

Abstract
Summary
Background
Ultraviolet (UV) radiation constitutes an important risk factor for malignant melanoma, but the wavelength responsible for the initiation of this disease is not fully elucidated. Solar UV induces multiple signalling pathways that are critical for initiation of apoptotic cell death as a cellular defence against malignant transformation.
Objectives
To evaluate the involvement of the transcription factors nuclear factor (NF)-κB and activator protein (AP)-1 in the signalling pathways induced by UVA or UVB irradiation in human melanocytes.
Methods
Primary cultures of normal human melanocytes were irradiated with UVA or UVB, and the concomitant DNA damage and redox alterations were monitored. The resulting activation of the NF-κB and AP-1 signalling pathways and subsequent apoptosis were studied.
Results
UVB irradiation causes DNA damage detected as formation of cyclobutane pyrimidine dimers, while UVA induces increased levels of 8-hydroxydeoxyguanosine and lipid peroxidation. UVA and UVB initiate phosphorylation of c-Jun N-terminal protein kinase and extracellular signal-regulated kinase, and the apoptosis signalling pathways converge into a common mechanism. Downregulation of c-Jun suppresses AP-1-mediated signalling and prevents apoptosis upstream of lysosomal and mitochondrial membrane permeabilization, whereas inhibition of NF-κB by SN50 increases apoptosis.
Conclusions
We conclude that AP-1 induces proapoptotic signalling, whereas NF-κB is a key antiapoptotic/prosurvival factor in both UVA- and UVB-induced cellular damage in human melanocytes, which might in turn impact melanoma development and progression.
What's already known about this topic?
Melanocytes are the target cells of ultraviolet (UV) irradiation, and the cells from which melanoma originates.
The mitogen-activated protein kinase signalling pathway is important for regulation of UV-induced cellular responses.
Previous studies have strongly implicated the transcription factors activator protein (AP)-1 and nuclear factor (NF)-κB as mediators of the UV response in other cell types.
What does this study add?
The present study identifies NF-κB as an antiapoptotic/prosurvival factor and shows that AP-1 stimulates proapoptotic signalling during both UVA- and UVB-induced apoptosis in human melanocytes.
An improved understanding of cellular responses in UV-exposed melanocytes is essential to understanding and preventing the formation of melanoma, and might provide an opportunity to identify apoptotic regulators.
Introduction
The most severe malignant tumour of the skin is malignant melanoma, arising from the pigment-producing epidermal melanocytes. Although the aetiology of melanoma involves a complex interplay between genetics, host characteristics and environmental factors, it is generally agreed that exposure to sunlight is a major external aetiological agent.1 However, there is no consensus concerning the wavelengths responsible for melanoma development. DNA is the major chromophore that absorbs ultraviolet (UV)B irradiation (290–320 nm), whereas UVA (320–400 nm) is poorly absorbed by DNA. Instead, UVA generates oxidative stress and induces chemical modifications to DNA, likely triggered by photosensitization.2 The UVB component of sunlight penetrates only into the epidermis, while the longer UVA wavelengths penetrate deeper into the underlying dermis. UV-induced damage to both DNA and other cellular components has been found to activate transcription factor pathways in several cell types, although these reactions are less well characterized in melanocytes. The nuclear factor (NF)-κB and activator protein (AP)-1 families have been identified as central transcription factors involved in the processes of cell death, proliferation and differentiation, as well as cell survival.3
The NF-κB family of transcription factors regulates the activities of various signalling pathways, and its members are considered key regulators in inflammation, oncogenesis and apoptosis. NF-κB consists of dimeric complexes of the Rel protein family, which includes five members: p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2).4 Inactive NF-κB resides in the cytoplasm due to the masking of its nuclear localization signal by IκB (inhibitor of κB).5 A wide range of stimuli induce post-translational modifications of IκB, including phosphorylation and ubiquitination, thus enabling proteasomal degradation. NF-κB then translocates to the nucleus, where it activates gene transcription.6
The activity of the nuclear transcription factor AP-1, which is composed of dimers of Fos (c-Fos, FosB, Fra-1, Fra-2) and Jun (c-Jun, JunB, JunD) proteins, is controlled by mitogen-activated protein kinase (MAPK) signalling pathways. The molecular mechanisms leading to MAPK activation via the production of UV-induced reactive oxygen species (ROS) are not fully understood and may involve interactions between different signalling pathway intermediates.6 The MAPK signalling pathways include extracellular signal-regulated kinase (ERK), p38 MAPK and c-Jun N-terminal kinase (JNK).
Elimination of photodamaged melanocytes via apoptosis is crucial to preventing malignant transformation. Previously, we reported that in human melanocytes, physiological doses of both UVA and UVB, as well as higher doses applied in our experimental model, activate the intrinsic pathway of apoptosis mediated by lysosomal membrane permeabilization (LMP).7–9 However, little is known concerning the roles of different wavelengths within the UV spectrum in the modulation of intracellular signal transduction in these cells. Hence, the present study was conducted to characterize the underlying signalling that determines cell fate after exposure to different wavelengths within the UV spectrum. Herein, we compare the action of NF-κB and AP-1 to clarify the mechanisms that determine melanocyte survival and death.
Materials and methods
Cell cultures and conditions
All experiments were performed according to the ethical principles of the Declaration of Helsinki and approved by the ethical committee at Linköping University, Linköping, Sweden. Melanocytes were obtained from fair-skinned donors (aged 0–3 years) by means of foreskin circumcisions, and pure cultures were established as previously described.10 The melanocytes were cultured in Medium 199 with supplements11 and incubated at 37 °C in a humidified atmosphere of 5% CO2 in air. The purity of the culture system has been confirmed via electron microscopy.12 The experiments were conducted between passages 2 and 7, and no cells were cultured for more than 3 weeks in total.
In some experiments, the cell-permeable inhibitor SN50 (20 μmol L−1, stock in H2O; Merck Biosciences, Darmstadt, Germany) was used to block NF-κB activity.
Ultraviolet irradiation
The UVB source consisted of two Philips TL20W/12 tubes (Philips, Eindhoven, the Netherlands) emitting in the spectral range of 280–370 nm, with a main output at 305–320 nm. For UVA irradiation, a Medisun 2000-L tube (Dr Gröbel UV-Elektronik GmbH, Ettlingen, Germany; 340–400 nm) was used. A Schott WG 305 cut-off filter (50% absorption below 305 nm; Schott, Mainz, Germany) was employed, and the UVA output was 80 mW cm−2, while that of UVB was 1·44 mW cm−2. Measurements were conducted with an RM-12 (Dr Gröbel UV-Elektronik GmbH) and a PUVA Combi Light dosimeter (UVee Therapy Systems; Leuven, Belgium).
To achieve 30–40% apoptosis with a minimum of necrotic cell death, the irradiation doses were titrated (UVA, 2–100 J cm−2 and UVB, 20–1000 mJ cm−2). This resulted in an experimental model using 60 J cm−2 UVA and 500 mJ cm−2 UVB. We performed further parallel studies in each experiment using doses resembling a more physiological situation for both UVA (6 J cm−2) and UVB (60 mJ cm−2) and detected similar trends (Figs S1–S3; see Supporting Information).
UV exposure was performed in culture dishes containing prewarmed phosphate-buffered saline. No increase in temperature was noted during irradiation. Following UV irradiation, fresh culture medium was added. Unirradiated cultures were also analysed at each time point.
Nuclear morphology and caspase activation
The cultures were fixed in 4% neutral buffered formaldehyde and mounted in Vectashield® Mounting Medium (Vector Laboratories, Burlingame, CA, U.S.A.) with 4' ,6-diamidino-2-phenylindole (DAPI; 1·5 μg mL−1). The nuclear morphology was evaluated in 200 randomly selected cells using a fluorescence microscope (× 60, λex 350 nm; Nikon, Tokyo, Japan). To analyse caspase-3 activity, the cells were collected in lysis buffer (10 mmol L−1 Tris-HCl at pH 7·5, 130 mmol L−1 NaCl, 1% Triton X-100, 10 mmol L−1 sodium pyrophosphate, 10 mmol L−1 NaH2PO4/NaHPO4) and incubated with the substrate Ac-DEVD-AMC according to the manufacturer's recommendations (BD Biosciences, San Jose, CA, U.S.A.). The concentration of the proteolytically released AMC substrate (7-amino-4-methylcoumarin) was analysed in a Shimadzu RF-540 spectrofluorometer (λex380/λem435; Shimadzu, Kyoto, Japan). Protein concentrations were determined using the BioRad DC Protein Assay System (BioRad Laboratories, Hercules, CA, U.S.A.), and caspase activity is expressed as arbitrary units per microgram protein per hour.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 20 min at 4 °C and processed for immunocytochemistry. The cells were permeabilized with 0·1% saponin and incubated overnight at 4 °C with one of the following monoclonal antimouse primary antibodies: NF-κB p50 (Santa Cruz Biotechnology, Santa Cruz, TX, U.S.A.) or cytochrome c (BD Biosciences); or polyclonal antirabbit primary antibodies: cathepsin D (Athens Research and Technology, Athens, GA, U.S.A.), c-Fos, c-Jun or NF-κB p65 (all Santa Cruz Biotechnology), followed by incubation with a secondary antimouse Alexa Fluor 488 conjugate or an antirabbit Alexa Fluor 488 conjugate (Molecular Probes, Eugene, OR, U.S.A.) for 1 h at room temperature. The samples were then mounted in Vectashield® Mounting Medium (Vector Laboratories) with DAPI and inspected using a Nikon E600W fluorescence confocal microscope (Nikon). In each culture dish, 200 cells were randomly selected, and the localization of the proteins was analysed. Negative controls, incubated without a primary antibody, showed no staining.
Cytosolic extraction
Cytosol was extracted through incubation with a digitonin (Sigma, St Louis, MO, U.S.A.) extraction buffer [250 mmol L−1 sucrose, 20 mmol L−1 HEPES, 10 mmol L−1 KCl, 1·5 mmol L−1 MgCl2, 1 mmol L−1 ethylene glycol tetraacetic acid, 1 mmol L−1 ethylenediaminetetraacetic acid (EDTA), 1 mmol L−1 Pefabloc, 8 mmol L−1 dithiothreitol, pH 7·5] on ice for 12 min. The concentration of digitonin was titrated to permeabilize the plasma membrane, but not membranes of intracellular organelles, determined via the analysis of lactate dehydrogenase activity13 and β-N-acetylglucosaminidase as previously described.14 The cytosol was centrifuged, and the proteins in the supernatant were precipitated with trichloroacetic acid (50%), followed by incubation on ice for 10 min and subsequent pelleting via centrifugation. To perform the Western blot analysis, the pellet was resuspended in a urea lysis buffer (6 mol L−1 urea, 150 mmol L−1 NaCl, 1% Triton X-100, 0·1% sodium dodecylsulfate, 50 mmol L−1 Tris pH 8·0, 5 mmol L−1 EDTA), a sample buffer [5% β-mercaptoethanol in Laemmli sample buffer (BioRad Laboratories) (1 : 1)] and 1 mol L−1 NaOH.
Western blot analysis
The protein samples were loaded onto a Ready gel (BioRad Laboratories) and transferred to a Hybond™-P blotting membrane (GE Healthcare, Little Chalfont, U.K.). After saturation with 5% nonfat dry milk, immunodetection was performed with a polyclonal primary antibody against c-Fos, c-Jun (both Santa Cruz Biotechnology), phospho-c-Jun (Sigma), cathepsin D (Athens Research and Technology) or NF-κB p65 (Santa Cruz Biotechnology), or with a monoclonal primary antibody against phospho-JNK, phospho-ERK (both Santa Cruz Biotechnology), cytochrome c (BD Biosciences), cathepsin D (Nuclea Biotechnologies, Pittsfield, MA, U.S.A.), or NF-κB p50 (Santa Cruz Biotechnology). Then, the corresponding horseradish peroxidase-conjugated secondary antibody was added [mouse, rabbit (Dako, Glostrup, Denmark) or goat (Santa Cruz Biotechnology)]. The membranes were developed using the enhanced ECL-Plus Western blotting detection system and detected on Hyperfilm ECL™ (both from GE Healthcare). The membranes were reprobed with glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Biogenesis, Poole, U.K.) or β-actin (Santa Cruz Biotechnology), as an internal control. Densiometric quantification of the bands was performed with Gel-Pro Analyzer 3.1 (Media Cybernetics, Rockville, MD, U.S.A.).
Small interfering RNA transfection
Human melanocytes were seeded at 60–70% confluence 1 day prior to transfection and transfected for 8 h with 1 μg of JUN small interfering (si)RNA (no. 1, AAAGATGGAAACGACCTTCTA or no. 2, AAGAAGTGTCCGAGAACTAAA; Qiagen, Venlo, the Netherlands) and 6 μL of RNAiFect Transfection Reagent (Qiagen) according to the manufacturer's recommendations. Alexa Fluor 555-labelled nonsilencing siRNA, with a scrambled sequence showing no homology to mammalian genes (AATTCTCCGAACGTGTCACGT; Qiagen) was used as negative control. siRNA targeting lamin A/C (AACTGGACTTCCAGAAGAACA; Qiagen) served as the positive control.
Analysis of DNA damage and lipid peroxidation
The OxiSelect™ Oxidative DNA Damage ELISA Kit was used to measure the formation of 8-hydroxydeoxyguanosine (8-OHdG) and cyclobutane pyrimidine dimers (CPDs), and pyrimidine (6–4) pyrimidone photoproducts were detected using the OxiSelect™ Cellular UV-induced DNA Damage ELISA Kit. The lipid peroxidation products malondialdehyde and 4-hydroxyalkenals were measured using the colorimetric Bioxytech LPO-586 assay (all from Oxis International, Beverly Hills, CA, U.S.A.).
Intracellular reduced glutathione
Melanocytes were collected in 0·5 mol L−1 HClO4 containing 1 mmol L−1 EDTA, and reduced glutathione content was analysed with high-performance liquid chromatography and electrochemical detection, as described elsewhere.15
Whole-human-genome microarray analysis
Microarray analyses of melanocytes from three different donors (sorted into apoptotic annexin V-positive cells or nonapoptotic annexin V-negative cells 4 h postirradiation with UVA, UVB or no irradiation; Fig. S4; see Supporting Information) were performed to compare gene expression differences. Briefly, total RNA was prepared using standard RNA extraction protocols (TRIzol; Sigma), and quality checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). Labelling and hybridization were performed with the Affymetrix Human Genome Microarray (WT GeneTitan ST1.1; Plate type, HuGene-1_1-st-v1-16; Array type, HuGene-1_1-st-v1) according to the manufacturer's protocols (Affymetrix, Santa Clara, CA, U.S.A.), at the Bioinformatics and Expression Analysis Core Facility, Karolinska Institute, Sweden. The data were summarized (PLIER algorithm) and normalized to the global median, and the cut-off fold change was ± 2 to be noted (P < 0·05; q < 0·05). The raw CEL files generated from this analysis were processed using the Qlucore Omics Explorer software package (Qlucore, Lund, Sweden). Data were quality assessed before and after normalization through the inspection of hierarchical clustering trees and principal component analysis plots.
Statistics
All experiments were repeated at least three times using melanocytes from different donors, and the results are presented as the means and SDs of independent samples. Statistical evaluation was performed via one-way anova, followed by Bonferroni's multiple comparison post-test for comparisons between groups. Differences were considered significant at P ≤ 0·05 and are indicated with asterisks in the figures.
Results
Human melanocytes were exposed to UVA (60 J cm−2) or UVB (500 mJ cm−2), which induced apoptotic cell death in approximately 50% of the cells, as estimated by examining nuclear fragmentation and activation of caspase-3 (Fig.1a,b). We confirmed that UVA causes ROS-induced oxidative modification of DNA, detected as an increase in 8-OHdG (Fig.1d). In contrast, UVB irradiation caused increased formation of CPDs, but no increase in lipid peroxidation products or 8-OHdG (Fig.1c–e). The enhanced production of ROS induced by UVA was further verified based on increased lipid peroxidation of specific aldehydes and a decrease in reduced glutathione, whereas these levels were unaffected after UVB irradiation (Fig.1e,f). Parallel studies using doses resembling a more physiological situation showed similar results after both UVA (6 J cm−2) and UVB (60 mJ cm−2) exposure (Fig. S1; see Supporting Information).
Figure 1.
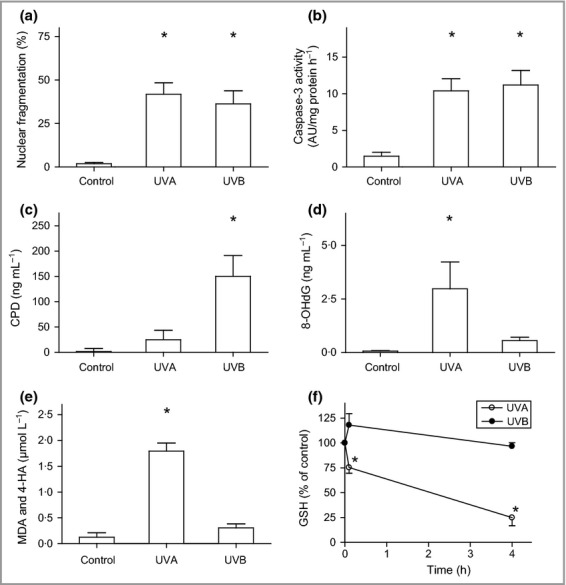
Oxidative damage induced by ultraviolet (UV) irradiation. Human melanocytes were exposed to UVA (60 J cm−2) or UVB irradiation (500 mJ cm−2). (a) Nuclear fragmentation quantified 24 h after irradiation and (b) caspase-3 activation detected 16 h after irradiation. AU, arbitrary units. Production of (c) cyclobutane pyrimidine dimers (CPDs) and (d) 8-hydroxydeoxyguanosine (8-OHdG) directly after irradiation. (e) Levels of the lipid peroxidation products malondialdehyde (MDA) and 4-hydroxyalkenals (4-HA) determined directly after irradiation. (f) Concentration of reduced glutathione (GSH) calculated as nmol mg−1 protein and expressed as the percentage of unirradiated cells from the same donor (set as 100%). Results are presented as the mean ± SD (n = 4). *P ≤ 0·05 vs. control.
Following UV irradiation, apoptotic melanocytes were isolated through cell sorting via flow cytometry and then subjected to whole-genome microarray analysis. Table1 presents selected results related to MAPK gene expression. We observed no variation in the expression of the MAPK1 (encoding ERK) and MAPK8 (encoding JNK) genes 4 h after UVA or UVB exposure, and accordingly no changes in the levels of their encoded proteins were detected during a period of 24 h (Fig.2a–d). However, both UVA and UVB induced phosphorylation of ERK and JNK (Fig.2a–d). UVA caused persistent phosphorylation of both JNK and ERK for 6 and 24 h, respectively. Following UVB exposure, JNK and ERK phosphorylation peaked after 2 h and then decreased. The mRNA levels of JUN and FOSB were significantly elevated 4 h after treatment with both UVA and UVB, and JUND was increased following UVA treatment (Table1). However, these alterations could not be verified at the protein level (Fig.2e,f). The UVA-induced phosphorylation of c-Jun peaked within 2 h, while UVB-irradiated melanocytes showed the highest level of c-Jun phosphorylation after 4–6 h (Fig.2e,f). Significantly, both c-Jun and c-Fos were translocated to the nucleus following UVA or UVB exposure (Fig.3a,b). A similar translocation was also detected when using physiological doses of UVA and UVB (Fig. S2; see Supporting Information).
Table 1.
Melanocytes were exposed to ultraviolet (UV)A and UVB irradiation and, after 4 h, the apoptotic fraction of cells was isolated and the mRNA expression compared with that of controls. The list presents selected genes of the nuclear factor (NF)-κB and activator protein (AP)-1 signalling pathways. The fold-change threshold is ± 2 (P < 0·05)
| UVA (dead) vs. control | UVB (dead) vs. control | ||||
|---|---|---|---|---|---|
| Fold change | P-value | Fold change | P-value | ||
| MAPK1 (ERK) | ns | ns | |||
| MAPK8 (JNK) | ns | ns | |||
| AP-1 | JUN (c-Jun) | 5·3 | 0·009 | 2·5 | < 0·001 |
| JUND | 2·6 | 0·011 | ns | ||
| FOS | ns | ns | |||
| FOSB | 7·86 | 0·019 | 3·27 | 0·021 | |
| NF-κB | NFKB1 (p50/p105) | ns | ns | ||
| NFKB2 (p52/p100) | ns | ns | |||
| RELA (p65) | ns | ns | |||
| RELB | ns | ns | |||
| REL (c-Rel) | ns | −2·5 | 0·042 | ||
| IκB | NFKBIA | ns | ns | ||
| TP53 (p53) | ns | ns | |||
ERK, extracellular signal-regulated kinase; IκB, inhibitor of κB; JNK, c-Jun N-terminal kinase; ns, not significant.
Figure 2.
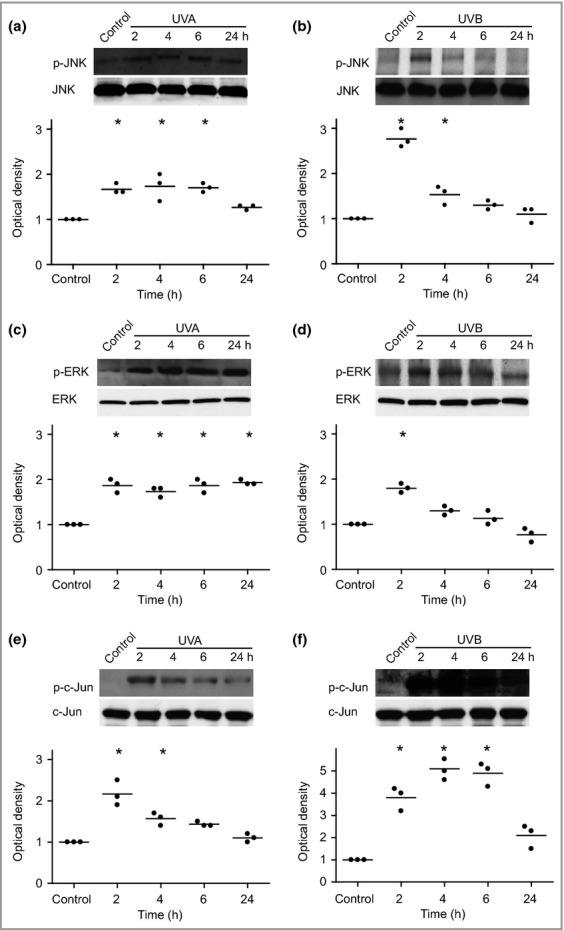
Ultraviolet (UV)-induced phosphorylation of c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and c-Jun. Human melanocytes were exposed to UVA (60 J cm−2) or UVB (500 mJ cm−2) and the resultant levels of phosphorylation were analysed by Western blotting at 2, 4, 6 and 24 h postirradiation. One representative Western blot out of four and the corresponding optical density of UVA-induced (a) phosphorylated JNK (p-JNK) and JNK, (c) phosphorylated ERK (p-ERK) and ERK, and (e) phosphorylated c-Jun (p-c-Jun) and c-Jun. UVB-induced (b) p-JNK and JNK, (d) p-ERK and ERK, and (f) p-c-Jun and c-Jun. The results are presented as the mean ± SD (four different donors). *P ≤ 0·05 vs. control.
Figure 3.
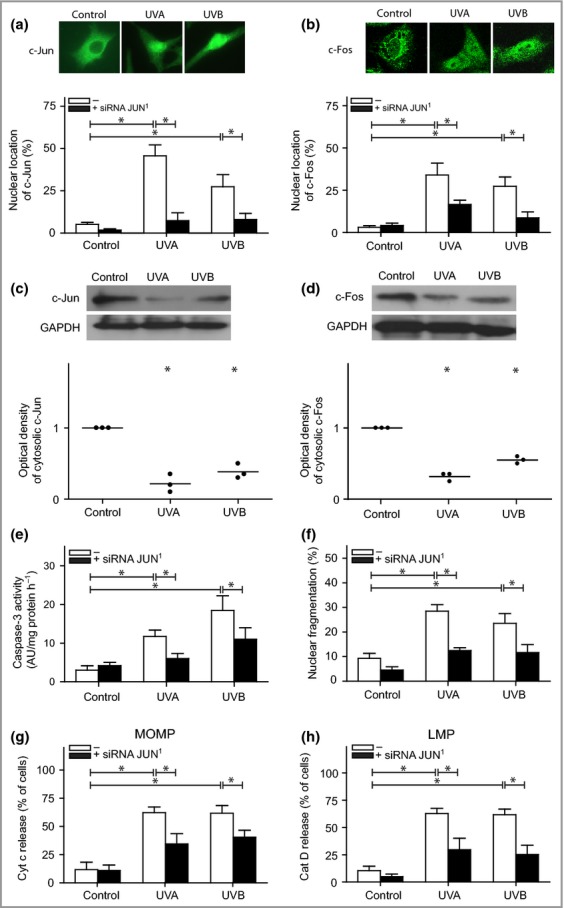
Activator protein-1 signalling in melanocytes following ultraviolet (UV) exposure. Melanocytes pretreated with JUN small interfering (si)RNA (black bars) 24 h prior to UVA (60 J cm−2) or UVB (500 mJ cm−2) treatment. Quantification of melanocytes showing nuclear staining of (a) c-Jun and (b) c-Fos 4 h after UV exposure. The images were selected to display the characteristic appearance of the nuclear and cytosolic location, respectively. The protein levels and corresponding optical density of (c) c-Jun and (d) c-Fos in digitonin-extracted cytosolic fractions are shown from one representative Western blot out of four. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. (e) Caspase-3 activation was analysed based on the cleavage of the substrate Ac-DEVD-AMC, 16 h postirradiation [expressed as arbitrary unites (AU) per mg protein and hour] and (f) apoptosis was quantified through microscopic inspection of nuclear morphology in 4',6-diamidino-2-phenylindole (DAPI)-stained cells after 24 h. UV-induced (g) mitochondrial outer membrane permeabilization (MOMP), as detected by cytochrome c release, and (h) lysosomal membrane permeabilization (LMP), as detected by the release of cathepsin D (cat D) to the cytosol. The results are presented as the mean ± SD (n = 4). *P ≤ 0·05 vs. control.
To evaluate the role of AP-1 in UV-induced apoptosis, melanocytes were transfected with siRNA to inhibit JUN expression. c-Jun protein expression was reduced by 79%, 24 h after transfection (Fig. S5; see Supporting Information). When silencing JUN, we observed a significantly reduced number of cells with nuclear-localized c-Jun and c-Fos following UV exposure (Fig.3a,b). The translocation of these proteins was verified in isolated cytosol (Fig.3c,d). Cells depleted of c-Jun showed a lower frequency of apoptosis, as verified based on reduced caspase-3 activity and a decreased number of fragmented nuclei (Fig.3e,f). No differences were detected between UVA- and UVB-induced cell death. Furthermore, analysis of upstream apoptosis signalling revealed decreased release of cytochrome c from the mitochondria (Fig.3g).
Participation of lysosomal-mediated signalling pathways in apoptosis in melanocytes has been reported to be upstream of cytochrome c release.9 Here, LMP was studied through assessing the translocation of the lysosomal protease cathepsin D from the lysosome to the cytosol, and immunofluorescence analysis revealed cytosolic staining 4 h after UVA or UVB treatment. In JUN-silenced cells, LMP was reduced (Fig.3h). These results were verified in isolated cytosolic fractions with Western blotting (not shown). Thus, proapoptotic signalling of the AP-1 complex operates upstream of LMP.
Neither any of the examined NF-κB genes nor IκB showed significant increases in gene expression following exposure to UVA or UVB (Table1). Immunofluorescence staining of p65 and p50 revealed a time-dependent increase in the nuclear translocation of the subunits after treatment with UVA and UVB (Fig.4a). Accordingly, cytosolic fractions showed a reduced amount of p65 following UV exposure, suggesting translocation to another subcellular location (Fig.4b). To elucidate the role of NF-κB in UV-induced apoptosis, we used the cell-permeable inhibitor SN50, which blocks the p50 protein of the NF-κB dimer. Treatment with SN50 resulted in reduced nuclear localization of both p50 and p65 after UV exposure (Fig.4c,d). When NF-κB was inhibited, release of cytochrome c from the mitochondria, caspase activation and nuclear fragmentation were all increased, suggesting that NF-κB promotes cell survival in human melanocytes following UVA or UVB exposure (Fig.4e–g). Interestingly, analysis of upstream signalling revealed increased LMP after UVA exposure, but not after exposure to UVB (Fig.4h). When irradiating melanocytes with physiological doses of UVA and UVB in parallel studies, we found similar trends in the results when using the NF-κB inhibitor SN50, although they were not statistically significant (Fig. S3; see Supporting Information).
Figure 4.
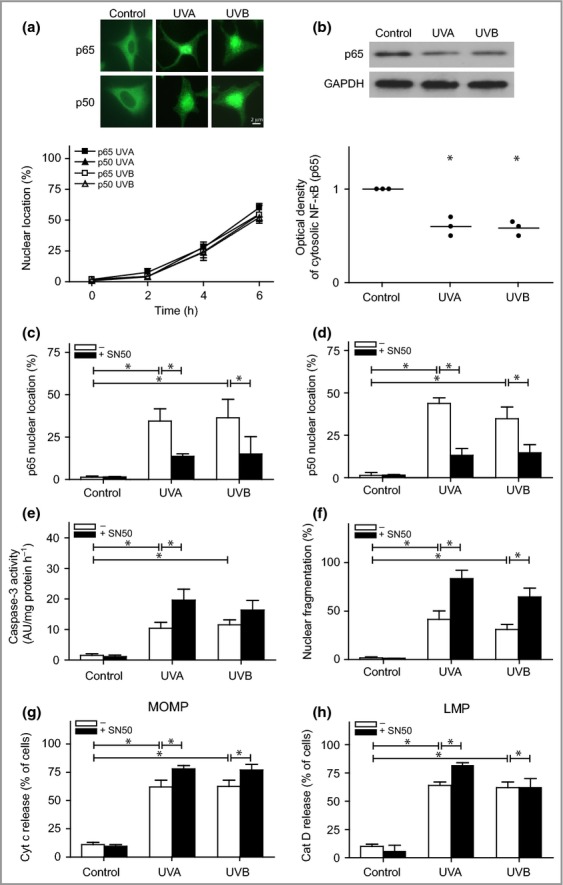
Nuclear factor (NF)-κB signalling in ultraviolet (UV)-irradiated melanocytes. Melanocytes were exposed to UVA (60 J cm−2) or UVB (500 mJ cm−2). (a) Immunostaining to determine the localization of the p50 and p65 subunits of the NF-κB dimer after irradiation. Images are selected to display the characteristic appearance of cytosolic and nuclear location, respectively. (b) One representative Western blot out of four and the corresponding optical density, showing the levels of the p65 subunit in digitonin-extracted cytosolic fractions. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Nuclear localization of the (c) p65 and (d) p50 subunits in melanocytes pretreated with an inhibitor of the p50 subunit of the NF-κB dimer (SN50, 20 μmol L−1; black bars) 3 h prior to irradiation. (e) Caspase-3 activation was analysed based on the cleavage of the substrate Ac-DEVD-AMC, 16 h postirradiation [expressed as arbitary units (AU) per mg protein and hour], and (f) apoptosis was quantified via microscopic analysis of nuclear morphology in 4',6-diamidino-2-phenylindole (DAPI)-stained cells after 24 h. UV-induced (g) mitochondrial outer membrane permeabilization (MOMP), as detected by cytochrome c release, and (h) lysosomal membrane permeabilization (LMP), as detected by the release of cathepsin D (cat D). The results are presented as the mean ± SD (n = 4). *P ≤ 0·05 vs. control.
Finally, we studied whether cross-talk or a compensatory mechanism connects AP-1 and NF-κB signalling in melanocytes. As shown in Fig.5 (a,b), inhibition of NF-κB using SN50 did not affect the phosphorylation pattern or intracellular location of c-Jun. Similarly, silencing of JUN did not alter the expression or cellular localization of p65 following exposure to UVA or UVB (Fig.5c,d). Instead, the data presented herein indicate that the individual expression and locations of the various transcription factors are important for melanocyte fate.
Figure 5.
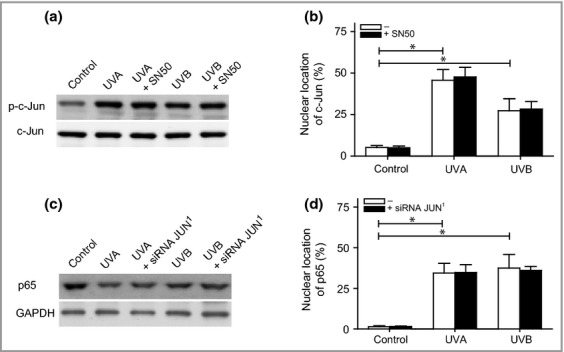
Crosstalk between ultraviolet (UV)-induced activator protein-1 and nuclear factor (NF)-κB signalling. Melanocytes were exposed to UVA (60 J cm−2) or UVB (500 mJ cm−2). (a) Pretreatment with an inhibitor of the p50 subunit of the NF-κB dimer (SN50, 20 μmol L−1; black bars) 3 h prior to irradiation. The intensity of c-Jun phosphorylation and c-Jun was analysed via Western blotting 4 h postirradiation. One representative Western blot out of four is presented. (b) Nuclear localization of c-Jun assessed via microscopic inspection 4 h postirradiation. (c) Pretreatment with JUN small interfering (si)RNA (black bars) 24 h prior to irradiation. One representative Western blot out of four showing the levels of the p65 subunit in digitonin-extracted cytosolic fractions 4 h postirradiation. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. (d) Nuclear localization of p65 assessed via microscopic inspection 4 h postirradiation. The results are presented as the mean ± SD (n = 4). *P ≤ 0·05 vs. control.
Discussion
The components of MAPK signalling are targets of UV irradiation and important for regulation of UV-induced cellular responses. Previous studies have strongly implicated the transcription factors AP-1 and NF-κB as mediators of the UV response in several different cell types.3,16 The present study identifies NF-κB as an antiapoptotic/prosurvival factor, whereas AP-1 stimulates proapoptotic signalling during both UVA- and UVB-induced apoptosis in human melanocytes (Fig.6).
Figure 6.
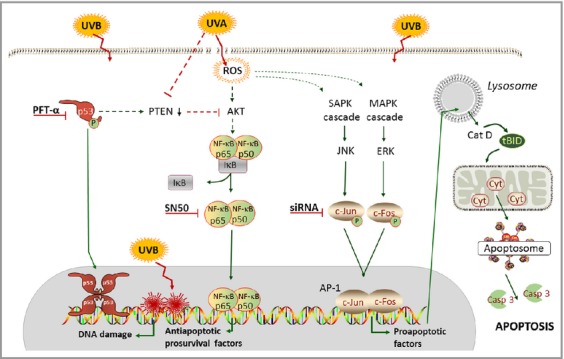
Suggested intracellular pathways for ultraviolet (UV)A- and UVB-induced signalling in human melanocytes. UVA induces plasma membrane damage and reactive oxygen species (ROS)-induced oxidative modification of DNA, whereas UVB causes direct damage to the DNA without any ROS or plasma membrane alterations. Even though the damage to DNA and induction of oxidative stress differ after irradiation with UVA and UVB in melanocytes, the apoptosis signalling converges into a common mechanism. The transcription factor nuclear factor (NF)-κB was identified as being important for the prosurvival reactions of the cell, and activator protein (AP)-1 as an important proapoptotic factor during both UVA- and UVB-induced apoptosis in human melanocytes. Casp, caspase; Cat, cathepsin; Cyt, cytochrome; ERK, extracellular signal-regulated kinase; IκB, inhibitor of κB; MAPK, mitogen-activated protein kinase; PFT, pifithrin; PTEN, phosphatase and tensin homologue; SAPK, stress-activated protein kinase; siRNA, small interfering RNA; tBID, truncated BH3-interacting-domain death agonist.
Previous studies using different cell types have suggested that wavelength-specific stimulation of ERK, p38 and JNK kinases occurs on UV exposure.17,18 However, the results are not consistent, and the observed variations are likely related to the applied doses and times of exposure, as well as cell type.16 Furthermore, only a few investigations have compared the effects of different wavelengths. In melanocytes and melanoma, UV has been shown to induce phosphorylation of the p38 and JNK/stress-activated protein kinase pathways, whereas NF-κB remains at constantly high expression.19–24 The B-Raf/ERK pathway has been suggested as the central pathway involved in melanoma development and progression. However, advances in the understanding of the signalling pathways in melanoma have identified altered expression of especially NF-κB, and also AP-1, among others, to promote tumour development.25 Moreover, evidence suggests that the inhibition/activation of other MAPK and NF-κB pathways, together with chemotherapeutic intervention, causes cytotoxicity in melanoma cells,26,27 although antitumorigenic activities have also been reported.28–31 Therefore, in addition to the B-Raf/ERK pathway, the activation of NF-κB or inactivation of AP-1 may play a role in melanoma development.21,25
Our study identified significantly increased levels of phosphorylated ERK and JNK after exposure to both UVA and UVB. The phosphorylation persisted for a longer time with UVA. Both the ERK and JNK kinase pathways have been implicated in apoptosis.3 In the present study, we found that the phosphorylation of c-Jun was increased for at least 6 h after UVB treatment, whereas only a twofold increase was observed following UVA, which decreased after 2 h. This finding indicates that the c-Jun signal is stronger after DNA damage (UVB) than after general oxidative stress (UVA). Immunocytochemical analysis showed that UV-induced translocation of both c-Jun and c-Fos into melanocyte nuclei occurs with both physiological doses and higher experimental doses. Similarly, in human keratinocytes, activated AP-1 signalling has been detected following UV exposure.32
Silencing of the JUN subunit of AP-1 results in decreased cell death, which suggests that AP-1 is part of the proapoptotic process in melanocytes following UVA/B irradiation. Furthermore, downregulation of JUN abolished nuclear localization of c-Fos, revealing a close codependence of these proteins in the AP-1 complex in melanocytes. The cells with phosphorylated c-Jun also showed nuclear fragmentation. Previous studies examining rat sympathetic neurons undergoing apoptosis upon withdrawal of nerve growth factor showed increased levels of both c-Jun mRNA and protein, as well as phosphorylation, which resulted in enhanced transactivation.33,34 Furthermore, neurons undergoing apoptosis induced by hypoxia–ischaemia show increased c-Jun protein levels in vivo.35
The molecular mechanism by which AP-1 triggers apoptosis has not been fully elucidated. We demonstrated that silencing of JUN reduces apoptosis, and that AP-1 operates upstream of LMP and release of cytochrome c from the mitochondria. The upstream signalling of LMP is poorly defined. Our results suggest that AP-1-mediated signalling participated in the regulation of lysosomal stability, as depletion of JUN prevents the cytosolic appearance of lysosomal cathepsin D. In a previous study, we found that downregulation of JNK prevented apoptosis by modulating the degradation of the antiapoptotic protein myeloid cell leukaemia-1,36 thus raising the possibility that c-Jun may activate apoptosis by regulating expression of members of the B-cell lymphoma-2 gene family.
Both UVA and UVB induced NF-κB activation and translocation to the nucleus in melanocytes at both physiological and higher doses. Production of ROS has been shown to enhance the signal transduction pathway leading to NF-κB activation and translocation to the nucleus.37 To evaluate the role of NF-κB after UV exposure in melanocytes, we used the cell-permeable inhibitor SN50, which effectively blocks the p50 subunit of NF-κB. Consequently, caspase activation and nuclear fragmentation were increased, indicating that NF-κB has prosurvival/antiapoptotic properties in human melanocytes after UVA and UVB irradiation. The intracellular redox state was substantially altered after exposure to UVA but not UVB, but such differences in the redox state did not seem to affect the transactivation of NF-κB in our system, which has been suggested of others.26
Our analysis of gene expression in UV-irradiated apoptotic melanocytes revealed altered expression of genes belonging to the AP-1 and NF-κB complex, but none of these changes was confirmed at the protein level. However, we observed nuclear translocation of AP-1 and NF-κB complex proteins, suggesting that the level of transcription factors already present in the cell was sufficient to mediate UVA- and UVB-induced signals via change in location. Similarly, in a previous study, we showed p53 relocation to the mitochondria after UVA and to the nucleus after UVB, and these events were important for apoptosis signalling.38 Moreover, inhibition of NF-κB and downregulation of c-Jun did not affect the nuclear translocation of each other, suggesting no cross-talk or compensatory effect between these transcription factors. Instead, the balance between their different signalling pathways seems to be of importance for the outcome in melanocytes.
In conclusion, we show that even though the damage to DNA and induction of oxidative stress differs after irradiation with UVA and UVB in melanocytes, the apoptosis signalling pathways converge into a common mechanism. This is true both in the experimental model used and in cultures irradiated with lower UV doses that resemble physiological conditions. We identified NF-κB as being important for the prosurvival reactions of the cell, and AP-1 as an important proapoptotic factor during UVA- or UVB-induced apoptosis in human melanocytes. An improved understanding of cellular responses in UV-exposed melanocytes is essential to understand and prevent the formation of melanoma, and might provide an opportunity to identify apoptotic regulators and future therapeutic targets.
Supporting Information
Figure S1. Apoptosis and oxidative damage induced by physiological ultraviolet irradiation.
Figure S2. Activator protein-1 signalling in melanocytes following physiological ultraviolet exposure.
Figure S3. Nuclear factor-κB signalling induced by physiological ultraviolet irradiation.
Figure S4. Representative cell sorting scheme for (a) unirradiated control melanocytes and (b) ultraviolet (UV)A- and (c) UVB-irradiated melanocytes.
Figure S5. Gene silencing of JUN.
References
- Chen ST, Geller AC, Tsao H. Update on the epidemiology of melanoma. Curr Dermatol Rep. 2013;2:24–34. doi: 10.1007/s13671-012-0035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrot L, Meunier JR. Skin DNA photodamage and its biological consequences. J Am Acad Dermatol. 2008;58(Suppl. 2):S139–48. doi: 10.1016/j.jaad.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Cooper SJ, Bowden GT. Ultraviolet B regulation of transcription factor families: roles of nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr Cancer Drug Targets. 2007;7:325–34. doi: 10.2174/156800907780809714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- Muthusamy V, Piva TJ. The UV response of the skin: a review of the MAPK, NFκB and TNFα signal transduction pathways. Arch Dermatol Res. 2010;302:5–17. doi: 10.1007/s00403-009-0994-y. [DOI] [PubMed] [Google Scholar]
- Larsson P, Andersson E, Johansson U, et al. Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis. Exp Dermatol. 2005;14:117–23. doi: 10.1111/j.0906-6705.2005.00238.x. [DOI] [PubMed] [Google Scholar]
- Larsson P, Ollinger K, Rosdahl I. Ultraviolet (UV)A- and UVB-induced redox alterations and activation of nuclear factor-κB in human melanocytes – protective effects of alpha-tocopherol. Br J Dermatol. 2006;155:292–300. doi: 10.1111/j.1365-2133.2006.07347.x. [DOI] [PubMed] [Google Scholar]
- Bivik CA, Larsson PK, Kågedal KM, et al. UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J Invest Dermatol. 2006;126:1119–27. doi: 10.1038/sj.jid.5700124. [DOI] [PubMed] [Google Scholar]
- Andersson E, Vahlquist A, Rosdahl I. Beta-carotene uptake and bioconversion to retinol differ between human melanocytes and keratinocytes. Nutr Cancer. 2001;39:300–6. doi: 10.1207/S15327914nc392_21. [DOI] [PubMed] [Google Scholar]
- Gilchrest BA, Vrabel MA, Flynn E, Szabo G. Selective cultivation of human melanocytes from newborn and adult epidermis. J Invest Dermatol. 1984;83:370–6. doi: 10.1111/1523-1747.ep12264638. [DOI] [PubMed] [Google Scholar]
- Rosdahl I, Andersson E, Kågedal B, Törmä H. Vitamin A metabolism and mRNA expression of retinoid-binding protein and receptor genes in human epidermal melanocytes and melanoma cells. Melanoma Res. 1997;7:267–74. doi: 10.1097/00008390-199708000-00001. [DOI] [PubMed] [Google Scholar]
- Vanderlinde RE. Measurement of total lactate dehydrogenase activity. Ann Clin Lab Sci. 1985;15:13–31. [PubMed] [Google Scholar]
- Leaback DH, Walker PG. Studies on glucosaminidase. 4. The fluorimetric assay of N-acetyl-β-glucosaminidase. Biochem J. 1961;78:151–6. doi: 10.1042/bj0780151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger CG, Langemann H, Krenger W, Kempf A. Liquid chromatographic determination of common water-soluble antioxidants in biological samples. J Chromatogr. 1989;487:463–8. doi: 10.1016/s0378-4347(00)83056-x. [DOI] [PubMed] [Google Scholar]
- Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE. 2003;2003:RE2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- Ariizumi K, Bergstresser PR, Takashima A. Wavelength-specific induction of immediate early genes by ultraviolet radiation. J Dermatol Sci. 1996;12:147–55. doi: 10.1016/0923-1811(95)00474-2. [DOI] [PubMed] [Google Scholar]
- Kabuyama Y, Homma MK, Sekimata M, Homma Y. Wavelength-specific activation of MAP kinase family proteins by monochromatic UV irradiation. Photochem Photobiol. 2001;73:147–52. doi: 10.1562/0031-8655(2001)073<0147:wsaomk>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Choi BY, Choi HS, Ko K, et al. The tumor suppressor p16INK4a prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity. Nat Struct Mol Biol. 2005;12:699–707. doi: 10.1038/nsmb960. [DOI] [PubMed] [Google Scholar]
- Abdel-Malek ZA, Kadekaro AL, Swope VB. Stepping up melanocytes to the challenge of UV exposure. Pigment Cell Melanoma Res. 2010;23:171–86. doi: 10.1111/j.1755-148X.2010.00679.x. [DOI] [PubMed] [Google Scholar]
- Muthusamy V, Piva TJ. UVB-stimulated TNFα release from human melanocyte and melanoma cells is mediated by p38 MAPK. Int J Mol Sci. 2013;14:17029–54. doi: 10.3390/ijms140817029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yang D, Minemoto Y, et al. NF-κB is required for UV-induced JNK activation via induction of PKCδ. Mol Cell. 2006;21:467–80. doi: 10.1016/j.molcel.2005.12.020. [DOI] [PubMed] [Google Scholar]
- Yanase H, Ando H, Horikawa M, et al. Possible involvement of ERK 1/2 in UVA-induced melanogenesis in cultured normal human epidermal melanocytes. Pigment Cell Res. 2001;14:103–9. doi: 10.1034/j.1600-0749.2001.140205.x. [DOI] [PubMed] [Google Scholar]
- Tada A, Pereira E, Beitner-Johnson D, et al. Mitogen- and ultraviolet-B-induced signaling pathways in normal human melanocytes. J Invest Dermatol. 2002;118:316–22. doi: 10.1046/j.0022-202x.2001.01694.x. [DOI] [PubMed] [Google Scholar]
- Bennett DC. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- Enzler T, Sano Y, Choo MK, et al. Cell-selective inhibition of NF-κB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer Discov. 2011;1:496–507. doi: 10.1158/2159-8290.CD-11-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuling AM, Andrew SE, Tron VA. Inhibition of p38 MAPK enhances ABT-737-induced cell death in melanoma cell lines: novel regulation of PUMA. Pigment Cell Melanoma Res. 2010;23:430–40. doi: 10.1111/j.1755-148X.2010.00698.x. [DOI] [PubMed] [Google Scholar]
- Alexaki VI, Javelaud D, Mauviel A. JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigment Cell Melanoma Res. 2008;21:429–38. doi: 10.1111/j.1755-148X.2008.00466.x. [DOI] [PubMed] [Google Scholar]
- Denkert C, Siegert A, Leclere A, et al. An inhibitor of stress-activated MAP-kinases reduces invasion and MMP-2 expression of malignant melanoma cells. Clin Exp Metastasis. 2002;19:79–85. doi: 10.1023/a:1013857325012. [DOI] [PubMed] [Google Scholar]
- Ivanov VN, Fodstad O, Ronai Z. Expression of ring finger-deleted TRAF2 sensitizes metastatic melanoma cells to apoptosis via up-regulation of p38, TNFα and suppression of NF-κB activities. Oncogene. 2001;20:2243–53. doi: 10.1038/sj.onc.1204314. [DOI] [PubMed] [Google Scholar]
- Kannaiyan R, Manu KA, Chen L, et al. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3K/Akt signaling pathways. Apoptosis. 2011;16:1028–41. doi: 10.1007/s10495-011-0629-6. [DOI] [PubMed] [Google Scholar]
- Djavaheri-Mergny M, Mergny JL, Bertrand F, et al. Ultraviolet-A induces activation of AP-1 in cultured human keratinocytes. FEBS Lett. 1996;384:92–6. doi: 10.1016/0014-5793(96)00294-3. [DOI] [PubMed] [Google Scholar]
- Reddy CE, Albanito L, De Marco P, et al. Multisite phosphorylation of c-Jun at threonine 91/93/95 triggers the onset of c-Jun pro-apoptotic activity in cerebellar granule neurons. Cell Death Dis. 2013;4:e852. doi: 10.1038/cddis.2013.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, et al. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–39. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, van der Kooij MA, van Bel F, et al. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav Immun. 2010;24:812–21. doi: 10.1016/j.bbi.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Bivik C, Ollinger K. JNK mediates UVB-induced apoptosis upstream lysosomal membrane permeabilization and Bcl-2 family proteins. Apoptosis. 2008;13:1111–20. doi: 10.1007/s10495-008-0240-7. [DOI] [PubMed] [Google Scholar]
- Gloire G, Piette J. Redox regulation of nuclear post-translational modifications during NF-κB activation. Antioxid Redox Signal. 2009;11:2209–22. doi: 10.1089/ars.2009.2463. [DOI] [PubMed] [Google Scholar]
- Waster PK, Ollinger KM. Redox-dependent translocation of p53 to mitochondria or nucleus in human melanocytes after UVA- and UVB-induced apoptosis. J Invest Dermatol. 2009;129:1769–81. doi: 10.1038/jid.2008.421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Apoptosis and oxidative damage induced by physiological ultraviolet irradiation.
Figure S2. Activator protein-1 signalling in melanocytes following physiological ultraviolet exposure.
Figure S3. Nuclear factor-κB signalling induced by physiological ultraviolet irradiation.
Figure S4. Representative cell sorting scheme for (a) unirradiated control melanocytes and (b) ultraviolet (UV)A- and (c) UVB-irradiated melanocytes.
Figure S5. Gene silencing of JUN.