures that are normally buried inside of a natively folded protein render an oxidatively damaged protein susceptible to proteolytic degradation mediated by the 20S proteasome. Those exposed hydrophobic structures are proposed to be responsible for proteasomal substrate recognition [16]. In any case, the transition from a natively folded protein to a slightly oxidized unfolded proteasomal substrate is often not a “one hit” event, but has to be considered as the accumulation of several single events.
Fig. 2.
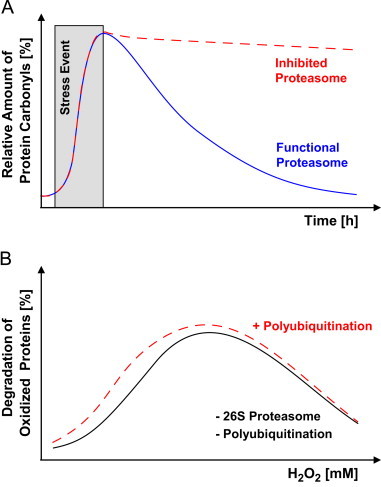
Degradation of oxidized proteins is mainly mediated by the 20S proteasome. Panel A shows the effect of proteasomal inhibition on the degradation of oxidized proteins. If cells are exposed to ROS/RNS (time of exposure highlighted in gray) a dramatic increase in the formation of oxidatively modified proteins takes place. These oxidized proteins (here measured as protein carbonyls) have to be recognized and degraded in order to maintain the functionality of the cell. According to experimental results, 80%–90% of all oxidatively damaged proteins are degraded via the 20S proteasomal pathway in an ATP-independent way [1,45–47]. Consequently, if the proteasome is inhibited such oxidized proteins are not removed from the protein pool (red dashed line) [5]. Experimental data show, that the 20S proteasome is a very effective system that reduced the amount of protein carbonyls in a cell within several hours to the level found in unexposed/unstressed cells (blue continuous line) [5,41]. If the 20S proteasome is inhibited or knocked down by siRNA or Antisense nucleotides (red dashed line), the amount of protein carbonyls might still decrease over time, but at a much lower rate, since the other cellular systems (for example the large diversity of lysosomal cathepsins) might be capable of degrading oxidized proteins, too, but show a much lower performance, or the proteasomal inhibition is not complete. Thus, proteasomal inhibition, a decrease of proteasomal activity or the loss of proteasomes promotes the accumulation of oxidized proteins. Panel B displays the contribution of the 26S proteasome or of the ubiquitination to the degradation of oxidized proteins. The 26S proteasome might be blocked by siRNA and ubiquitination by the usage of conditionally ubiquitin deficient cell lines [48,49]. After an E1-enzyme inactivation in such conditionally ubiquitin deficient cell lines, the amount of substrate polyubiquitination and thus labeling for 26S proteasomal degradation [1] significantly decreases. Neither of these modulations shows any significant impact on the degradation of oxidized proteins. In this panel the theoretical proteolytic response of cells to a given ROS concentration (here hydrogen peroxide) is demonstrated [12,15,48,50,51]. Elimination of the 26S proteasome or of the ubiquitination capacities do not have any influence on this response. Taken together, this demonstrates that the 20S proteasome is the main system removing oxidized proteins from the cellular protein pool, and that oxidized proteins do not have to be polyubiquitinylated in order to be degraded. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
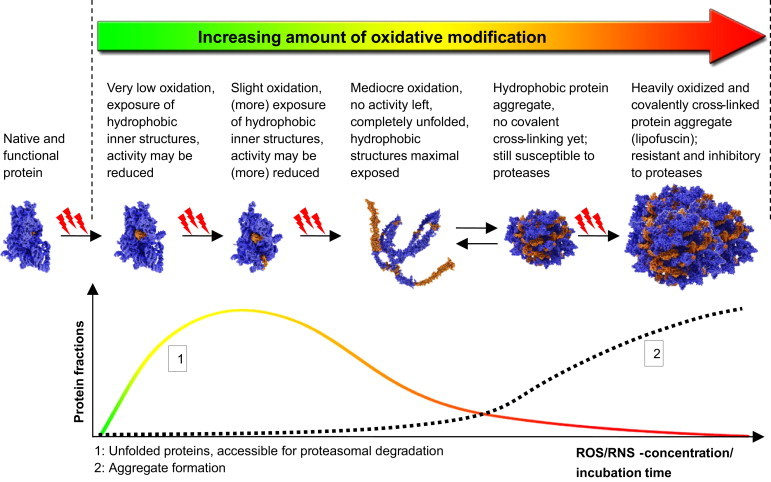
Increasing levels of oxidative protein damage affects the proteasomal degradation. The upper part of this figure shows the continuum of oxidative protein damage and the resulting modification and unfolding (according to Grune et al. [16]). Important to note, that oxidative modification of a protein is a continuous process covering a very broad range of successively increasing damage. An undamaged, natively folded protein (shown on the left in the upper row of images) is usually not recognized as a substrate by the 20S proteasome. After slight oxidative damage a protein might show only insignificant changes in folding or activity. Here the solubility of the protein increases due to additional charges that are brought in by oxidation. After further oxidative damage, hydrophobic sequences, normally buried in the inside of the protein are exposed more and more to its surface and thus the aqueous environment (third image from the left). The damage-induced conformational change causes significant loss of activity until the protein is completely unfolded (fourth image from the left). Such proteins are ideal substrates for proteasomal degradation and most of those proteins are actually recognized and removed via the 20S proteasomal pathway. However, the exposure of hydrophobic moieties to the protein surface holds the risk of a thermodynamically driven protein aggregate formation. So initial aggregate formation is mainly driven by thermodynamics: the exposed hydrophobic sequences of the damaged proteins tend to form clusters in order to minimize their interaction with the aqueous environment. These aggregates can still decompose again and be degraded as single unfolded proteins mainly by the 20S proteasome. Here, chaperones might play a role, but this is still not demonstrated conclusively. Though, under oxidizing conditions the probability of further protein oxidation and thus covalent cross-linking of the aggregated proteins increases. Once they are covalently cross-linked, they become resistant to (proteasomal) proteolytic degradation. Such material has been shown to be resistant even to the most unspecific proteases [17]. Such a cross-linking might take place by direct protein–protein interaction, e.g. tyrosine radicals and dityrosine formation or protein carbonyl and amino groups via a Schiff's-base formation. Furthermore, this cross-linking may be facilitated by products of lipid-peroxidation, mainly bifunctional aldehydes like MDA or HNE. Heavily cross-linked protein aggregates may show an autofluorescence that may cover a very broad spectrum of the visible range. One of the few already identified fluorescent structures is the pyridinium bisretinoid A2E, also a secondary product of lipid peroxidation [52] that can be excited using an Argon laser (458 nm) [53]; further structures are 1,4-dihydropyridine, that results from the reaction of MDA with glycine [54–56] as well as 2-hydroxy-3-imino-1,2-dihydropyrrol derivatives that result from the cyclization of lysine-HNE Michael adducts [57]. This heavily oxidized material, containing both lipids and proteins, as well as low amounts of sugars is termed in the literature as “lipofuscin” [17,18,58–62]. Lipofuscin accumulates during aging, especially in postmitotic aging cells. However, lipofuscin can be formed with an increased rate during different pathologies, termed as “lipofuscinoses” or “ceroid-lipofuscinoses” [63,64]. These protein aggregates can incorporate redox-active transition metals (in mammalian cells mainly Fe2+) [45], thus catalyzing radical formation via Fenton-reaction, again increasing the rate of protein oxidation and lipofuscin-formation [59]. Interestingly, lipofuscin has been shown to be a potent proteasomal inhibitor [17], Lipofuscin, the long-term product of oxidative stress, accumulates mainly in the lysosomal system, while only a very low amount (about 1%) is found free in the cytosol [18], where it may interact with the proteasomal system, slowing down the degradation of lipofuscin-precursor-material. The lower part of the figure summarizes the different effects of the exposure-time or the amount of oxidative stress applied to a cells on the proteolytic response. Curve 1 shows the amount of oxidized proteins that are accessible for proteasomal degradation. Over time or with increasing ROS/RNS-concentration this amount first increases due to a rising number of damaged proteins in the cell, but then decreases again, due to the rising amount of non-degradable covalently-cross-linked material formed, that is very resistant to proteolytic attacks. (compare to Fig. 2B). Curve 2 depicts this increasing amount of protein aggregate formation that decreases the amount of damaged proteins accessible to proteasomal degradation shown in Curve 1.
Proteolytic degradation of an oxidized protein has different aspects in the protection of the normal function of a cell. Furthermost, is the avoidance of the formation of highly oxidized, cross-linked protein aggregates, that may grow [17,18] become further oxidized and covalently cross-linked. Such aggregates are largely resistant to enzymatic degradation. Therefore, it is of key importance to reduce the amount of oxidatively damaged proteins after a stress event. Several regulatory events, as the release of 20S proteasome by disintegration of the 26S proteasome and the release of its regulators [19] and the induction of the immunoproteasome, are the consequence of oxidative stress. Interestingly an induction of the immunoproteasomal and 11S subunits was observed after exposure to modified proteins or oxidative stress [20,21]. Most interestingly, the immunoproteasome in combination with the 11S do have a higher activity towards the oxidatively modified proteins (Fig. 4) and degrade them with a higher efficiency. Since the induction of the immunoproteasome is fast compared with the turnover of the constitutive proteasome [22], the immunoproteasomal form might significantly contribute to the degradation of oxidized proteins. Therefore, this form of the proteasome is not only involved in antigen presentation (hence the name ‘immunoporteasome’), but seems to be involved in several cellular processes as an inducible proteasomal form.
Fig. 4.
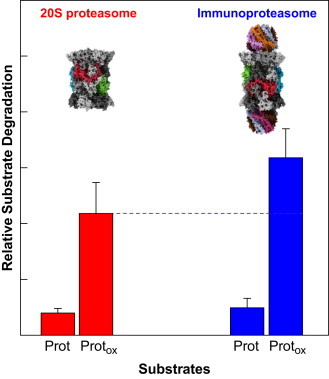
Proteasome and immunoproteasome are involved in the degradation of oxidized proteins. This figure compares the proteolytic capacities of both the constitutive 20S proteasome (left side, red columns) and its inducible form, the immunproteasome or i20S, respectively (shown on the right side, blue barns). The immunoproteasome and its 11S regulator can be induced by TNF-α, interferon-γ, lipopolysaccharides, AGEs or just oxidative stress, as recently shown [20,65–68]. The column “Prot” shows the insignificant degradation of a natively folded protein, “Protox” the degradation of the same protein after oxidative damage by an oxidative agent. Thus, both proteasomal forms are able to recognize oxidized proteins more effectively [21]. However the immunoproteasome seems to be more efficient compared to the constitutive 20S proteasome (purple dashed line). Both 20S and i20S degrade unfolded proteins in an ATP-independent way. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Summary
The efficient degradation and removal of oxidized proteins is an absolute requirement for the maintenance of the cellular metabolism. It was demonstrated that the 20S proteasome is in an ATP- and ubiquitin-independent way responsible for the degradation of oxidized proteins. Furthermore, the substrate susceptibility is dependent on the degree of its oxidation. Interestingly, lately an involvement of the immunoproteasome and the 11S proteasomal regulator was proposed to play a role in the degradation of oxidized proteins.
References
- 1.Jung T., Grune T. The proteasome and the degradation of oxidized proteins: Part I – structure of proteasomes. Redox Biol. 2012;1:178–182. doi: 10.1016/j.redox.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volarevic V., Ljujic B., Stojkovic P., Lukic A., Arsenijevic N., Stojkovic M. Human stem cell research and regenerative medicine – present and future. Br. Med. Bull. 2011;99:155–168. doi: 10.1093/bmb/ldr027. [DOI] [PubMed] [Google Scholar]
- 3.Jung T., Hohn A., Catalgol B., Grune T. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Arch. Biochem. Biophys. 2009;483:127–135. doi: 10.1016/j.abb.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Jung T., Engels M., Kaiser B., Grune T. Distribution of oxidized and HNE-modified proteins in U87 cells. Biofactors. 2005;24:165–170. doi: 10.1002/biof.5520240120. [DOI] [PubMed] [Google Scholar]
- 5.Jung T., Engels M., Kaiser B., Poppek D., Grune T. Intracellular distribution of oxidized proteins and proteasome in HT22 cells during oxidative stress. Free Radic. Biol. Med. 2006;40:1303–1312. doi: 10.1016/j.freeradbiomed.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 6.Ngo J.K., Pomatto L.C., Davies K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013;1:258–264. doi: 10.1016/j.redox.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rigas S., Daras G., Tsitsekian D., Hatzopoulos P. The multifaceted role of Lon proteolysis in seedling establishment and maintenance of plant organelle function: living from protein destruction. Physiol. Plant. 2012;145:215–223. doi: 10.1111/j.1399-3054.2011.01537.x. [DOI] [PubMed] [Google Scholar]
- 8.Grimm S., Hohn A., Grune T. Oxidative protein damage and the proteasome. Amino Acids. 2012;42:23–38. doi: 10.1007/s00726-010-0646-8. [DOI] [PubMed] [Google Scholar]
- 9.Jung T., Grune T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life. 2008;60:743–752. doi: 10.1002/iub.114. [DOI] [PubMed] [Google Scholar]
- 10.Sitte N., Merker K., Grune T. Proteasome-dependent degradation of oxidized proteins in MRC-5 fibroblasts. FEBS Lett. 1998;440:399–402. doi: 10.1016/s0014-5793(98)01495-1. [DOI] [PubMed] [Google Scholar]
- 11.Grune T., Reinheckel T., Davies K.J. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- 12.Grune T., Reinheckel T., Davies K.J. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. J. Biol. Chem. 1996;271:15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- 13.Davies K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 14.Shang F., Taylor A. Oxidative stress and recovery from oxidative stress are associated with altered ubiquitin conjugating and proteolytic activities in bovine lens epithelial cells. Biochem. J. 1995;307(Pt 1):297–303. doi: 10.1042/bj3070297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinheckel T., Sitte N., Ullrich O., Kuckelkorn U., Davies K.J., Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 1998;335(Pt 3):637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasch P., Petras T., Ullrich O., Backmann J., Naumann D., Grune T. Hydrogen peroxide-induced structural alterations of RNAse A. J. Biol. Chem. 2001;276:9492–9502. doi: 10.1074/jbc.M008528200. [DOI] [PubMed] [Google Scholar]
- 17.Hohn A., Jung T., Grimm S., Catalgol B., Weber D., Grune T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radic. Biol. Med. 2011;50:585–591. doi: 10.1016/j.freeradbiomed.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Hohn A., Sittig A., Jung T., Grimm S., Grune T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic. Biol. Med. 2012;53:1760–1769. doi: 10.1016/j.freeradbiomed.2012.08.591. [DOI] [PubMed] [Google Scholar]
- 19.Grune T., Catalgol B., Licht A., Ermak G., Pickering A.M., Ngo J.K. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 2011;51:1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimm S., Ott C., Horlacher M., Weber D., Hohn A., Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem. J. 2012;448:127–139. doi: 10.1042/BJ20120298. [DOI] [PubMed] [Google Scholar]
- 21.Pickering A.M., Koop A.L., Teoh C.Y., Ermak G., Grune T., Davies K.J. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010;432:585–594. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka K., Ichihara A. Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem. Biophys. Res. Commun. 1989;159:1309–1315. doi: 10.1016/0006-291x(89)92253-5. [DOI] [PubMed] [Google Scholar]
- 23.Petrushanko I.Y., Yakushev S., Mitkevich V.A., Kamanina Y.V., Ziganshin R.H., Meng X. S-glutathionylation of the Na,K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012;287:32195–32205. doi: 10.1074/jbc.M112.391094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrini S., Passarelli C., Pastore A., Tozzi G., Coccetti M., Colucci M. Protein glutathionylation in cellular compartments: a constitutive redox signal. Redox Rep. 2012;17:63–71. doi: 10.1179/1351000212Y.0000000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastore A., Piemonte F. S-Glutathionylation signaling in cell biology: progress and prospects. Eur. J. Pharm. Sci. 2012;46:279–292. doi: 10.1016/j.ejps.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Hohn A., Konig J., Grune T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteomics. 2013;92:132–159. doi: 10.1016/j.jprot.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Ott C., Grune T. Protein oxidation and proteolytic signalling in aging. Curr. Pharm. Des. 2013 doi: 10.2174/13816128113196660709. [DOI] [PubMed] [Google Scholar]
- 28.Kastle M., Grune T. Protein oxidative modification in the aging organism and the role of the ubiquitin proteasomal system. Curr. Pharm. Des. 2011;17:4007–4022. doi: 10.2174/138161211798764898. [DOI] [PubMed] [Google Scholar]
- 29.Catalgol B., Grune T. Protein pool maintenance during oxidative stress. Curr. Pharm. Des. 2009;15:3043–3051. doi: 10.2174/138161209789058129. [DOI] [PubMed] [Google Scholar]
- 30.Jung T., Bader N., Grune T. Oxidized proteins: intracellular distribution and recognition by the proteasome. Arch. Biochem. Biophys. 2007;462:231–237. doi: 10.1016/j.abb.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 31.Jeong W., Bae S.H., Toledano M.B., Rhee S.G. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic. Biol. Med. 2012;53:447–456. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 32.Monboisse J.C., Braquet P., Randoux A., Borel J.P. Non-enzymatic degradation of acid-soluble calf skin collagen by superoxide ion: protective effect of flavonoids. Biochem. Pharmacol. 1983;32:53–58. doi: 10.1016/0006-2952(83)90651-2. [DOI] [PubMed] [Google Scholar]
- 33.Guptasarma P., Balasubramanian D., Matsugo S., Saito I. Hydroxyl radical mediated damage to proteins, with special reference to the crystallins. Biochemistry. 1992;31:4296–4303. doi: 10.1021/bi00132a021. [DOI] [PubMed] [Google Scholar]
- 34.Lindner H., Helliger W. Age-dependent deamidation of asparagine residues in proteins. Exp. Gerontol. 2001;36:1551–1563. doi: 10.1016/s0531-5565(01)00140-1. [DOI] [PubMed] [Google Scholar]
- 35.Grimm S., Horlacher M., Catalgol B., Hoehn A., Reinheckel T., Grune T. Cathepsins D and L reduce the toxicity of advanced glycation end products. Free Radic. Biol. Med. 2012;52:1011–1023. doi: 10.1016/j.freeradbiomed.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 36.Nedic O., Rattan S.I., Grune T., Trougakos I.P. Molecular effects of advanced glycation end products (AGEs) on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013;47:28–38. doi: 10.3109/10715762.2013.806798. [DOI] [PubMed] [Google Scholar]
- 37.Baynes J.W., Thorpe S.R. Glycoxidation and lipoxidation in atherogenesis. Free Radic. Biol. Med. 2000;28:1708–1716. doi: 10.1016/s0891-5849(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 38.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 39.Lambert A.J., Portero-Otin M., Pamplona R., Merry B.J. Effect of ageing and caloric restriction on specific markers of protein oxidative damage and membrane peroxidizability in rat liver mitochondria. Mech. Ageing Dev. 2004;125:529–538. doi: 10.1016/j.mad.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Berlett B.S., Stadtman E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 41.Jung T., Engels M., Klotz L.O., Kroncke K.D., Grune T. Nitrotyrosine and protein carbonyls are equally distributed in HT22 cells after nitrosative stress. Free Radic. Biol. Med. 2007;42:773–786. doi: 10.1016/j.freeradbiomed.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 42.Sohal R.S. Role of oxidative stress and protein oxidation in the aging process. Free Radic. Biol. Med. 2002;33:37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 43.van d V., Eiserich J.P., O’Neill C.A., Halliwell B., Cross C.E. Tyrosine modification by reactive nitrogen species: a closer look. Arch. Biochem. Biophys. 1995;319:341–349. doi: 10.1006/abbi.1995.1303. [DOI] [PubMed] [Google Scholar]
- 44.Uchida K., Kawakishi S. 2-Oxo-histidine as a novel biological marker for oxidatively modified proteins. FEBS Lett. 1993;332:208–210. doi: 10.1016/0014-5793(93)80632-5. [DOI] [PubMed] [Google Scholar]
- 45.Jung T., Catalgol B., Grune T. The proteasomal system. Mol. Aspects Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Bader N., Jung T., Grune T. The proteasome and its role in nuclear protein maintenance. Exp. Gerontol. 2007;42:864–870. doi: 10.1016/j.exger.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 47.Grune T., Botzen D., Engels M., Voss P., Kaiser B., Jung T. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch. Biochem. Biophys. 2010;500:181–188. doi: 10.1016/j.abb.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shringarpure R., Grune T., Mehlhase J., Davies K.J. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 49.Poppek D., Keck S., Ermak G., Jung T., Stolzing A., Ullrich O. Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem. J. 2006;400:511–520. doi: 10.1042/BJ20060463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kastle M., Reeg S., Rogowska-Wrzesinska A., Grune T. Chaperones, but not oxidized proteins, are ubiquitinated after oxidative stress. Free Radic. Biol. Med. 2012;53:1468–1477. doi: 10.1016/j.freeradbiomed.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 51.Kastle M., Grune T. Proteins bearing oxidation-induced carbonyl groups are not preferentially ubiquitinated. Biochimie. 2011;93:1076–1079. doi: 10.1016/j.biochi.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 52.Hammer M., Richter S., Kobuch K., Mata N., Schweitzer D. Intrinsic tissue fluorescence in an organotypic perfusion culture of the porcine ocular fundus exposed to blue light and free radicals. Graefes Arch. Clin. Exp. Ophthalmol. 2008;246:979–988. doi: 10.1007/s00417-008-0789-4. [DOI] [PubMed] [Google Scholar]
- 53.Marmorstein A.D., Marmorstein L.Y., Sakaguchi H., Hollyfield J.G. Spectral profiling of autofluorescence associated with lipofuscin, Bruch’s Membrane, and sub-RPE deposits in normal and AMD eyes. Investig. Ophthalmol. Vis. Sci. 2002;43:2435–2441. [PubMed] [Google Scholar]
- 54.Fang C., Peng M., Li G., Tian J., Yin D. New functions of glucosamine as a scavenger of the lipid peroxidation product malondialdehyde. Chem. Res. Toxicol. 2007;20:947–953. doi: 10.1021/tx700059b. [DOI] [PubMed] [Google Scholar]
- 55.Yin D.Z., Brunk U.T. Microfluorometric and fluorometric lipofuscin spectral discrepancies: a concentration-dependent metachromatic effect? Mech. Ageing Dev. 1991;59:95–109. doi: 10.1016/0047-6374(91)90076-c. [DOI] [PubMed] [Google Scholar]
- 56.Li L., Li G., Sheng S., Yin D. Substantial reaction between histamine and malondialdehyde: a new observation of carbonyl stress. Neuro Endocrinol. Lett. 2005;26:799–805. [PubMed] [Google Scholar]
- 57.Tsai L., Szweda P.A., Vinogradova O., Szweda L.I. Structural characterization and immunochemical detection of a fluorophore derived from 4-hydroxy-2-nonenal and lysine. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7975–7980. doi: 10.1073/pnas.95.14.7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sitte N., Merker K., Grune T., von Z.T. Lipofuscin accumulation in proliferating fibroblasts in vitro: an indicator of oxidative stress. Exp. Gerontol. 2001;36:475–486. doi: 10.1016/s0531-5565(00)00253-9. [DOI] [PubMed] [Google Scholar]
- 59.Hohn A., Jung T., Grimm S., Grune T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic. Biol. Med. 2010;48:1100–1108. doi: 10.1016/j.freeradbiomed.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 60.Jung T., Hohn A., Grune T. Lipofuscin: detection and quantification by microscopic techniques. Methods Mol. Biol. 2010;594:173–193. doi: 10.1007/978-1-60761-411-1_13. [DOI] [PubMed] [Google Scholar]
- 61.Jung T., Bader N., Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann. N. Y. Acad. Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 62.Hohn A., Grune T. Lipofuscin: formation, effects and role of macroautophagy. Redox Biol. 2013;1:140–144. doi: 10.1016/j.redox.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jolly R.D., Dalefield R.R., Palmer D.N. Ceroid lipofuscin and the ceroid-lipofuscinoses (Batten disease) J. Inherit. Metab. Dis. 1993;16:280–283. doi: 10.1007/BF00710265. [DOI] [PubMed] [Google Scholar]
- 64.Jolly R.D., Shimada A., Dopfmer I., Slack P.M., Birtles M.J., Palmer D.N. Ceroid-lipofuscinosis (Batten's disease): pathogenesis and sequential neuropathological changes in the ovine model. Neuropathol. Appl. Neurobiol. 1989;15:371–383. doi: 10.1111/j.1365-2990.1989.tb01236.x. [DOI] [PubMed] [Google Scholar]
- 65.Husom A.D., Peters E.A., Kolling E.A., Fugere N.A., Thompson L.V., Ferrington D.A. Altered proteasome function and subunit composition in aged muscle. Arch. Biochem. Biophys. 2004;421:67–76. doi: 10.1016/j.abb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 66.Ferrington D.A., Husom A.D., Thompson L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- 67.Ferrington D.A., Hussong S.A., Roehrich H., Kapphahn R.J., Kavanaugh S.M., Heuss N.D. Immunoproteasome responds to injury in the retina and brain. J. Neurochem. 2008;106:158–169. doi: 10.1111/j.1471-4159.2008.05345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas S., Kotamraju S., Zielonka J., Harder D.R., Kalyanaraman B. Hydrogen peroxide induces nitric oxide and proteosome activity in endothelial cells: a bell-shaped signaling response. Free Radic. Biol. Med. 2007;42:1049–1061. doi: 10.1016/j.freeradbiomed.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]