

Abstract
A series of novel bis-tetrahydropyran 1,4-triazole analogues based on the acetogenin framework display low micromolar trypanocidal activities towards both bloodstream and insect forms of Trypanosoma brucei, the causative agent of African sleeping sickness. A divergent synthetic strategy was adopted for the synthesis of the key tetrahydropyran intermediates to enable rapid access to diastereochemical variation either side of the 1,4-triazole core. The resulting diastereomeric analogues displayed varying degrees of trypanocidal activity and selectivity in structure activity relationship studies.
Keywords: Trypanosomiasis, acetogenins, neglected diseases, natural products
Introduction
Neglected tropical diseases are a continuing health concern in developing countries due to the lack of effective prevention methods and therapeutic agents.[1] One of these prevalent Neglected tropical diseases which has been slowly attracting attention over the past few years is African sleeping sickness or Human African Trypanosomiasis (HAT), caused by the protozoan parasite Trypanosoma brucei that is transmitted by the bite of the Tsetse fly. HAT is a major health concern in sub-Saharan Africa threatening more than 60 million people. The annual mortality rate of HAT estimated by the World Health Organization (WHO) stands at approximately 8000 with less than 30,000 new cases per year based on recent reported cases.[2] At present the treatment for HAT is extremely limited and dependent upon the diseasre stage of HAT infection i.e. the lymphatic first stage or whether the T. brucei parasite has crossed the blood brain barrier. Four drugs are used in the treatment of HAT (Figure 1), namely suramin (1) and pentamidine (2 for stage one HAT and melarsoprol (3) and eflornithine (4) for stage two. These drugs are difficult to administer to patients often requiring lengthy infusion rates, have varying degrees of human toxicity and resistance is becoming a significant problem.[3] In an effort to reduce the costs associated with these drugs, improve the logistics of their administration and reduce drug resistance (a common problem associated with parasitic diseases) a combinatorial therapy (NECT) consisting of eflornithine (4) and nifurtimox (5) (used to treat Chagas disease) has been recently introduced for stage 2 HAT by the WHO.[4] The use of NECT against HAT has proven to be just as effective as the treatment with eflornithine (4) alone. However, there have been reported cases of drug resistance to this combination therapy. Hence, the lack of new effective therapeutic agents and an emerging drug resistance to current therapies highlights the urgent demand for the development of new drug-like molecules and their clinical implementation in effective drug therapies against HAT.
Figure 1.

Current drugs for treatment of Human African trypanosomiasis.
The acetogenins are a class of polyketide natural products isolated from the annonaceae plant species found in the tropical regions of West Africa and South America.[5,6] Chamuvarinin (6) was isolated in 2004 by Laurens et. al. from the roots of the bush banana plant Uvaria chamae and showed significant cytotoxicity against the KB 3-1 cervix cancer cell line (IC50 value of 0.8 nM), shown in Figure 2.[7] In 2011 we reported the total synthesis of chamuvarinin, we showed that 6 and a series of synthetic derivatives exhibited low micromolar activities towards both the bloodstream and procyclic forms of T. brucei.[8] These encouraging preliminary results and the limited investigations into trypanocidal activity of the acetogenins[9] prompted our initial interest in designing simplified analogues of 6 which retain the important structural features of the acetogenin family of natural products. In an effort to reduce structural complexity it was hypothesized that a 1,4-triazole motif could form an effective spatial mimic of the central C20-C23 tetrahydrofuran (THF) motif found in chamuvarinin (6). The synthesis and incorporation of THF motifs are notoriously difficult, hence, the C16-19 THF ring system of 6 was substituted with the readily accessible and manageable tetrahydropyran (THP) motif, 7, Figure 2. The heterocyclic spacer was envisioned to arise from the “click” reaction of two THP subunits, whilst the butenolide moiety could be appended to the ether core in an analogous manner to 6 by a suitable length alkyl spacer.
Figure 2.
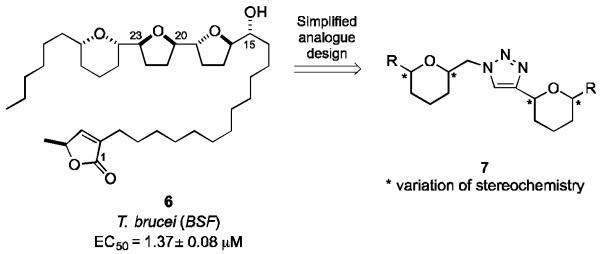
Rationale for simplified analogue design.
Results and Discussion
Chemistry
As outlined in Scheme 1, intermediate THP alcohols 8a and 9a could be readily assembled in three steps from (S)-epoxyoctane (10a)[10] via Cu(I)-promoted ring opening of 10a with homoallyl magnesium bromide followed by m-CPBA epoxidation and subsequent in situ acid mediated ring closure to provide a readily separable mixture of diastereomeric alcohols 8a and 9a. This divergent approach provided an excellent opportunity to introduce structural variation within the THP scaffolds. With the alcohols in hand, both diastereomers could be independently converted to the azide by Mitsunobu reaction (DPPA, DIAD, iPr2NEt, PPh3) of alcohols 8a and 9a to give 11a and 12.[11] The enantiomeric syn azide (13) could be accessed in an analogous manner from (R)-epoxyoctane.[10] For the alkyne subunits, syn-8a and anti-9a alcohols were oxidized under Swern conditions to afford aldehydes 14a and 15a in excellent yields and subsequent exposure of 14a to Ohira-Bestmann homologation[12] gave exclusively syn-16a in 72% yield. Due to epimerization of aldehyde 15a under the mildly basic conditions an alternative two-step Corey-Fuchs homologation[13] approach was adopted, giving 17a in 70% yield. The incorporation of terminal oxygenation as a functional handle for extended elaboration was achieved by synthesis of the corresponding benzylated and silylated series of azides 11b-c and alkynes 16b and 17b from epoxides 10b and 10c.
Scheme 1.

Synthesis of azides 11a-c, 12 and 13 and alkynes 16a-b and 17a-b: a) CH2CHCH2CH2MgBr, CuI, THF, −40 °C ✶ RT; b) mCPBA, CH2Cl2, 0 °C ✶ RT; then (±)-CSA (20 mol%), RT; c) PPh3, iPr2NEt, DIAD, DPPA, 0 °C ✶ RT; d) (COCl)2, DMSO, CH2Cl2, Et3N, −78 °C ✶ RT; e) dimethyldiazo-2-oxopropylphosphonate, K2CO3, MeOH, RT; f) CBr4, PPh3, CH2Cl2, 0 °C then −78 °C; g) n-BuLi, THF, −78 °C. mCPBA = 3-chloroperbenzoic acid, (±)-CSA = (±)-camphorsulfonic acid, DIAD = diisopropyl azodicarboxylate, DPPA = diphenyl phosphoryl azide, DMSO = dimethyl sulfoxide.
The central 1,4-triazole scaffold was conveniently installed by application of the Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition (Scheme 2).[14] Thus, diastereomeric analogues of the alkyl, benzylated and silylated bis-THP triazole motifs were synthesized by treatment of the corresponding azides 11a-c, 12, 13 and alkynes 16a-b, 17a-b under “click” reaction conditions (CuSO4, Na ascorbate) to afford triazole products 18a-d, 19a-b, 20a-b, 21a-b and 22 in good yields.
Scheme 2.

Synthesis of triazole analogues 18a-d, 19a-b, 20a-b, 21a-b and 22: a) CuSO4·5H2O, Na ascorbate, H2O, t-BuOH, RT.
Benzylated 18b-c, 19b, 20b, 21b and 22 analogues, outlined in Scheme 3, were deprotected under atmospheric hydrogenolysis (20% Pd(OH)2/C, H2) to the corresponding alcohols 23-28 (entries 1-6) in excellent yields.
Scheme 3.

Synthesis of alcohols 23-28: a) 20% Pd(OH)2/C, H2 (1 atm), EtOH, RT.
Having previously established SAR data for the advanced analogues of chamuvarinin, it was observed that incorporation of the butenolide side chain impacted positively on the trypanocidal activity. Thus, installation of the butenolide motif to successfully mimic chamuvarinin was accomplished in four steps from lead alcohol 23, outlined in Scheme 4. Alcohol 23 was manipulated to sulfone 29 via a two-step Mitsunobu reaction and catalytic Mo[6] oxidation protocol.[15] Subsequent Julia-Kocienski olefination[16] with aldehyde 30[8,17] and diimide reduction[18] (TsNHNH2, NaOAc) provided advanced analogue 31. To ascertain whether the triazole analogue would bind in a directional manner to the unknown protein target, analogue 32 was prepared in an analogous manner to 31 from alcohol 28 via sulfone 33.
Scheme 4.

Synthesis of triazoles 31 and 32: a) 1H-mercaptophenyltetrazole, PPh3, DIAD, 0 °C; b) (NH4)6Mo7O24·4H2O, H2O2, EtOH, 0 °C → RT; c) NaHMDS, THF, −78 °C; then 30, THF −78 → −20 °C; d) TsNHNH2, NaOAc, DME, H2O, 100 °C. PT = phenyl tetrazole, NaHMDS = sodium hexamethyldisilylazide, DME = dimethoxyethane.
Having successfully installed the butenolide motif and in an effort to further extend the SAR data, bis-directional analogue 34, outlined in Scheme 5, was synthesized in six steps from advanced triazole 18d. Silyl deprotection of 18d to mono alcohol 35, followed by a Mitsunobu/oxidation protocol gave sulfone 36. Julia-Kocienski olefination of sulfone 36 with 30 and subsequent diimide reduction gave triazole 37. Benzyl deprotection of 37 with boron trichloride furnished alcohol 34 in 23% yield. Diol analogue 38 was accessed in excellent yield from alcohol 35 under atmospheric hydrogenolysis.
Scheme 5.

Synthesis of advanced alcohol 34: a) TBAF, THF, 0 °C → RT; b) 1H-mercaptophenyltetrazole, PPh3, DIAD, 0 °C; c) (NH4)6Mo7O24·4H2O, H2O2, EtOH, 0 °C → RT; d) NaHMDS, THF, −78 °C; then 30, THF −78 → −20 °C; e) TsNHNH2, NaOAc, DME, H2O, 100 °C; f) BCl3·SMe2, CH2Cl2, −78 °C → RT; g) 20% Pd(OH)2/C, H2 (1 atm), EtOH, RT. TBAF = tetrabutylammonium fluoride.
Structure-activity relationship summary
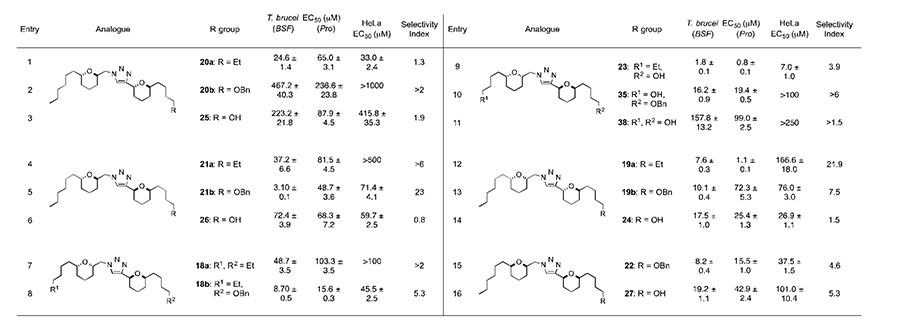
The triazole based analogues were then screened against the bloodstream (BSF) and procyclic (Pro) forms of T. brucei and the mammalian HeLa cell line,[19] providing some insight into the structural features required for potent parasitic inhibition, as shown in Table 1. From the data it was evident that the stereochemistry surrounding the THP ring systems and the functionalization of the terminal motifs impacted on the T. brucei inhibition profile. In comparison to chamuvarinin (6), the initial set of diastereomeric alkyl triazoles 20a, 21a and 18a (entries 1, 4, 7) were >17 times less potent than 6 and indicated that stereochemistry had little effect on the inhibition profile. Surprisingly analogue 19a (entry 12) displayed good levels of T. brucei inhibition with an EC50 value of 7.6 ± 0.3 μM and moderate levels of parasite selectivity (SI = 21.9). Introduction of terminal oxygenation as a handle for further manipulation to one side of the alkyl side chain, analogues 21b, 18b, 19b and 22 (entries 5, 8, 13, 15), resulted in good micromolar activities with EC50 values <10 μM. Interestingly, the anti-anti analogue 20b (entry 2) was devoid of HeLa and T. brucei activity. By comparison, analogue 21b (entry 5) displayed good levels of parasite inhibition and selectivity with an SI of 23. For analogues 24 and 27 (entries 14 and 16), removal of the benzyl group resulted in ~2-fold loss in parasite activity. Removal of the benzyl group from analogue 21b (entry 5) to give 26 (entry 6) led to a significant loss in parasitic activity and selectivity, EC50 values of 3.1 ± 0.1 and 72.4 ± 3.9 μM and SI’s of 23 and 0.8, respectively. The anti-anti analogue 25 (entry 3) was still devoid of parasitic activity, despite transformation to the free hydroxyl. Gratifyingly, introducing the free hydroxyl to analogue 23 (entry 9) led to a slight increase in T. brucei inhibition and 23 has shown comparable levels of parasitic activity to that of chamuvarinin (6), EC50 values of 1.80 ± 0.10 and 1.4 ± 0.1 μM, respectively. On the basis of 23, the bis-directional analogue 35 (entry 10) was tested in the hope of further improving the activity. Unfortunately, 35 was 9 times less potent than 23 and subsequent removal of the benzyl group to give diol 38 (entry 11) resulted in significantly diminished activities. Comparison of the biological data for the ethyl, benzyl and alcohol triazole series, imply that the benzyl group may be potentially interacting with residues at a protein target site, which an alcohol group cannot accommodate. Alternatively, or as well as the benzyl group may be able to readily insert itself into the lipophilic membrane, whereas the hydrophilic alcohol incurs a greater penalty for membrane insertion.
Table 1.
SAR data for ethyl analogues 20a, 21a, 18a, 19c, benzyl analogues 20b, 21b, 18b, 35, 19b, 22 and alcohol analogues 23-27, 38, selectivity index refers to BSF parasitic inhibition versus mammalian HeLa inhibition; chamuvarinin (6), T. brucei (BSF) EC50 = 1.4 ± 0.1 μM.
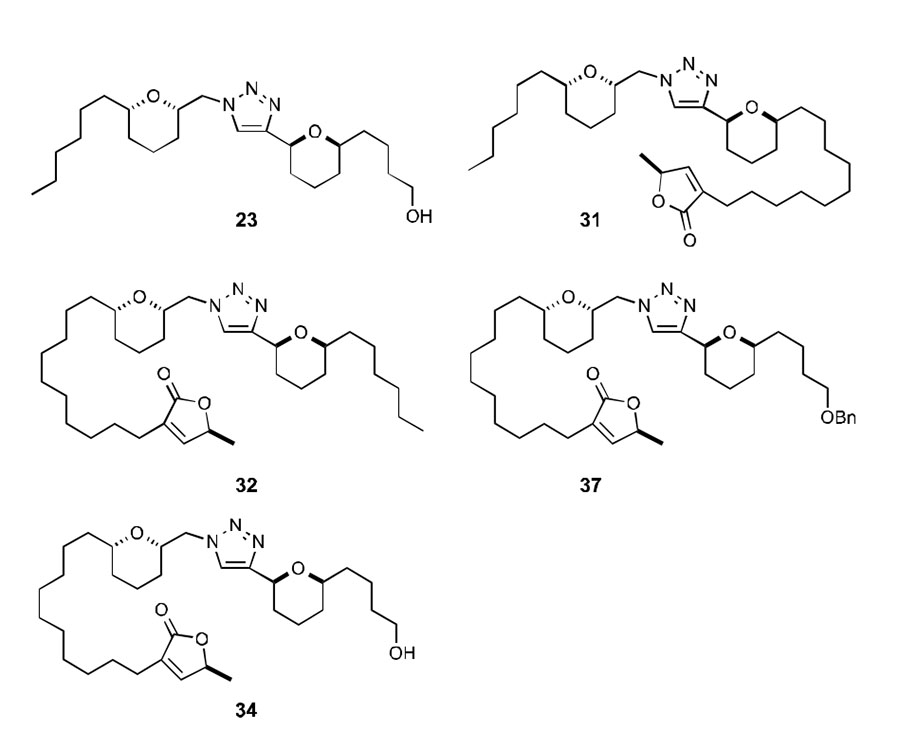
The synthesis of triazole analogues with terminal oxygenation has provided a functional handle for further elaboration (Table 2). The non-natural chamuvarinin-like analogues previously reported revealed that introduction of the butenolide side chain resulted in a >5-fold increase in T. brucei activity. Encouraged by the potent activity of lead alcohol 23 (entry 1), it was decided to incorporate the butenolide moiety based on this structure. Analogues 31 and 32 (entries 2 and 3) clearly highlight the importance of the spatial orientation of the pendent butenolide sidechain. While 31 was essentially inactive, 32 displayed low micromolar selective activities against both forms of T. brucei with EC50 values of 3.2 ± 0.1 and 5.7 ± 0.6 μM respectively and a selectivity index of 15.8. This suggests that although the structures are only subtly different at face value, their binding/interaction is highly specific and indicative of a protein target, rather than the biophysical properties of the compounds.
Table 2.
Biological profiles for analogues 23, 31-32, 37 and 34, selectivity index refers to BSF parasitic inhibition versus mammalian HeLa inhibition.
| |||||
|---|---|---|---|---|---|
| Entry | Analogue | T.brucei(BSF) EC50(μM) | T.brucei(Pro) EC50(μM) | Hela EC50(μM) | Selectivity Index |
| 1 | 23 | 1.8 ± 0.1 | 0.8±0.1 | 7.0±1.0 | 3.9 |
| 2 | 31 | >500 | >1000 | >1000 | - |
| 3 | 32 | 3.2±0.1 | 5.7±0.6 | 50.8±3.7 | 15.8 |
| 4 | 37 | 5.2±0.3 | 19.2±0.8 | 2.1±0.4 | 0.4 |
| 5 | 34 | 28.5±4.6 | 5.6±0.6 | 8.3±1.11 | 0.3 |
On the basis of the SAR data, incorporation of both the butenolide moiety and a benzyl-protected alcohol in place of the alkyl side chain indicated that the benzyl analogue 37 (entry 4) had similar activity to 32. Removal of the benzyl group to reveal the free hydroxyl analogue 34 (entry 5), was found to diminish activity and was 9 times less active than 32, with EC50 values of 28.5 ± 4.6 and 3.2 ± 0.1 μM, respectively. This may indicate that a non-hydrophilic arm on the opposite side of the butenolide moiety is required for maximum protein/analogue/lipid interaction.
Lipophilic efficiency
A theoretical comparison of the triazole analogues molecular properties was conducted in an effort to highlight the lipophilicity of these compounds and provide an indication as to whether they would be suitable drug candidates. As outlined in Table 3, the data indicated that our lead compound alcohol 23 (entry 1) displayed a suitable log P of 5.163, had a molecular weight less than 500 and had a suitable number of hydrogen bond donors and acceptors, in accordance with Lipinski’s rule of 5. Introduction of the butenolide moiety on one end of the THP ring system and an alkyl side chain on the opposite side in analogue 32 (entry 2), led to an increased log P value of 9. Exchange of the hydrophobic alkyl side chain with either a benzyl group (37, entry 4) or a free hydroxyl group (34, entry 3) decreased log P values while increasing the potential for hydrogen bond donation. Chamuvarinin itself (6, entry 5) displayed a high log P value of 9. This estimation for 6 was surprising, as the acetogenins have been shown to cross the blood-brain barrier. The lipophilic efficiency (LipE) is a combination of the calculated lipophilicity (clogP) and the potency (pEC50) of analogues to estimate the drug-likeness (LipE >5) of compounds. As highlighted in Table 3, triazoles 23, 32, 34, 37 and chamuvarinin 6 exhibited values ranging from +0.6 to −3.7, which were lower than the optimal LipE for potential drug candidates. In 2012, Ecker et. al. reported the calculated lipophilic efficiencies of marketed drugs such as cyclosporine A (known to inhibit the P-glycoprotein expressed at the blood-brain barrier) and found that these drugs had LipE values ranging from +2 to −9, which do not meet the specific requirements for drug candidates.[20] The LipE values of our triazole compounds are comparable with known drugs associated with crossing the blood-brain barrier, implying that they are potentially suitable drug candidates and reiterates that the triazoles may be targeting an enzymatic site rather than relying on the effects of the biophysical properties of the molecules.
Table 3.
Theoretical data of molecular properties for triazole analogues 6, 23, 32, 34 and 37.
| Entry | Compound | T.brucei(BSF) EC50(μM) | pEC50 | clog P | LipE | MW | No of H bond donors | No of H bond acceptors |
|---|---|---|---|---|---|---|---|---|
| 1 | 23 | 1.8±0.1 | 5.75 | 5.163 | 0.59 | 407.6 | 1 | 6 |
| 2 | 32 | 3.2±0.1 | 5.49 | 9.056 | −3.57 | 571.8 | 0 | 7 |
| 3 | 34 | 28.5±4.6 | 4.55 | 7.336 | −2.79 | 559.8 | 1 | 8 |
| 4 | 37 | 5.2±0.3 | 5.29 | 8.935 | −3.65 | 649.9 | 1 | 8 |
| 5 | 6 | 1.4±0.1 | 5.86 | 9.208 | −3.35 | 604.9 | 1 | 6 |
Molecular modelling
Our initial rationale for the design and synthesis of simplified triazole analogues was based on the molecular modelling of the central tricyclic core of chamuvarinin (6), as outlined in Figure 3a, indicating that 6 adopts a “U-shape” conformation.[21] We hypothesized that a 5-membered heterocyclic spacer (Figures 3b and 3c) would act as an effective spatial mimic for the central THF core of 6 and thus retain potent biological activity. Modelling highlights that the lowest energy conformation of the syn-syn bis-THP rings acts a better mimic of this ‘U-shaped’ conformation than the corresponding anti-anti and this would appear to be broadly born out by the biological results.[21] The encouraging trypanocidal activity of the triazole compounds discussed in this paper has now formed the basis of further design iterations and lead structure optimization. The evolving structure-activity-relationship (SAR) will be enabled by cycles of iterative design towards the development of more potent trypanocidal compounds.
Figure 3.

Molecular modelling of the lowest energy conformations for a) chamuvarinin (6), b) syn-syn triazole (18a) and c) anti-anti triazole (20a).
Conclusion
A focused library of 1,4-triazole-based acetogenin analogues have been synthesized and have demonstrated low micromolar trypanocidal activities against both bloodstream and procyclic forms of T. brucei. Analogue 23 has displayed comparable levels of T. brucei inhibition to that of natural product chamuvarinin (6), EC50 values of 1.8 ± 0.1 and 1.4 ± 0.1 μM, respectively demonstrating that this series of triazole analogues are potential lead compounds for the development of an effective therapeutic agent for HAT. The acetogenins have long been established as mitochondrial Complex I inhibitors within mammalian cells, therefore, it is postulated that the analogues are targeting a protein within the mitochondrion of the parasite. The specific protein target and mode of action for parasite inhibition is currently undetermined for these analogues. However, analogues 31 and 32 clearly highlighs that spatial orientation of the butenolide side chain impacts heavily on T. brucei inhibition. Current focus is to establish whether the analogues are disrupting mitochondrial functions through the incorporation of fluorescent and affinity tags to lead triazole compounds in order to isolate specific proteins of interest, allowing them to be genetically and chemically validated.
Experimental Section
See supporting information for full experimental details
Supplementary Material
Acknowledgements
This work was funded by the Royal Society, the Leverhulme Trust, the EPSRC (GJF) and the Wellcome Trust (TKS, WT 093228). We thank the EPSRC National Mass Spectrometry Service Centre, Swansea, UK for mass spectrometry services.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
Contributor Information
Dr Gordon J. Florence, EaStCHEM School of Chemistry, Biomedical Science Research Complex, University of St Andrews, North Haugh, St Andrews, Fife, KY16 9ST, U. K..
Professor Terry K. Smith, EaStCHEM School of Chemistry, Biomedical Science Research Complex, University of St Andrews, North Haugh, St Andrews, Fife, KY16 9ST, U. K..
References
- [1].Zhou L, Stewart G, Rideau E, Westwood NJ, Smith TK. J. Med. Chem. 2013;56:796–806. doi: 10.1021/jm301215e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Trypanosomiasis, African. World Health Organization; Geneva: [Accessed 19 June, 2014]. 2012. http://www.who.int/topics/trypanosomiasis_african/en/ [Google Scholar]
- [3].a) Steverding D. Parasites & Vectors. 2010;3:15. doi: 10.1186/1756-3305-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Delespaux V, de Koning HP. Drug Resistance Updates. 2007;10:30–50. doi: 10.1016/j.drup.2007.02.004. [DOI] [PubMed] [Google Scholar]
- [4].Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Baudin E, Buard V, Kazadi-Kyanza S, Ilunga M, Mutangala W, Pohlig G, Schmid C, Karunakara U, Torreele E, Kande V. Lancet. 2009;374:56–64. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- [5].a) Alali FQ, Liu X-X, McLaughlin JL. J. Nat. Prod. 1999;62:504–540. doi: 10.1021/np980406d. [DOI] [PubMed] [Google Scholar]; b) Zafra-Polo M-C, Figadère B, Gallardo T, Tormo JR, Cortes D. Phytochemistry. 1998;48:1087–1117. [Google Scholar]; c) Zafra-Polo MC, González MC, Estornell E, Sahpaz S, Cortes D. Phytochemistry. 1996;42:253–271. doi: 10.1016/0031-9422(95)00836-5. [DOI] [PubMed] [Google Scholar]; d) Zeng L, Ye Q, Oberlies NH, Shi G, Gu Z-M, He K, McLaughlin JL. Nat. Prod. Rep. 1996;13:275–306. doi: 10.1039/np9961300275. [DOI] [PubMed] [Google Scholar]; e) Rupprecht JK, Hui YH, McLaughlin JL. J. Nat. Prod. 1990;53:237–278. doi: 10.1021/np50068a001. [DOI] [PubMed] [Google Scholar]
- [6].Bermejo A, Figadère B, Zafra-Polo MC, Barrachina I, Estornell E, Cortes D. Nat. Prod. Rep. 2005;22:269–303. doi: 10.1039/b500186m. [DOI] [PubMed] [Google Scholar]
- [7].a) Fall D, Duval RA, Gleye C, Laurens A, Hocquemiller R. J. Nat. Prod. 2004;67:1041–1043. doi: 10.1021/np030521a. [DOI] [PubMed] [Google Scholar]; b) Derbré S, Poupon E, Gleye C, Hocquemiller R. J. Nat. Prod. 2007;70:300–303. doi: 10.1021/np060376b. [DOI] [PubMed] [Google Scholar]
- [8].a) Florence GJ, Morris JC, Murray RG, Vanga RR, Osler JD, Smith TK. Org. Lett. 2011;13:514–517. doi: 10.1021/ol1028699. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Florence GJ, Morris JC, Murray RG, Vanga RR, Osler JD, Smith TK. Chem. Eur. J. 2013;19:8309–8320. doi: 10.1002/chem.201204527. [DOI] [PubMed] [Google Scholar]
- [9].a) Sahpaz S, Bories C, Loiseau PM, Cortès D, Hocquemiller R, Laurens A, Cavé A. Planta. Med. 1994;60:538–540. doi: 10.1055/s-2006-959566. [DOI] [PubMed] [Google Scholar]; b) Waechter A-I, Yaluff G, Inchausti A, Rojas de Arias A, Hocquemiller R, Cavé A, Fournet A. Phytother. Res. 1998;12:541–544. [Google Scholar]; c) Hoet S, Opperdoes F, Brun R, Quetin-Leclercq J. Nat. Prod. Rep. 2004;21:353–364. doi: 10.1039/b311021b. [DOI] [PubMed] [Google Scholar]
- [10].a) Paddon-Jones GC, McErlean CSP, Hayes P, Moore CJ, Konig WA, Kitching W. J. Org. Chem. 2001;66:7487–7495. doi: 10.1021/jo0159237. [DOI] [PubMed] [Google Scholar]; b) Schaus SE, Brandes AD, Larrow JF, Tokunga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- [11].Scott JP, Alam M, Bremeyer N, Goodyear A, Lam T, Wilson RD, Zhou G. Org. Process Res. Dev. 2011;15:1116–1123. [Google Scholar]
- [12].a) Müller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521–522. [Google Scholar]; b) Roth GJ, Liepold B, Müller SG, Bestmann HJ. Synthesis. 2004:59–62. [Google Scholar]
- [13].Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769–3772. [Google Scholar]
- [14].a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b) Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- [15].Schultz HS, Freyermuth H,B, Buc SR. J. Org. Chem. 1963;28:1140–1142. [Google Scholar]
- [16].a) Blakemore PR, Cole WJ, Kocieński PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]; b) Blakemore PR. J. Chem. Soc. Perkin Trans. 2002;1:2563–2585. [Google Scholar]; c) Dumeunier RI, Markó IE. In: Modern Carbonyl Olefination: Methods and Applications. Takeda T, editor. Wiley-VCH; Weinheim: 2004. pp. 104–150. [Google Scholar]
- [17].The butenolide aldehyde 30 was synthesized in 5 steps from enantio-enriched (S)-propylene oxide, see Ref 8a.
- [18].a) Marshall JA, Chen M. J. Org. Chem. 1997;62:5996–6000. [Google Scholar]; b) Crimmins MT, Zhang Y, Diaz FA. Org. Lett. 2006;8:2369–2372. doi: 10.1021/ol060704z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Biological testing was performed using the Alamar Blue viability test as described in: Mikus J, Steverding D. Parasitol. Int. 2000;48:265–269. doi: 10.1016/s1383-5769(99)00020-3.
- [20].Jabeen I, Pleban K, Rinner U, Chiba P, Ecker GF. J. Med. Chem. 2012;55:3261–3273. doi: 10.1021/jm201705f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schrodinger Maestro (v9.7.014) and Macromodel (v10.4) were used for 10000 step Monte-Carlo searches using the MMFF force field.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.