

Li et al. report ADP-ribosylation as a new post-translational modification of the tumor suppressor PTEN. Tankyrases interact with and ribosylate PTEN, which promotes the recognition of PTEN by a PAR-binding E3 ubiquitin ligase, RNF146, leading to PTEN ubiquitination and degradation. Tankyrases were up-regulated and negatively correlated with PTEN expression in human colon carcinomas.
Keywords: PTEN, PARsylation, tankyrase, RNF146, ubiquitination
Abstract
PTEN [phosphatidylinositol (3,4,5)-trisphosphate phosphatase and tensin homolog deleted from chromosome 10], a phosphatase and critical tumor suppressor, is regulated by numerous post-translational modifications, including phosphorylation, ubiquitination, acetylation, and SUMOylation, which affect PTEN localization and protein stability. Here we report ADP-ribosylation as a new post-translational modification of PTEN. We identified PTEN as a novel substrate of tankyrases, which are members of the poly(ADP-ribose) polymerases (PARPs). We showed that tankyrases interact with and ribosylate PTEN, which promotes the recognition of PTEN by a PAR-binding E3 ubiquitin ligase, RNF146, leading to PTEN ubiquitination and degradation. Double knockdown of tankyrase1/2 stabilized PTEN, resulting in the subsequent down-regulation of AKT phosphorylation and thus suppressed cell proliferation and glycolysis in vitro and tumor growth in vivo. Furthermore, tankyrases were up-regulated and negatively correlated with PTEN expression in human colon carcinomas. Together, our study revealed a new regulation of PTEN and highlighted a role for tankyrases in the PTEN–AKT pathway that can be explored further for cancer treatment.
PTEN [phosphatidylinositol (3,4,5)-trisphosphate phosphatase and tensin homolog deleted from chromosome 10] is an important tumor suppressor gene that is frequently lost due to deletions involving chromosome 10q23 (Li and Sun 1997; Li et al. 1997; Steck et al. 1997). Germline mutations of PTEN have been linked to Cowden syndrome (Liaw et al. 1997) and other disorders (Hobert and Eng 2009), indicating a role for PTEN in tumor suppression and other physiological functions. Studies of Pten-deficient mice substantiated the functions of Pten in embryonic development and tumor suppression (Di Cristofano et al. 1998; Suzuki et al. 1998).
PTEN is involved in sundry cellular processes. PTEN inhibits cell growth and apoptosis by suppressing the PI3K–AKT pathway; therefore, inhibition of PTEN leads to the activation of AKT and AKT-dependent downstream events (Manning and Cantley 2007). As an important regulator of many cellular processes, it is not surprising that PTEN is subjected to a variety of post-translational modifications that regulate its phosphatase activity as well as its subcellular localization. These modifications include phosphorylation (Torres and Pulido 2001), ubiquitination (Wang et al. 2007; Maddika et al. 2011), acetylation (Okumura et al. 2006), oxidation (Kwon et al. 2004), and SUMOylation (Huang et al. 2012; Bassi et al. 2013).
Tankyrase1 (TNKS1)/PARP5a and TNKS2/PARP5b are members of the poly(ADP-ribose) polymerase (PARP) family and are known to have diverse functions (Smith et al. 1998; Ye and de Lange 2004; Kim et al. 2011; Ozaki et al. 2012). Tankyrase-induced ribosylation of TRF1 targets it for proteasomal degradation, which leads to telomere elongation (Chang et al. 2003). TNKS1 can also PARsylate 3BP2, leading to the degradation of 3BP2 and thus facilitating normal bone formation (Levaot et al. 2011). More interestingly, tankyrases have been reported to regulate the Wnt signaling pathway by ribosylating Axin and promoting Axin degradation (Huang et al. 2009), which directly links tankyrase function to potential anti-cancer therapy with the discovery of tankyrase inhibitor XAV939 (Huang et al. 2009).
In this study, we identified PTEN as a novel tankyrase-binding protein. We demonstrated that tankyrases ribosylate PTEN and promote PTEN degradation, which is mediated by the E3 ubiquitin ligase RNF146. In addition, inhibition of tankyrase suppresses cell proliferation, glycolysis, colony formation, and tumor growth. Up-regulation of tankyrases correlate with down-regulation of PTEN in human colon cancer tissues. This new finding supports the further development of tankyrase inhibitors as anti-cancer agents.
Results
PTEN is a tankyrase-binding protein
To investigate the role of tankyrase inhibitor XAV939 in cell proliferation, we used four colorectal cancer cell lines: SW480, DLD1, RKO, and HCT116. As previously reported (Huang et al. 2009), XAV939 suppressed cell proliferation in SW480 and DLD1 cells, both of which express wild-type Axin and β-catenin. Interestingly, in HCT116 cells, which express a degradation-resistant β-catenin mutant (Morin et al. 1997), and RKO cells, which have undetectable β-catenin levels because of the wild-type APC, XAV939 also inhibited cell proliferation, albeit less effectively (Supplemental Fig. S1A). When we knocked down β-catenin in the Axin/β-catenin wild-type SW480 and 293T cells, XAV939 still suppressed growth in these cells (Supplemental Fig. S1B). These results suggested that the tankyrase inhibitor XAV939 can also regulate tumor cell proliferation in an Axin/β-catenin-independent manner, which prompted us to search for new tankyrase substrates that are involved in tumor cell proliferation. Excitingly, we found that the very N terminus of PTEN contains a motif similar to the RXXXDG tankyrase-binding motif (RYQEDG) (Fig. 1A). This motif is highly conserved (Fig. 1B), indicating that it may be important for PTEN function and/or regulation. Indeed, coimmunoprecipitation results showed that TNKS1 and TNKS2 were associated with PTEN (Fig. 1C). NEDD4 and WWP2, which are known PTEN-interacting proteins (Wang et al. 2007; Maddika et al. 2011), were included as positive controls. Moreover, the interaction between endogenous TNKS1 and PTEN was observed (Fig. 1D,E), further confirming that PTEN is a tankyrase-binding protein. To determine whether the RYQEDG motif at the N terminus of PTEN is critical for PTEN/TNKS1 interaction, we generated a mutant PTEN (PTEN△TBM) deleted of the putative tankyrase-binding motif (Supplemental Fig. S2A). Unlike wild-type PTEN, this mutant failed to bind either TNKS1 or TNKS2 (Supplemental Fig. S2B). To confirm this observation, we generated a site-directed point mutagenesis of the tankyrase-binding motif of PTEN (PTEN-AA) (Fig. 1A); this mutant also was unable to interact with overexpressed or endogenous TNKS1/2 (Fig. 1F; Supplemental Fig. S2C). Together, these data suggest that PTEN is a novel tankyrase-binding protein.
Figure 1.

PTEN is a tankyrase-binding protein. (A) Schematic representation of wild-type and the double point mutation of PTEN. The putative tankyrase-binding motif RYQEDG is indicated. (B) Alignment of the N terminus of PTEN in different species. The identical RYQEDG motif is indicated in red. (C) TNKS1 and TNKS2 interact with PTEN. 293T cells were transfected with plasmids encoding GFP-tagged PTEN together with vector or plasmids encoding myc-tagged NEDD4, WWP2, TNKS1, or TNKS2. Immunoprecipitation (IP) was carried out using anti-myc antibody and then subjected to Western blotting using the indicated antibodies. (D,E) Interaction between endogenous PTEN and TNKS1. Lysates from HCT116 cells were subjected to immunoprecipitation/Western analysis using the indicated antibodies. (F) The tankyrase-binding motif of PTEN is required for TNKS1/PTEN interaction. 293T cells were cotransfected with constructs encoding myc-tagged TNKS1/TNKS2 together with vector alone or construct encoding SFB-tagged wild-type or the tankyrase-binding motif point mutant of PTEN (PTEN-AA). Immunoprecipitation/Western analysis was conducted using the indicated antibodies.
PTEN is ADP-ribosylated by tankyrases in vitro and in vivo
To determine whether PTEN can be ribosylated, we used anti-PAR antibody to immunoprecipitate the PARylated proteins in cells. As a positive control, we detected ribosylated TNKS1 (Fig. 2A). Similarly, PTEN was also ribosylated (Fig. 2A). To eliminate the possibility of indirect binding of PTEN to PARylated proteins, we performed a denaturing immunoprecipitation of PTEN followed by immunoblotting for PAR and confirmed that PTEN is ribosylated in vivo (Fig. 2B), indicating that there is a new post-translational modification of PTEN that has never been reported. Next, we tested whether PTEN could be ribosylated by tankyrases. We performed an in vitro ribosylation reaction using recombinant TNKS1/2/PTEN and biotin-labeled NAD+. The reaction products were resolved by SDS-PAGE, and anti-biotin antibody was used to detect the ribosylated proteins. As shown in Figure 2C, TNKS1 and TNKS2 ribosylated not only themselves but also PTEN directly. The tankyrase inhibitor XAV939 sufficiently suppressed the ribosylation of TNKS1 and PTEN even at very low doses (Fig. 2D). To eliminate the possibility that other PARPs (PARP1/2/3) may PARylate PTEN, we performed an in vitro ribosylaltion assay and showed that PTEN was not the substrate of PARP1 (Supplemental Fig. S3A), and XAV939 had no effect on PARP1 ribosylation compared with the PARP1/2 inhibitor olaparib (Supplemental Fig. S3B). In addition, olaparib had a very mild effect on the ribosylaltion of TNKS1 and PTEN, but three different inhibitors of tankyrases (XAV939, JW55, and Wiki4) blocked TNKS1 and PTEN ribosylation significantly (Supplemental Fig. S3C). As PARP1/2/3 are DNA damage-activated PARPs, we also checked whether DNA damage would affect PTEN PARylation, and the result indicated that the PTEN/tankyrase interaction and ribosylation status of PTEN were not influenced by DNA damage (Supplemental Fig. S3E,F). All of these data suggest that ribosylation of PTEN is carried out directly by tankyrases but not other PARPs. Next, we generated a catalytically inactive mutant of TNKS1 (TNKS1-H1184A/E1291A, also known as TNKS1-PD [PARP-dead]). Wild-type TNKS1, but not TNKS1-PD, ribosylated itself and PTEN (Fig. 2E), again validating that PTEN ribosylation depends on the catalytic activity of TNKS1. Moreover, as shown in Figure 2F, the PTEN-AA mutant could not be ribosylated. Together, these data suggest that PTEN is a substrate of tankyrases.
Figure 2.
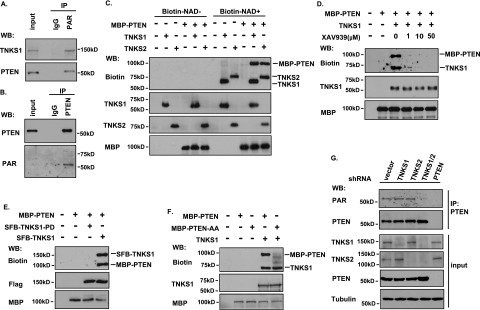
PTEN is ADP-ribosylated by tankyrases in vitro and in vivo. (A,B) PTEN is PARylated in vivo. (A) HCT116 cells were lysed with NETN buffer containing PARG inhibitor ADP-HPD (5 μM) and protease inhibitor. Lysates were then immunoprecipitated using control or anti-PAR antibodies and immunoblotted using the indicated antibodies. (B) HCT116 cells were lysed with NETN denaturing buffer containing PARG inhibitor ADP-HPD (5 μM) and protease inhibitor. Lysates were then immunoprecipitated using control or anti-PTEN antibodies followed by Western blotting as indicated. (C) Ribosylation of PTEN by TNKS1 and TNKS2 in vitro. Recombinant TNKS1, TNKS2, and PTEN were subjected to in vitro ribosylation assays in the absence or presence of biotin-labeled NAD+. The recombinant proteins were detected by the indicated antibodies, and the ribosylated proteins were determined with anti-biotin antibody. (D) The ribosylation of PTEN by TNKS1 is diminished by tankyrase inhibitor XAV939. The recombinant MBP-PTEN and TNKS1 were subjected to an in vitro ribosylation reaction as described above in the absence or presence of the indicated concentrations of XAV939. (E) The catalytic activity of TNKS1 is required for PTEN ribosylation. The MBP-PTEN and immunoprecipitated SFB-tagged wild-type or the catalytically inactive mutant of TNKS1 (TNKS1-PD) were subjected to an in vitro ribosylation reaction followed by Western blotting as indicated. (F) The tankyrase-binding motif of PTEN is required for its ribosylation by TNKS1. The recombinant MBP-PTEN, MBP-PTEN-AA, and TNKS1 were subjected to an in vitro ribosylation assay and analyzed by Western blotting as indicated. (G) Only double knockdown of TNKS1/2 diminishes the ribosylation of PTEN in vivo. HCT116-derived cells with stable knockdown of TNKS1, TNKS2, TNKS1/2, or PTEN were collected and immunoprecipitated using anti-PTEN antibody. The input and immunoprecipitated proteins were analyzed by Western blotting using the indicated antibodies, and the ribosylation of endogenous PTEN was detected by anti-PAR antibody.
We further confirmed PTEN ribosylation by tankyrases in vivo. We first stably knocked down TNKS1, TNKS2, PTEN, or TNKS1/2 using shRNAs in HCT116 cells and then immunoprecipitated endogenous PTEN using anti-PTEN antibody. Ribosylation of PTEN was detected using the anti-PAR antibody. Knockdown of TNKS1 or TNKS2 alone did not cause significant changes in PTEN ribosylation (Fig. 2G). However, double knockdown of TNKS1 and TNKS2 abolished PTEN ribosylation (Fig. 2G). Knockdown of PTEN was included as a negative control (Fig. 2G) to demonstrate that the signal detected by the anti-PAR antibody was indeed due to PTEN ribosylation. Thus, both TNKS1 and TNKS2 are involved in PTEN ribosylation in vivo.
To investigate which residues on PTEN are PARylated by tankyrases, we performed an in vitro ribosylation assay of PTEN by TNKS1 and TNKS2 and identified several tankyrase-dependent ribosylation sites on PTEN by mass spectrometry analysis. Three candidate sites with the higher scores (E40, E150, and D326) were ribosylated by both TNKS1 and TNKS2 (Supplemental Table S1), indicating that these are likely the major ribosylation sites on PTEN. Indeed, we generated a 3A point mutant construct (Supplemental Fig. S4A) and showed that although this mutant was still able to bind to tankyrases (Supplemental Fig. S4B), its ribosylation by tankyrases was notably reduced both in vitro (Supplemental Fig. S4C) and in vivo (Supplemental Fig. S4D), revealing that these are major ribosylation sites on PTEN by tankyrases.
Tankyrases regulate PTEN degradation and ubiquitination
Knockdown of both TNKS1 and TNKS2 not only abolished PTEN ribosylation but also led to a significant increase in PTEN protein levels (Fig. 2G). Since ribosylation of some tankyrase substrates is known to lead to their degradation via a proteasome-dependent pathway (Huang et al. 2009; Kim et al. 2011; Levaot et al. 2011), we decided to test whether tankyrases could affect PTEN ubiquitination and degradation.
Overexpression of TNKS1 or TNKS2 led to PTEN down-regulation that was reversed by treatment with the protease inhibitor MG132 (Fig. 3A), indicating that tankyrases indeed promote PTEN degradation via a proteasome-dependent pathway. This regulation of PTEN degradation by tankyrases requires the catalytic activity of tankyrases, since XAV939 treatment also prevented PTEN degradation promoted by TNKS1 overexpression (Fig. 3B). Likewise, the TNKS1 catalytically inactive mutant (TNKS1-PD) also failed to promote PTEN degradation (Fig. 3C). We also used colorectal cancer cell lines SW480, DLD1, RKO, and HCT116 to test the effect of tankyrase inhibition on endogenous PTEN. As expected, inhibition of tankyrases by XAV939 led to the up-regulation of Axin1 and the corresponding down-regulation of its downstream effector, β-catenin, in SW480 and RKO cells, but this treatment did not change the β-catenin protein level in HCT116 cells (Supplemental Fig. S6A) due to the expression of the degradation-resistant β-catenin mutant in these cells. XAV939 also stabilized Axin1 in RKO cells, but β-catenin was hard to detect in these cells. Regardless of the status of the Wnt/β-catenin pathway, we consistently observed that XAV939 increased the PTEN level and decreased Akt phosphorylation in all four cell lines (Supplemental Fig. S6A). We further observed that all different tankyrase inhibitors (XAV939, JW55, and Wiki4), but not the PARP1/2 inhibitor olaparib, increased PTEN and decreased p-Akt levels in HCT116 cells (Supplemental Fig. S3D). These data again support a role for tankyrases in promoting PTEN degradation. In addition, tankyrase inhibition led to increased levels of TNKS1 and wild-type PTEN but not that of PTEN-AA (the tankyrase-binding motif point mutant of PTEN) (Fig. 3D) or PTEN-3A (Supplemental Fig. S4E), indicating that the tankyrase-binding motif and the ribosylation sites of PTEN are required for its regulation by tankyrases. Moreover, we used shRNA to knock down TNKS1, TNKS2, or both. Only double depletion of endogenous TNKS1/2, but not single depletion, increased the PTEN level and decreased the downstream Akt phosphorylation (Fig. 3E), suggesting that both TNKS1 and TNKS2 are involved in PTEN regulation.
Figure 3.

Tankyrases regulate PTEN protein stability through a ubiquitination-dependent pathway. (A) TNKS1 and TNKS2 promote the degradation of PTEN. 293T cells were transfected with constructs encoding GFP vector, GFP-tagged PTEN, and myc-tagged TNKS1 or TNKS2. Twenty-four hours after transfection, cells were treated with DMSO or 10 μM protease inhibitor MG132 for 6 h. Cell lysates were immunoblotted with the indicated antibodies. (B) TNKS1-mediated PTEN degradation is abolished by tankyrase inhibitor XAV939. 293T cells were transfected with constructs encoding GFP-tagged PTEN and myc-tagged TNKS1. Twenty-four hours after transfection, cells were treated with DMSO, 10 μM MG132 for 6 h, or 5 μM XAV939 for 6 h, and cell extracts were examined by immunoblotting using antibodies as indicated. (C) TNKS1 catalytic activity is required for PTEN degradation. 293T cells were transfected with constructs encoding Myc-tagged PTEN and SFB-tagged TNKS1 or TNKS1-PD. Cell lysates were subjected to Western blotting with the indicated antibodies. (D) Tankyrase inhibition stabilizes wild-type PTEN but not the tankyrase-binding motif double point mutant of PTEN. 293T cells transfected with constructs encoding myc-tagged TNKS1, PTEN, or PTEN-AA were treated with 5 μM tankyrase inhibitor XAV939 for 6 h. Cell extracts were examined by Western blotting as indicated. (E) Double knockdown of TNKS1/2 increases the protein level of PTEN. TNKS1, TNKS2, TNKS1/2 stable knockdown cells and control HCT116 cells were harvested, and cell lysates were analyzed by immunoblotting using the indicated antibodies. (F) TNKS1 and TNKS2 promote the polyubiquitination of wild-type but not tankyrase-binding motif point mutant PTEN in vivo. 293T cells were transfected with constructs encoding HA-ubiquitin, Myc-PTEN, and Myc-PTEN-AA together with constructs encoding SFB-TNKS1 or TNKS2. Cells were treated with 10 μM MG132 for 6 h. Whole-cell lysates were subjected to immunoprecipitation with anti-HA antibody followed by Western blotting using the indicated antibodies. (G) The catalytic activity of TNKS1 is critical for TNKS1-mediated PTEN ubiquitination. 293T cells were transfected with constructs encoding HA-ubiquitin and Myc-PTEN together with constructs encoding SFB-TNKS1 or TNKS1-PD. Cells were treated with MG132, and cell lysates were subjected to immunoprecipitation/Western analysis using the indicated antibodies. (H) Double knockdown of TNKS1/2 suppresses PTEN ubiquitination in vivo. TNKS1, TNKS2, TNKS1/2 stable knockdown cells and control HCT116 cells were transfected with HA-ubiquitin and treated with 10 μM MG132 for 6 h. Cell lysates were immunoprecipitated with anti-HA antibody, and Western blotting was carried out using the indicated antibodies.
To determine whether this newly discovered post-translational modification affects PTEN cellular localization, we performed experiments to show that overexpression of TNKS1/2 or XAV939 treatment does not affect PTEN localization (Supplemental Fig. S5A,B). In addition, the PTEN-AA mutant, which is unable to bind to TNKS1/2 and thus cannot be ribosylated, showed localization similar to that of wild-type PTEN (Supplemental Fig. S5C), indicating that ribosylation of PTEN does not affect its subcellular localization.
Experiments using the proteasome inhibitor MG132, as shown in Figure 3A,B, implied that tankyrases may promote PTEN degradation via a ubiquitylation-dependent pathway. Indeed, an in vivo ubiquitination assay showed that both TNKS1 and TNKS2 can efficiently promote the ubiquitination of wild-type PTEN but not the PTEN-AA mutant (Fig. 3F). This requires tankyrase activity, since the TNKS1 catalytically inactive mutant (TNKS1-PD) failed to promote PTEN ubiquitination (Fig. 3G). Additionally, inhibition of tankyrase by XAV939 significantly diminished PTEN polyubiquitination (Supplemental Fig. S6B).
To determine the ubiquitination of endogenous PTEN, we used the TNKS1, TNKS2, and TNKS1/2 knockdown HCT116 cells and found that only double knockdown of TNKS1 and TNKS2 reduced the polyubiquitination of endogenous PTEN (Fig. 3H). Together, these data indicate that both tankyrases are involved in the regulation of PTEN ubiquitination and degradation.
The E3 ligase RNF146 is required for tankyrase-mediated PTEN ubiquitination and degradation
Recent studies reported that the E3 ligase RNF146 is responsible for the ubiquitination and degradation of the tankyrase substrates Axin and 3BP2 (Huang et al. 2009; Levaot et al. 2011). We thus wanted to test whether RNF146 would also act as an E3 ubiquitin ligase for PTEN.
Endogenous PTEN and RNF146 interacted with each other; this interaction was diminished by treatment with tankyrase inhibitor XAV939 (Fig. 4A), and XAV939 treatment did not diminish the association between PTEN and the other two E3 ligases, NEDD4 and WWP2 (Supplemental Fig. S7A). Only double knockdown of TNKS1 and TNKS2 inhibited the PTEN/RNF146 interaction (Supplemental Fig. S7B), implying that the association between PTEN and RNF146 may be enhanced by the ribosylation of PTEN. Given that the WWE domain of RNF146 is required for its interaction with ribosylated proteins (Zhang et al. 2011), we generated the RNF146-△WWE and control RNF146-△RING deletion mutants. We observed that wild-type RNF146 and the RNF146-△RING mutant associated with PTEN, but the RNF146-△WWE mutant failed to do so (Fig. 4B). While the expression of wild-type RNF146 led to reduced PTEN protein level, which was completely reversed by XAV939 (Fig. 4C), the expression of the RNF146-△WWE or RNF146-△RING deletion mutants had no effect on PTEN protein level (Fig. 4C), suggesting that PTEN needs to be ribosylated before it can be recognized by RNF146 for ubiquitination and degradation. We also performed cycloheximide treatment experiments and showed that only wild-type RNF146, but not the RNF146-△WWE or RNF146-△RING deletion mutants, diminished the half-life of PTEN (Fig. 4D). Moreover, only wild-type RNF146 greatly promoted the ubiquitination of PTEN (Fig. 4E), indicating that both the PAR recognition domain (WWE) and the E3 ligase activity domain (RING) of RNF146 are required for RNF146-mediated PTEN ubiquitination and degradation.
Figure 4.

The E3 ligase RNF146 is involved in tankyrase-mediated PTEN ubiquitination and degradation. (A) Interaction between PTEN and RNF146. HCT116 cells were treated without or with 5 μM XAV939 for 6 h. Cell lysates were immunoprecipitated with anti-PTEN or control antibody followed by Western blotting using the indicated antibodies. (B) The WWE domain of RNF146 is required for its binding to PTEN. 293T cell were transfected with constructs encoding GFP-tagged PTEN together with vector alone or constructs encoding myc-tagged wild-type RNF146, the RNF146-△WWE mutant, or the RNF146-△RING mutant. Immunoprecipitation reactions were conducted using anti-myc antibody and followed by Western blotting analysis. (C) Both the WWE and RING domains of RNF146 are required for PTEN degradation. 293T cells were transfected with constructs as described in B. Twenty-four hours after transfection, cells were treated with DMSO, 10 μM MG132, or 5 μM XAV939 for 6 h. Cell lysates were examined by Western blotting using the indicated antibodies. (D) Overexpression of RNF146, but not of the RNF146-△WWE mutant or the RNF146-△RING mutant, destabilizes PTEN. 293T transfection was conducted as described in B. Cells were treated with 100 μg/mL cycloheximide (CHX) for the indicated times. Protein levels were analyzed by immunoblotting using antibodies as indicated (left panel) and quantified (right panel). (E) RNF146 ubiquitinates PTEN in vivo. 293T cells were transfected with constructs encoding HA-tagged ubiquitin, GFP-tagged PTEN, and myc-tagged wild-type RNF146, the △WWE mutant of RNF146, or the △RING mutant of RNF146. Cells were treated with MG132 before being collected and analyzed by immunoprecipitation/Western blotting using the indicated antibodies. (F) Depletion of RNF146 stabilizes PTEN in vivo. RNF146 stable knockdown cells and control HCT116 cells were harvested, and cell lysates were examined by Western blotting and RT–PCR. (G) Knockdown of RNF146 suppresses PTEN ubiquitination in vivo. RNF146 stable knockdown cells and control cells were transfected with HA-ubiquitin and treated with 10 μM MG132 for 6 h. Cell lysates were immunoprecipitated with anti-HA antibody, and Western blotting was carried out using the indicated antibodies.
To determine the effect of the tankyrase-binding motif of PTEN on RNF146-mediated PTEN ubiquitination and degradation, we cotransfected wild-type PTEN or the PTEN-AA mutant with WWP2 or RNF146. WWP2 promoted the degradation of both wild-type PTEN and the PTEN-AA mutant, but RNF146 facilitated only the degradation of wild-type PTEN (Supplemental Fig. S7C). Similarly, in vivo ubiquitination assays revealed that RNF146 triggered the ubiquitination of only wild-type PTEN but not that of the PTEN-AA mutant (Supplemental Fig. S7D), again supporting the idea that RNF146-mediated PTEN ubiquitination and degradation depend on the ribosylation of PTEN.
To further investigate the effect of RNF146 on PTEN, we generated three HCT116-derived cell lines with stable RNF146 knockdown using three RNF146 shRNAs. As expected, deletion of endogenous RNF146 led to PTEN stabilization (Fig. 4F). Additionally, knockdown of RNF146 diminished the ubiquitination of PTEN (Fig. 4G), suggesting that RNF146 is an E3 ligase that promotes PTEN ubiquitination and degradation.
To identify the lysine residues that are required for PTEN ubiquitination by RNF146, we generated 10 point mutants of PTEN, changing different lysine residues. In vivo ubiquitination assays showed that Lys342, Lys344, and Lys349 are likely the major sites for RNF146-mediated PTEN ubiquitination (Supplemental Fig. S8).
Tankyrases regulate tumor cell proliferation in a PTEN-dependent manner
PTEN is a well-known tumor suppressor that is critical for cell survival and growth. As demonstrated above, we found that TNKS1/2 negatively regulated the protein level of PTEN. Based on these findings, we proposed that TNKS1/2 may have a proproliferation function. To exclude the known function of TNKS1/2 in the regulation of the Axin/β-catenin pathway, we used HCT116 cells (which express a degradation-resistant mutant form of β-catenin) and RKO cells (which have no detectable level of β-catenin). We stably knocked down TNKS1, TNKS2, and TNKS1/2 in these cells and found that only the double-knockdown cells significantly decreased cell proliferation (Fig. 5A). This inhibition was confirmed by Ki67 and BrdU staining (Fig. 5B; Supplemental Fig. S9A,B). Importantly, although PTEN depletion led to increased cell proliferation in both HCT116 and RKO cells (Fig. 5C), double knockdown of TNKS1/2 in PTEN-depleted cells did not significantly reduce proliferation (Fig. 5C), suggesting that tankyrases control cell proliferation at least in part in a PTEN-dependent manner.
Figure 5.

Tankyrases regulate tumor cell proliferation in a PTEN-dependent manner. (A,B) Double knockdown of TNKS1/2 suppresses cell proliferation. TNKS1, TNKS2, and TNKS1/2 stable knockdown cells and control cells were examined to determine their rate of cell proliferation in HCT116 and RKO cells. (A) Data are means ± SD (n = 3 independent experiments), and knockdown efficiency was validated by Western blot. (B) Ki67 and BrdU staining of these control and HCT116/RKO knockdown cells was performed (top panel), and the percentages of positive cells were summarized (bottom panel). Data are means ± SD (n = 3 independent experiments). (C) Tankyrases regulate cell proliferation in a PTEN-dependent manner. PTEN, TNKS1/2, and PTEN/TNKS1/2 triple stable knockdown cells and control cells were examined to determine their rate of cell proliferation in HCT116 and RKO cells. Data are means ± SD (n = 3 independent experiments), and knockdown efficiency was validated by Western blot. (D,E) Lactate production (D) and glucose uptake (E) in HCT116 and RKO cells by knockdown of TNKS1/2/PTEN. Data are means ± SD (n = 3 independent experiments). (F) Inhibition of cell proliferation by depletion of TNKS1/2 was restored by the expression of wild-type TNKS1 but not the catalytically inactive mutant of TNKS1. shRNA-resistant inducible TNKS1 and TNKS1-PD were transduced into TNKS1/2-depleted HCT116 cells, and cell proliferation rate was measured. Data are means ± SD (n = 3 independent experiments). (G) PTEN protein level in TNKS1/2 knockdown cells was rescued by expression of TNKS1 but not TNKS1-PD. Protein levels in the cells shown in F were determined by Western blotting as indicated. (H) Resistance of PTEN-depleted cells to XAV939 was reversed by the expression of wild-type PTEN but not the PTEN-AA mutant. shRNA-resistant inducible PTEN and PTEN-AA were transduced into PTEN-depleted HCT116 cells, and cell number was determined at day 6 (n = 3). (I) Protein levels in the cells shown in H were determined by Western blotting.
Because tankyrases regulate Akt activation through PTEN ribosylation/degradation, Akt is reported to play key roles in regulating glycolysis (Elstrom et al. 2004). We investigated whether tankyrase can regulate glycolysis through PTEN. Indeed, double knockdown of TNKS1 and TNKS2 reduced lactate production and glucose uptake, and this regulation was PTEN-dependent (Fig. 5D,E).
While double knockdown of TNKS1/2 suppressed cell proliferation, reintroduction of shRNA-resistant TNKS1, but not that of TNKS1-PD, into TNKS1/2 knockdown cells reversed cell growth inhibition (Fig. 5F). Western blot analysis also confirmed that shRNA-resistant TNKS1, but not TNKS1-PD, could reverse the PTEN stabilization caused by TNKS1/2 down-regulation (Fig. 5G). XAV939 treatment greatly reduced cell proliferation in HCT116 cells (Fig. 5H). Depletion of PTEN increased cell proliferation; however, these PTEN knockdown cells became resistant to XAV939 treatment (Fig. 5H). Reintroduction of shRNA-resistant PTEN and PTEN-AA into PTEN-depleted cells reversed cell proliferation to a normal level; however, XAV939 treatment suppressed proliferation only in cells expressing wild-type PTEN but not those expressing the PTEN-AA mutant (Fig. 5H). Similar results were obtained by measuring the protein levels of PTEN and Akt phosphorylation (Fig. 5I), indicating that PTEN is a target of tankyrase inhibitor XAV939.
Tankyrases are required for tumor growth in a PTEN-dependent manner
As shown in Figure 6A, only double knockdown of TNKS1/2 significantly reduced the colony formation of HCT116 and RKO cells. However, double knockdown of TNKS1/2 did not suppress colony formation in PTEN-depleted cells (Fig. 6B). Reintroduction of shRNA-resistant TNKS1, but not TNKS1-PD, reversed the reduction in colony formation caused by TNKS1/2 knockdown (Fig. 6C). Similarly, reintroduction of shRNA-resistant PTEN and PTEN-AA in PTEN-depleted cells reversed colony formation to a normal level; however, XAV939 treatment could inhibit colony formation only in cells expressing wild-type PTEN but not those expressing the PTEN-AA mutant (Fig. 6D). Moreover, we tested the potential of tankyrase depletion in preventing tumor growth in vivo using xenograft models. Mice injected with TNKS1/2 double-knockdown cells showed significantly reduced tumor growth; however, this effect was abolished in PTEN-depleted cells (Figure 6E,F), indicating that tankyrase inhibition may be effective only in treating tumor cells expressing wild-type PTEN.
Figure 6.

Tankyrases are required for tumor growth in a PTEN-dependent manner. (A) Double knockdown of TNKS1/2 suppresses colony formation of HCT116 and RKO cells. TNKS1, TNKS2, and TNKS1/2 stable knockdown cells and control cells were tested in colony formation assays. Colonies viable after 2 wk were counted and analyzed (n = 3). (B) Tankyrases regulate tumor cell colony formation in a PTEN-dependent manner. PTEN, TNKS1/2, and PTEN/TNKS1/2 stable knockdown cells and control cells were tested in colony formation assays. Colonies viable after 2 wk were counted and analyzed (n = 3). (C) Inhibition of colony formation by depletion of TNKS1/2 was restored by the expression of TNKS1 but not TNKS1-PD. shRNA-resistant inducible TNKS1 and TNKS1-PD were transduced into TNKS1/2-depleted HCT116 cells, and cells were tested in colony formation assays (n = 3). (D) Resistance of PTEN-depleted cells to XAV939 was reversed by the expression of wild-type PTEN but not the PTEN-AA mutant as measured by colony formation assays. shRNA-resistant inducible PTEN and PTEN-AA were transduced into PTEN-depleted HCT116 cells, and colonies viable after 2 wk were measured (n = 3). (E) Tankyrases regulate tumor formation in a PTEN-dependent manner. Nude mice 4 to 6 wk old were injected with PTEN, TNKS1/2, and PTEN/TNKS1/2 stable knockdown cells or control cells. The tumor weights of each group are shown, presented as average weights ± SD (21 d after injection; n = 6 mice per group). (F) Immunoblotting of TNKS1, TNKS2, PTEN, p-Akt, and Tubulin in tumor lysates from E.
Tankyrase expression negatively correlates with PTEN expression in human colon tumors
We checked the association between PTEN and tankyrases in normal and tumor cell lines. The PTEN/tankyrase interaction was observed in 10 different colon cancer cell lines (Supplemental Fig. S10A), two breast epithelial cells, and three breast cancer cell lines (Supplemental Fig. S10B).
To determine the pathological relevance of PTEN regulation by tankyrases in human cancers, we first validated that the anti-TNKS1, anti-TNKS2, anti-PTEN, anti-Axin1, and anti-p-Akt antibodies were all suitable for immunohistochemistry (IHC) staining (Supplemental Fig. S11). Next, we performed IHC staining of colon cancer tissue arrays and determined the expression levels of TNKS1, TNKS2, Axin1, PTEN, and p-Akt in these samples (Fig. 7A; Supplemental Fig. S12A). Significantly, we observed that TNKS1, TNKS2, and p-Akt are up-regulated (39.0% vs. 56.9%, 45.8% vs. 64.4%, and 15.2% vs. 52.5%), while PTEN and Axin1 are down-regulated (69.5% vs. 31.3% and 62.7% vs. 38.1%) in colon cancers (Fig. 7B; Supplemental Fig. S12A). Moreover, a significant negative correlation between PTEN and TNKS1 (P = 0.0001) or PTEN and TNKS2 (P = 0.0013) was noticed (Fig. 7C,D). Similarly, another reported tankyrase substrate, Axin1, also showed a negative correlation with TNKS1 (P = 0.0150) and TNKS2 (P = 0.0057) (Fig. 7C,D). As a downstream target of PTEN, p-Akt showed a significant negative correlation (P = 0.0002) with PTEN (Supplemental Fig. S12C,D). In addition, p-Akt positively correlated with TNKS1 (P = 0.0284) and TNKS2 (P = 0.0262) (Supplemental Fig. S12C,D). We also noted a positive correlation between TNKS1 and TNKS2 (P < 0.0001) in these colon tumor samples (Supplemental Fig. S12D). These findings suggest that tankyrases may be up-regulated, which correlates with PTEN down-regulation in human colon carcinomas. Additionally, we performed the RNAscope assay (Xing et al. 2014) to check the mRNA levels of PTEN in these tissue arrays (Supplemental Fig. S12A). Compared with PTEN gene mutation, which is <20% in colon cancer cell lines (Dicuonzo et al. 2001) and colon cancer samples (Berg et al. 2010), PTEN mRNA levels are greatly reduced in colon carcinomas (50.6%) (Supplemental Fig. S12B). Interestingly, positive staining for PTEN mRNA levels is still much higher than that of PTEN protein levels (50.6% vs 31.3%), indicating that post-translational modification plays important roles in PTEN regulation. Together, these findings suggest that tankyrases may be up-regulated and thus contribute to PTEN down-regulation in human colon carcinomas.
Figure 7.

Tankyrases are up-regulated and negatively correlate with PTEN status in human colon tumors. (A) IHC staining of PTEN, Axin1, TNKS1, and TNKS2 in representative normal colon and colon carcinoma specimens on the US Biomax tissue microarrays. Brown staining indicates positive immunoreactivity. Bars, 50 μm. (B) PTEN, Axin1, TNKS1, and TNKS2 expression status in normal colon tissue and colon carcinoma specimens. (C) IHC staining of PTEN, Axin1, TNKS1, and TNKS2 of representative human colon tumor samples. Bars, 50 μm. (D) Correlation between expression status of PTEN/TNKS1, PTEN/TNKS2, Axin1/TNKS1, and Axin1/TNKS2 in human colon tumor samples.
Discussion
In this study, we demonstrated that PTEN is a novel tankyrase substrate. We showed that PTEN contains a conserved tankyrase-binding motif, through which tankyrases bind to and ribosylate PTEN in vitro and in vivo. The ribosylated PTEN is then recognized by the PAR-binding E3 ligase RNF146 and targeted for polyubiquitination and degradation. Importantly, this ability of regulating PTEN protein level is a key aspect of proproliferation functions of tankyrases in the cell.
As a well-studied tumor suppressor, PTEN is known to be the target of many post-translational modifications that contribute to the regulation of PTEN protein stability (Wang and Jiang 2008). Previous studies revealed two ubiquitin E3 ligases—NEDD4 and WWP2—that are involved in promoting PTEN ubiquitination and degradation (Wang et al. 2007; Maddika et al. 2011). Phosphorylation of PTEN has also been demonstrated to regulate its stability through affecting ubiquitin-mediated degradation (Vazquez et al. 2000; Torres and Pulido 2001; Maccario et al. 2007; Yim et al. 2009). In this study, we reported a new modification of PTEN and identified RNF146 as a new E3 ligase of PTEN that specifically regulates PTEN ubiquitination and degradation in a ribosylation-dependent manner. This ribosylation-linked regulation of PTEN provides yet another layer of PTEN regulation that was not previously recognized. How this pathway acts in coordination with other pathways involved in PTEN degradation remains to be resolved.
The multifunctional nature of tankyrases is likely due to the variety of substrates regulated by these proteins. TNKS1 knockout mice develop normally and exhibit only a metabolic disorder (Yeh et al. 2009), while TNKS2 deficiency results in reduced body weight (Chiang et al. 2006; Hsiao et al. 2006). However, TNKS1/2 double-knockout mice show embryonic lethality by day 10 (Chiang et al. 2008), suggesting that TNKS1 and TNKS2 have redundant functions during embryonic development. These findings are in agreement with our observations. Both TNKS1 and TNKS2 bind to and ribosylate PTEN. Only double knockdown of TNKS1/2 significantly suppressed PTEN ribosylation, ubiquitination, and degradation, which contribute to the roles of tankyrases in promoting cell proliferation and tumor growth.
Besides Axin, PTEN is likely another target of tankyrases involved in cell proliferation. Previous studies demonstrated that tankyrases regulate cell proliferation by targeting axin for ribosylation, ubiquitination, and degradation, thus suppressing the WNT signaling pathway (Huang et al. 2009; Kim et al. 2011; Levaot et al. 2011). Here, we showed that tankyrases could also influence cell proliferation in an Axin-independent but PTEN-dependent manner. Our findings provide a strong rationale for the development of tankyrase inhibitors for cancer treatment because these inhibitors will be useful for treating not only tumors with an intact WNT pathway but also tumors expressing wild-type PTEN. This is especially relevant for designing clinical trials using these tankyrase inhibitors, since we can use both the WNT pathway and PTEN status as biomarkers for selecting patients who could benefit from such treatment and therefore greatly enhance the potential of tankyrase-based therapies.
Materials and methods
Antibodies and reagents
Rabbit anti-TNKS2 antibody was raised by immunizing rabbits with GST-TNKS2 fusion protein containing the 527–776 amino acids of human TNKS2, and rabbit anti-MBP antibody was raised immunizing rabbits with bacterially expressed and purified MBP proteins. Monoclonal anti-TNKS1 (ab13587) was obtained from Abcam. Anti-α-tubulin (T6199), anti-HA (H9658), and anti-Flag (F1804) were purchased from Sigma-Aldrich. Anti-PTEN (9552S), anti-Axin1 (2074S), anti-AKT1 (9272S), anti-phosphor-AKT1 (S473) (9271S), and anti-biotin-HRP (5571S) were obtained from Cell Signaling Technology. Anti-GFP (sc-9996), anti-myc (sc-40), anti-PTEN (sc-7974), anti-TNKS (sc-8337), anti-RNF146 (sc-132440), and anti-GST (sc-138) were purchased from Santa Cruz Biotechnology. Anti-ubiquitin (04-263) and anti-PARP (MAB3290) were obtained from Millipore; anti-PAR (51-8114KC), anti-β-catenin (610154), and anti-Ki67 (550609) monoclonal antibodies was purchased from BD Bioscience; and anti-PAR (4335-MC-100) was purchased from Trevigen.
Cycloheximide, MG132, Wiki4, puromycin, G418, and doxycycline were purchased from Sigma-Aldrich; PAPR1/2 inhibitor Olaparib was from LC Laboratories; PARG inhibitor ADP-HPD and JW55 were from Millipore; and XAV939 was from Sigma-Aldrich and Selleckchem.
Constructs and shRNAs
The TNKS1, TNKS2, and TNKS1-PD plasmids were kindly provided by Dr. Susan Smith (New York University), and the NEDD4 plasmid was provided by Dr. Li Ma (M.D. Anderson Cancer Center). The PTEN, WWP2, and RNF146 constructs were obtained from the Human ORFeome version 5.1 collection. All constructs were subcloned into pDONOR201 vector and then into S-protein/Flag/SBP triple-tagged, GFP-tagged, HA-tagged, and Myc-tagged destination vectors using Gateway Technology (Invitrogen). The PTENΔTBM mutant was generated by deleting the first 30 amino acids of PTEN. The RNF146-ΔWWE and RNF146-ΔRING mutants were generated, respectively, by deleting amino acids 104–159 or amino acids 36–78 of the RNF146 coding region. The PTEN-AA mutant was generated by site-directed mutagenesis and verified by sequencing. The PTEN-3A mutant was synthesized by Integrated DNA Technologies.
The pLKO.1 TNKS1 shRNA was purchased from Sigma (TRCN0000040184). The following pGIPZ lentiviral shRNA sets were from the shRNA and ORFeome Core Facility at The University of Texas M.D. Anderson Cancer Center: TNKS2-shRNA (clone no. V3LHS_354387), PTEN-shRNA (clone no. V3LHS_312160), β-catenin-shRNA (clone no. V2LHS_151022), RNF146-shRNA-3# (clone no. V2LHS_12922), RNF146-shRNA-4# (clone no. V3LHS_395650), and RNF146-shRNA-5# (clone no. V3LHS_395653). Lentiviral packaging plasmids (pMD2.G and psPAX2) were kindly provided by Dr. Zhou Songyang (Baylor College of Medicine).
The shRNA-resistant expression constructs of TNKS1, TNKS1-PD, PTEN, and PTEN-AA were generated by mutating the nucleotide sequence but not changing the amino acid sequence. The original shRNA targeting sequence for TNKS1, 5′-CGACTCTTAGAGGCATCTAAA-3′, was changed to 5′-AGGTTACTGGAAGCGTCTAAA-3′, and the original shRNA targeting sequence for PTEN, 5′-CGGGAAGACAAGTTCATGT-3′, was changed to 5′-AGAGAGAATAAATTTATGT-3′ and verified by sequencing.
Cell culture and cell transfection
The HEK293T, HCT116, RKO, DLD1, SW480, SW620, HCT15, HT29, LOVO, MCF-10A, MCF-12A, MDA-MB-231, MDA-MB-435, and MDA-MB-453 cells were purchased from American Type Culture Collection (ATCC) and cultured under conditions specified by the manufacturer. The SW1463, SKCO1, LS513, and VACO5 cells were kindly provided by Dr. E. Scott Kopetz (M.D. Anderson Cancer Center) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. Plasmid transfection was performed using Lipofectamine 2000 or polyethyleneimine reagent according to the manufacturer’s protocol.
Immunoprecipitation, Western blotting, and immunofluorescence staining
Cells were lysed with NETN buffer (20 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) containing protease inhibitors, and the whole-cell lysates obtained by centrifugation were incubated with protein A agarose beads coupled with 2 μg of the indicated antibodies for 2 h at 4°C. The immunocomplexes were washed three times with NETN buffer, applied to SDS-PAGE, and transferred to PVDF membrane, and immunoblotting was carried out with the indicated antibodies.
For immunofluorescence staining assays, cells were cultured on coverslips overnight and fixed with 4% paraformaldehyde for 10 min at room temperature followed by permeabilization with 0.5% Triton X-100 in PBS for 5 min. Cells were then blocked with 5% goat serum in PBS for 30 min and incubated with primary antibodies in 5% goat serum for 60 min. Cells were washed with PBS and incubated with rhodamine or FITC-conjugated secondary antibodies for 60 min, and then coverslips were mounted on slides using anti-fade mounting medium with DAPI. Immunofluorescence images were acquired on a Nikon Eclipse E800 fluorescence microscope with a Nikon Plan Fluor 10× objective lens. Each experiment was repeated three times or more unless otherwise noted.
Cell proliferation and colony formation assays
For proliferation assays, equal numbers of cells were plated in 6-cm dishes. The next day, cells were collected and subsequently counted every 2 d.
For colony formation assays, cells were seeded at 500 cells per well into six-well plates in triplicates. Twelve hours after seeding, cells were treated with XAV939 with the indicated concentration. Medium was replenished every 2 d, and cells were incubated for 14 d. Resulting colonies were fixed and stained with Coomassie blue. The numbers of colonies were counted with a GelDoc with Quantity One software (Bio-Rad).
In vitro PARsylation assay
Samples containing 0.2 μg of recombinant baculovirus-derived TNKS1 or TNKS2 (purchased from Sigma-Aldrich) and 2 μg of recombinant Escherichia coli-derived MBP-PTEN, MBP-PTEN-AA, or MBP-PTEN-3A were incubated in 50 μL of PARP reaction buffer (50 mM Tris-HCl at pH 8.0, 4 mM MgCl2, 0.2 mM dithiothreitol) with or without 25 μM biotinylated NAD+ (Trevigen) for 30 min at 25°C. Reactions were terminated by addition of 2× sample buffer and analyzed by immunoblotting using an anti-biotin HRP antibody.
For the in vitro ribosylation assay of PARP1, samples containing 0.2 μg of recombinant PARP1 (purchased from Trevigen) and 2 μg of recombinant E. coli-derived GST-PTEN were incubated in 50 μL of PARP reaction buffer (20 mM Tris-HCl at pH 7.5, 50 mM NaCl, 7.5 mM MgCl2, 0.2 mM TCEP) with 1 μM dsDNA (5′-GAGTGTTGCATTCCTCTCTGGGCGCCGGGCAGGTACCTGCTG-3′) and 5 mM NAD+ for 30 min at room temperature. The reactions were stopped by the addition of SDS loading buffer containing 0.1 M EDTA, and samples were analyzed by immunoblotting using an anti-PAR antibody.
In vivo ubiquitination assay
293T cells were transfected with various combinations of constructs, as indicated, along with HA-tagged ubiquitin. Twenty-four hours after transfection, cells were treated with 10 μM MG132 for 6 h. Cells were then harvested, and lysates were subjected to immunoprecipitation using the indicated antibodies. The ubiquitination of PTEN was detected by Western blotting.
Lactate production assay
Cells were seeded in culture dishes, and the medium was changed after 6 h with serum-free DMEM. Cells were incubated for 12–16 h, and the culture medium was then determined using lactate test strips on a Lactate Plus meter (Nova Biomedical). The remaining cells were harvested and counted. The lactate production was calculated and determined as lactate concentration per 104 cells.
Glucose uptake assay
Cells were seeded in culture dishes, and the medium was changed after 6 h with glucose-free DMEM. Cells were incubated for 16 h and then incubated with high-glucose DMEM for an additional 24 h. The culture medium was collected for measurement of glucose concentrations using a glucose assay kit (BioVision) according to manufacturer’s instructions.
In vivo xenograft study
All animal experiments were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the M.D. Anderson Cancer Center. Using a power calculation, our experiments indicated that six mice per group can identify the expected effect of TNKS1/2 on tumor weight with 100% power. PTEN, TNKS1/2, PTEN/TNKS1/2 stable knockdown or control cells (5 × 106 in 100 μL of medium) were injected into 6- to 8-wk-old female BALB/c nude mice. Mice were euthanized when they met the institutional euthanasia criteria for tumor size and overall health. The tumors were removed, photographed, and weighed. Animals were randomly assigned to each cell line injected. A laboratory technician (Kathryn Aziz) who provided animal care and tumor collection was blinded to the group allocation during all animal experiments and outcome assessments.
Human tissue IHC analysis
All of the 219 cases of human colon cancer tissue microarrays were purchased from US Biomax. While BC051110a tissue microarray contained 110 cases of colon carcinoma and 10 cases of normal colonic tissue, the BC05118b tissue microarray contained 50 cases of colon carcinoma and 50 cases of cancer adjacent normal colonic tissue. Samples were deparaffinized and rehydrated. Antigen retrieval was done using 0.01 M sodium citrate buffer (pH 6.0) in a microwave oven. To block endogenous peroxidase activity, the sections were treated with 1% hydrogen peroxide in methanol for 30 min. After 1 h of preincubation in 10% normal goat serum to prevent nonspecific staining, the samples were incubated with anti-PTEN (1:50; Cell Signaling Technology, 9559S), anti-p-Akt (Ser473) (1:50; Cell Signaling Technology, 4060S), anti-Axin1 (1:50; Santa Cruz Biotechnology, sc14029), anti-TNKS1 (1:100; Abcam, ab13587), or anti-TNKS2 (1:100; raised by ourselves) overnight at 4°C. The sections were then incubated with a biotinylated secondary antibody (1:200; Vector Laboratories, PK-6101) and incubated with avidin–biotin peroxidase complex solution (1:100) for 30 min at room temperature. Color was developed with the 3-amino-9-ethylcarbazole (AEC) solution. Counterstaining was carried out using Mayer’s hematoxylin. All immunostained slides were scanned on the Automated Cellular Image System III (ACIS III, Dako) for quantification by digital image analysis. A total score of protein expression was calculated from both the percentage of immunopositive cells and the immunostaining intensity. High and low protein expressions were defined using the mean score of all samples as a cutoff point.
RNAscope assay
The RNAscope probe targeting PTEN mRNA was ordered from Advanced Cell Diagnostics, and detection of PTEN mRNA expression was performed using the RNAscope 2.0 High-Definition (HD)-BROWN assay in accordance with manufacturer’s instructions (Advanced Cell Diagnostics). The images were acquired by Zeiss Axioskop2 Plus microscope.
Statistical analysis
All experiments were repeated at least three times. We did not exclude any samples or animals from the analysis and used random assignment of samples or animals to different groups. Data were analyzed by the one-way ANOVA test and Pearson χ2 analysis. We used an F-test to compare variances, and the variances were not significantly different. A P-value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank all of our colleagues in J.C.’s laboratory for insightful discussions, and Kathryn Aziz for assistance with the animal study. This work was supported in part by an M.D. Anderson Start-up fund to J.C. J.C. is also a recipient of an Era of Hope Scholar Research award (W81XWH-09-1-0409). This work was also supported in part by M.D. Anderson’s Cancer Center Support Grant (CA016672). This work was also supported by grants from the University of Texas Southwestern Medical Center Endowed Scholar Program, the Cancer Prevention and Research Institute of Texas (CPRIT R1103), and the Welch Foundation (I-1800). Y.Y. is a Virginia Murchison Linthicum Scholar in Medical Research and a CPRIT Scholar in Cancer Research.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.251785.114.
References
- Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. 2013. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M, Danielsen SA, Ahlquist T, Merok MA, Agesen TH, Vatn MH, Mala T, Sjo OH, Bakka A, Moberg I, et al. 2010. DNA sequence profiles of the colorectal cancer critical gene set KRAS–BRAF–PIK3CA–PTEN–TP53 related to age at disease onset. PLoS ONE 5: e13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Dynek JN, Smith S. 2003. TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev 17: 1328–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YJ, Nguyen ML, Gurunathan S, Kaminker P, Tessarollo L, Campisi J, Hodes RJ. 2006. Generation and characterization of telomere length maintenance in tankyrase 2-deficient mice. Mol Cell Biol 26: 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YJ, Hsiao SJ, Yver D, Cushman SW, Tessarollo L, Smith S, Hodes RJ. 2008. Tankyrase 1 and tankyrase 2 are essential but redundant for mouse embryonic development. PLoS ONE 3: e2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. 1998. Pten is essential for embryonic development and tumour suppression. Nat Genet 19: 348–355. [DOI] [PubMed] [Google Scholar]
- Dicuonzo G, Angeletti S, Garcia-Foncillas J, Brugarolas A, Okrouzhnov Y, Santini D, Tonini G, Lorino G, De Cesaris M, Baldi A. 2001. Colorectal carcinomas and PTEN/MMAC1 gene mutations. Clin Cancer Res 7: 4049–4053. [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. 2004. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892–3899. [DOI] [PubMed] [Google Scholar]
- Hobert JA, Eng C. 2009. PTEN hamartoma tumor syndrome: an overview. Genet Med 11: 687–694. [DOI] [PubMed] [Google Scholar]
- Hsiao SJ, Poitras MF, Cook BD, Liu Y, Smith S. 2006. Tankyrase 2 poly(ADP-ribose) polymerase domain-deleted mice exhibit growth defects but have normal telomere length and capping. Mol Cell Biol 26: 2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. 2009. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461: 614–620. [DOI] [PubMed] [Google Scholar]
- Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J, et al. 2012. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun 3: 911. [DOI] [PubMed] [Google Scholar]
- Kim MK, Dudognon C, Smith S. 2011. Tankyrase 1 regulates centrosome function by controlling CPAP stability. EMBO Rep 13: 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. 2004. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci 101: 16419–16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levaot N, Voytyuk O, Dimitriou I, Sircoulomb F, Chandrakumar A, Deckert M, Krzyzanowski PM, Scotter A, Gu S, Janmohamed S, et al. 2011. Loss of tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 147: 1324–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DM, Sun H. 1997. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res 57: 2124–2129. [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. 1997. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275: 1943–1947. [DOI] [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. 1997. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16: 64–67. [DOI] [PubMed] [Google Scholar]
- Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR. 2007. PTEN is destabilized by phosphorylation on Thr366. Biochem J 405: 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S, Kavela S, Rani N, Palicharla VR, Pokorny JL, Sarkaria JN, Chen J. 2011. WWP2 is an E3 ubiquitin ligase for PTEN. Nat Cell Biol 13: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. 1997. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 275: 1787–1790. [DOI] [PubMed] [Google Scholar]
- Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. 2006. PCAF modulates PTEN activity. J Biol Chem 281: 26562–26568. [DOI] [PubMed] [Google Scholar]
- Ozaki Y, Matsui H, Asou H, Nagamachi A, Aki D, Honda H, Yasunaga S, Takihara Y, Yamamoto T, Izumi S, et al. 2012. Poly-ADP ribosylation of Miki by tankyrase-1 promotes centrosome maturation. Mol Cell 47: 694–706. [DOI] [PubMed] [Google Scholar]
- Smith S, Giriat I, Schmitt A, de Lange T. 1998. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 282: 1484–1487. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. 1997. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15: 356–362. [DOI] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, et al. 1998. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 8: 1169–1178. [DOI] [PubMed] [Google Scholar]
- Torres J, Pulido R. 2001. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 276: 993–998. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. 2000. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20: 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jiang X. 2008. Post-translational regulation of PTEN. Oncogene 27: 5454–5463. [DOI] [PubMed] [Google Scholar]
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, et al. 2007. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128: 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, Lin A, Li C, Liang K, Wang S, Liu Y, Park PK, Qin L, Wei Y, Hawke DH, et al. 2014. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 159: 1110–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JZ, de Lange T. 2004. TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 36: 618–623. [DOI] [PubMed] [Google Scholar]
- Yeh TY, Beiswenger KK, Li P, Bolin KE, Lee RM, Tsao TS, Murphy AN, Hevener AL, Chi NW. 2009. Hypermetabolism, hyperphagia, and reduced adiposity in tankyrase-deficient mice. Diabetes 58: 2476–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim EK, Peng G, Dai H, Hu R, Li K, Lu Y, Mills GB, Meric-Bernstam F, Hennessy BT, Craven RJ, et al. 2009. Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell 15: 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, et al. 2011. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13: 623–629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.