

Abstract
Tuberculosis is primarily a disease of the lung and dissemination is dependent upon productive infection of this critical organ. Upon aerosol infection with Mycobacterium tuberculosis (Mtb), the acquired cellular immune response is slow to be induced and to be expressed within the lung. This slowness allows infection to become established and forces the acquired response to be expressed in a context of an inflammatory site that has been initiated and modulated by the bacterium. Mtb has a variety of surface molecules that interact with the innate response and this interaction along with the auto-regulation of the immune response by several mechanisms results in less than optimal control of bacterial growth. To improve current vaccine strategies we must understand the factors that mediate induction, expression and regulation of the immune response in the lung. We must also determine how to induce both known and novel immunoprotective responses without inducing immunopathologic consequences.
Keywords: T cell, B cell, phagocyte, regulation, memory, inflammation
Introduction
The pathogenesis of tuberculosis is the product of the interaction between bacterial virulence and host resistance, which are two distinct and independent variables; this has been appreciated for a long time (1). However, modern tools have allowed us to define the role of specific bacterial and host components to an unprecedented degree in recent years. The desired end point of efforts to define bacterial and host components of this devastating disease is to identify virulence factors and drug targets within the bacterium as well identify components of the host’s immune system which can be augmented and indeed altered by vaccination. In this review I will touch only lightly on the classic literature of the early modern period as this work has been eloquently reviewed in the two most recent Annual Review articles (2, 3). I will focus more on the relatively recent literature, which has benefited from the maturation of the tools available as a result of the publication of the mouse and bacterial genome (4, 5). These tools, along with the ability to genetically alter the bacterium (6, 7), have resulted in an increased ability to manipulate the disease model in a directed manner.
The detrimental impact of tuberculosis on public health worldwide is generally appreciated. Indeed with the number of exposed individuals being approximated at one third of the world’s population, the fact that only 5% of those exposed eventually develop disease is not comforting when the case rate is 8 million new cases per year (8). When the ability of HIV infection to reduce immunity to Mycobacterium tuberculosis (Mtb) infection is considered, the consequences for spread of both drug sensitive and drug resistant tuberculosis are daunting (9, 10). While the public health impact of the disease is enormous and warrants the high level of interest shown by scientists worldwide, the disease and the immunopathologic lesions it evokes have also fascinated immunologists since the birth of the discipline.
Perhaps the key reason for the interest of immunologists is that both immunity and pathogenesis are mediated by the lymphocyte response to mycobacterial infection. Thus, while in the absence of an acquired cellular response there is limited to no immunity, the absence of this response also limits the generation of the classical caseation associated with transmission of the pathogen. This statement is perhaps best supported by considering the consequences of HIV infection on the development of tuberculosis. Tuberculosis is an index disease for HIV infected individuals and develops when CD4 numbers are still much higher than those predisposing to other opportunistic infections (11). However, when the immunopathologic consequences of Mtb infection in AIDS patients are assessed there is a much altered disease state (11) and an altered inflammatory response. Specifically, there is a dominant granulocytic infiltrate and necrosis but not the typical caseous necrosis seen in non-HIV infected tuberculosis granulomas (12). This strong tendency to granulocytic involvement is also see in the mouse model wherein the CD4 molecule is genetically disrupted (13). The acquired cellular response, as represented largely by CD4 T cells, provides therefore protective immunity while also promoting the development of mononuclear lesions and the caseous necrosis required for transmission. It is the duality of the role of the acquired cellular response that leads to the apparently contradictory presence of a strong cellular immune response at the site of unresolving disease.
The most important aspect of the acquired cellular response is the rapidity with which it is expressed. If the response is too slow, bacteria grow and reach a point where although a potentially protective response is being expressed, the environment is such that it is not effective. In this same vein it is clear that dose plays a role in the ability of the host to control bacteria. Specifically, if one is infected by too high a dose then the local bacterial burden may reach a level that interferes with the efficient expression of protective immunity. These ideas were brought together eloquently by Rich (1) using the lung histopathology from patients in the pre-drug era to describe the natural history of the disease. He suggested that the acquired cellular response was able to control bacterial growth but that it failed to do so in the face of high numbers of bacteria. To support this idea he observed that within the same patient, large lesions tend to progress while small ones are restrained in their growth. Further, the nature of metastatic lesions was different from the primary lesion in that they are generally circumscribed and bacterial growth is controlled. Finally, he reported that the large number of bacteria that arrive at a new site as a result of aspiration of large primary lesions usually results in a sizeable progressive lesion. This interpretation predates our understanding of much of the acquired cellular response but supports the importance of the kinetics of the response, the importance of the environment within which the response must occur and the potential for regulation of the response by either the bacteria or the acquired response itself.
The importance of lymphocytes in controlling tuberculosis was under appreciated in early work, as although these cells were clearly present in lesions their function was unknown. It was early mouse model work that demonstrated that T cells were required for anti-tuberculous immunity in systemic (14, 15) and aerosol (16) challenge models. That CD4 T cells were the primary mediators of anti-tuberculous immunity was shown as a result of transfer models and then later by the use of gene-deficient mice (17, 18). It was also gene-deficient mice that demonstrated the importance of cytokine mediated macrophage activation in the control of bacterial growth (19, 20). The relevance of these studies to the human condition was demonstrated by the observation that HIV-mediated loss of CD4 T cell rendered patients susceptible to tuberculosis (11) and that people genetically deficient for the cytokine-mediated macrophage activation pathway were also susceptible to tuberculosis (21).
While significant progress has been made in our understanding of what lymphocytes do during both primary and recall responses to mycobacterial infection, the vaccine that is currently in use, a derivative of Mycobacterium bovis called bacille Calmette Guerin (BCG), was first developed in 1921 when we had no idea what lymphocytes were doing. While the efficacy of this early vaccine is clear for limiting disseminated childhood tuberculosis (22) its efficacy is less obvious for pulmonary disease (23) and we are still limited in the rational design of vaccines by our lack of understanding of both the primary and recall acquired cellular response to tuberculosis.
In order for significant progress to be made in the development of vaccines we need to understand the interaction between the acquired cellular response and the pathogen from the beginning of infection throughout the disease process. There are several important issues to be addressed. We need to know whether the acquired cellular response that is expressed upon infection is truly optimal and to what extent it is modulated by the pathogen. We also need to know what types of effector cell are activated by infection and whether each sub type is required for immunity or whether it acts to mediate pathologic or regulatory consequences. Further, we need to know the extent to which lymphocytes induced by infection, modulate the function of other cells and indeed the extent to which the pathogen modulates the function of host cells. Indeed, we need to know the extent to which the inflammatory site itself impacts the ability of effector cells to mediate their protective function. Finally, we need to identify anti-bacterial activities that are not induced during natural infection but which can be augmented by vaccination and which do not lead to adverse pathologic consequences. This list of objectives is somewhat daunting but the field has made dramatic progress recently and in light of new tools and approaches further progress should result in significant breakthroughs (24).
Initiation of the T cell response
The initiation of the T cell response following mycobacterial infection has been studied indirectly for many years. The first modern analysis (17) demonstrated that lymphocytes induced by systemic infection were capable of mediating protection upon transfer by day 5 post primary infection. This measure of protective function was seen first in L3T4 (CD4) T cells and later in Ly2 (CD8) T cells and both populations could protect against aerosol challenge. The protective ability of cells produced in the first 25 days post challenge was sensitive to the effects of cyclophosphamide indicating that these cells were dividing cells and were likely what is now termed effector T cells (17). Later studies utilized the effector function of antigen-specific cells as an indicator of the initiation of the cellular response and demonstrated that dissemination of bacteria to the draining lymph node occurred following aerosol infection (25). It was also shown that the draining lymph node was the first site of expression of effector function followed by the spleen and the lung. The temporal correlation between lymphocyte activation and bacterial arrival in the lymph node was taken to mean that acquired cellular immunity was initiated in the draining lymph node by bacteria disseminating from the lungs via the lymphatic drainage and that further dissemination occurred systemically thereafter (25). This paper also highlighted the importance of accelerated kinetics in the induction of protective acquired cellular responses. Specifically, the dissemination of the bacteria from the lung and the induction of effector cell responses were compared for the resistant B6 mouse compared to the susceptible C3H mouse. The resistant strain had equivalent bacterial growth in the lung compared to the susceptible strain but the dissemination and induction of cellular responses were accelerated in this strain compared to the susceptible strain (25). Thus, it was clear that slow induction of the immune response was detrimental to the ultimate success of the response. While Chackerian and colleagues demonstrated that induction of effector function required dissemination of bacteria from the lung to the draining lymph node they did not directly address the issue of naive T cell activation. This is an important issue, as the mechanism resulting in the slow expression of effector function in the antigen-specific lymphocyte population was not defined. One hypothesis to explain this slow induction is that Mtb exerts a regulatory activity via either inhibition of migratory activity or by modulation of antigen presenting cell function. Alternatively, slow induction could simply be a consequence of the low number of bacteria being delivered into an organ that has a limited ability to initiate cellular responses and that the delay is due to lack of inflammation and antigen. To begin to address this issue it was necessary to determine when and where sufficient antigen is presented for the activation of naive T cells to occur. In order to do this one needs to utilize T cell receptor (TCR) transgenic mice wherein naive T cells express a TCR specific for Mtb antigens that are expressed early in infection. To this end, two groups have recently reported data that strongly supports the conclusion of Chackerian and colleagues that antigen-specific T cells are first activated within the draining lymph node of the lung and that this activation is slow to occur. In one study, proliferation of antigen-specific naive cells was examined and found to occur first in the draining lymph node and to be subsequent to the dissemination of bacteria to this organ (26). Proliferation is however a downstream effect of naive T cell activation and in order to detect the first events in activation, the induction of CD69 on naive antigen-specific T cells was examined in a second paper. In this study, CD69 expression was seen on T cells in the draining node first and was subsequent to the arrival of bacteria in this organ (27). Thus by any measure of naive T cell activation, it was clear that no T cell activation occurred within the host before day 9 following aerosol infection and that the lung draining lymph node was the first place where it was seen. In all studies the response was co-incident with the arrival of viable bacteria in the node demonstrating that it is very likely that dissemination is essential to priming of the response (Figure 1).
Figure 1.

Low dose aerosol infection, which approximates the natural delivery route for induction of tuberculosis, results in low numbers of Mtb (green) being deposited in the lower airways and the alveolar tissue. Bacteria do not disseminate from the lung until 9 days post infection when they can be detected in the draining lymph nodes. This dissemination coincides with the first activation of naive T cells (pink). The fact that bacteria do not disseminate rapidly, suggests that they either inhibit migration of dendritic cells or that the cells that they infect cannot migrate readily to the lymph node. Activation of naive T cells occurs in the presence of live bacteria and effector cells develop with expected kinetics. The effector cell phenotype will depend upon the availability of specific cytokines. These effector cells migrate to the lung in response to inflammation and mediate protection by activating infected phagocytes (red). The response takes 18–20 days to reach an effective level and to thereby stop bacterial growth.
Why then is the dissemination so slow? In all three of the above models the bacteria was delivered to the lung via an aerosol cloud containing particles of a size that can pass through the lower bronchioles and be deposited in the alveolar space. Once in this location it is most likely that the cells infect alveolar macrophages although this is impossible to test due to the low number of events. The deposition of bacteria in the alveolar space places it in the lung parenchyma rather than the mucosal tissue of the airways and there are distinct differences in the phenotype and function of the cells encountered in these two compartments of the lung (28). The events that occur following initial infection with a low dose of bacteria have not been defined, however after nine days and substantial expansion of the population, the bacteria enter the lymphatic drainage as this is when they are first detected in the draining lymph node (25–27). Prior to this time even the delivery of an external inflammatory mediator, which should mobilize any infected cells to the lymph node, fails to accelerate the dissemination of bacteria or the initiation of immunity (26). While a more than 10 fold increase in dose can accelerate dissemination it can only do so by one to two days suggesting that the location where the bacteria are deposited is a key aspect that contributes to the slowness of the response (27) (Figure 1).
The involvement of the dendritic cell in the migration of bacteria to the lymph node is assumed to be an essential element of the initiation of the response. It is possible therefore that the different functionality of these cells in the airways compared to the alveolar areas may define the kinetics of the response. It has been shown several times that Mtb-infected dendritic cells delivered to the lung via the intratracheal route are capable of migrating to the draining lymph node and initiating cellular responses (29–31). It is however unlikely that these cells are deposited in the alveolar space and indeed it is not known if they represent the functional aspects of the dendritic cells that occupy this unique environment. It is thought that conducting airway dendritic cells are active samplers of the mucosal environment and migrate readily to the draining node (32, 33). In contrast, dendritic cell within the alveolar tissue are in a regulated environment with surfactant protein and alveolar macrophages that may limit their ability to respond to infection and migrate to the lymph node as rapidly as would dendritic cell in the airway tissue (28) (Figure 1). The delay in dissemination of bacteria and the initiation of the cellular response may therefore reflect the infection of alveolar phagocytes (both macrophages and dendritic cells) that are resistant to migration and indeed activation. This resistance to migration and activation is likely crucial to the maintenance of the alveolar space as an air exchange surface as opposed to an inflammatory site. It appears therefore that the slow initiation of the acquired immune response by Mtb is a product of the slow growth of this bacteria and the relatively immunoprivileged nature of the alveolar tissue.
Development of cellular responses
Dendritic cells are currently considered to be the most efficient inducers of activation in naive T cells and to do this they provide not only the antigen-specific stimulus but also secondary and tertiary signals that promote efficient development of effector T cells. The impact of mycobacterial infection on dendritic cell function has been studied extensively. Indeed, the classic demonstration that immature dendritic cell are able to phagocytose particles and become efficient antigen presenting cells used BCG as the maturation agent (34). More recently however the ability of Mtb to interfere with T cell stimulation has been suggested by the fact that dendritic cells infected in vivo are less efficient at stimulating antigen-specific T cells than are equivalent uninfected dendritic cells (35).
As discussed above, once bacteria reach the draining lymph node initiation of naive T cell activation occurs. Whether this is as a result of direct interaction between lung-derived bacteria-infected dendritic cells has not been definitively proven. However in a model system wherein antigen-pulsed dendritic cells are delivered to the lung intratracheally, it is clear that those dendritic cells exposed to Mtb prior to delivery are better at migrating to the draining node and initiating T cell activation (29–31). It appears also that while IL-12p40 promotes this migration, IL-10 may limit it (29, 31, 36). These data suggest that simple exposure to Mtb is not sufficient to limit the ability of an infected dendritic cell to initiate T cell activation. Infected lung dendritic cells can be seen to migrate from the lung to the draining lymph node however it is difficult to see this migration before day 14 of infection and this is after the initial migration and initiation of T cell activation has occurred (35). Despite our inability to define the exact population delivering the bacteria to the lymph node it is clear that bacteria are in the lymph node when activation of naive T cells occurs and that therefore the bacteria could inhibit the process of activation once initiated. Importantly however, as the naive T cells become activated, they proliferate, upregulate CD44 and down regulate CD62L and begin to accumulate in the lung with the expected kinetics (27) (Figure 1). That this activation occurs with the expected kinetics indicates that the simple presence of the bacteria in the lymph node does not impact initial T cell activation; we do not know however whether the expansion and survival of the newly activated T cells is optimal. Indeed the observation that infection of the lymph node by either viral or bacterial pathogens results in altered chemokine expression in the T cell area and that this impacts generation of new antigen-specific responses (37, 38) suggests that infection of the draining lymph node with mycobacteria may impact naive T cell activation.
Once cells are activated they must migrate to the primary site of infection and this migration takes place 15–18 days post infection (27) (Figure 1). The question of whether this recruitment is delayed and indeed whether it is dependent upon antigen and or inflammation has not been definitively determined. However new real time imaging techniques are providing improved understanding of the factors modulating the generation of the inflammatory site. In particular, a recent paper has begun to determine how T cells and macrophages interact within the granuloma and shows that following systemic infection with BCG, tissue macrophages of the liver take up the bacteria and a granuloma forms as a result of migrating local uninfected macrophages and monocytes from the blood. T cells then enter the granuloma and remain within it using the myeloid cells as a framework for moving throughout the granuloma (39). Live imaging using the zebra fish model is also providing information about the kinetics and development of the cellular response to mycobacterial infection (40, 41). In particular, the ability to genetically modify both bacterium and host in this model will allow for novel insight into the factors impacting the development of granulomata (40–42).
New subsets of effector T cell
As discussed above, while we know that lymphocytes accumulate in the inflammatory lesion and that they probably mediate protective immunity by activating infected phagocytes we have not yet fully defined their function in vivo. Indeed, although it is widely stated that CD4 T cells making IFN-γ are required for protective immunity there is largely based on correlative data. In particular it is clear that cessation of bacterial growth correlates with arrival of IFN-γ-producing CD4 T cells (43–46) and that loss of CD4 T cells increases the likelihood of succumbing to tuberculosis (11). However, while we appreciate the role that CD4 T cells play in protecting against bacterial growth we have not yet fully defined their capacity to mediate protection or the mechanisms whereby they mediate immunity. It has become apparent recently that there is a variety of CD4 T cell effector subtypes ranging from early activated cells making just IL-2, through cells making IFN-γ and to multifunctional cells expressing IL-2, IFN and TNF and the presence of these multifunctional cells is associated with protection (47). There are also cytolytic CD4 T cells that have been identified in mice (43) and man (48). Multifunctional cells are seen at high frequency in tuberculosis patients (49) and also in those people in high incidence areas (50) and in vaccinated infants (51). Development and function of all effector cells depends upon the ability of dendritic cell within the draining node to prime and promote survival of these cells efficiently. This priming will require expression of antigen in the context of MHC, costimulatory molecules and the necessary cytokines that promote T cells polarization. In the case of IFN-γ-producing cells the role of IL-12p70 has been demonstrated extensively (52) however the conditions required to generate and maintain the multi-functional and cytolytic antigen-specific lymphocytes have not been fully defined (Figure 1).
Other subsets of functional T cell such as those producing IL-17 and IL-22 have also been identified recently and these cells have been seen in the mouse model and in humans exposed to tuberculosis (53). Their protective role in tuberculosis has yet to be demonstrated, however their recent discovery demonstrates that we are still learning about the cellular response to tuberculosis even in the well-defined mouse model. IL-17 producing antigen-specific cells are induced in mice following aerosol infection, these cells and indeed the majority of the IL-17 response in the lung is dependent upon the presence of IL-23 (54). γδ T cells are a source of IL-17 and produce this cytokine very early following a high dose intranasal challenge with BCG (55). A large portion of the IL-17 response in the mouse model is within the γδ T cell population (56). When IL-17 is blocked during a high dose challenge, neutrophil recruitment is hindered and this may alter subsequent development of inflammation (55). In the absence of IL-23 and therefore the majority of the IL-17 response in the mouse model, there is a modest alteration in the early inflammatory response (54). Whether these cells are protective or damaging we do not yet know, however when Mtb infected animals are repeatedly challenged with mycobacterial antigen they develop significantly increased lung lesions containing a higher frequency of granulocytes and necrosis (57, 58). Recently we investigated the role of IL-17 in this enhanced pathology and found that increased lesion size and increased granulocyte presence in the lesions was dependent upon IL-23 and could be ablated by the delivery of anti-IL-17 antibody (Cruz pers com). These data suggest that the nature of the inflammation that develops in response to chronic antigen exposure is dependent upon IL-23 and IL-17. It also demonstrates that there is the potential for a dual role for these two cytokines in the response to Mtb infection in the mouse. Whether this response is equally important in humans is as yet unknown, however it is clear that CD4+ antigen-specific IL-17 and IL-22 producing cells can be detected in humans exposed to Mtb although only IL-22 is detected in the lung (50).
Effector functions
Determining the lymphocyte response to mycobacterial infection is essential to rational vaccine development however the response of the phagocyte to both infection and activation is also crucial in this regard. Specifically, if infected phagocytes are resistant to activation, despite the presence of acquired cellular responses, then this is an important limitation of any vaccine-induced response. It is also known that infected phagocytes limit cellular responses as a result of feedback pathways involving products of phagocyte activation (59).
In most vertebrate infections, bacterial growth occurs logarithmically until acquired immunity is expressed, whereupon control of bacterial growth occurs (60). This pattern is true for the B6 mouse model (61) and in this model there is a clear inability to control bacterial growth if phagocyte activating cytokines such as IFN-γ (19, 20) and TNF (62) are missing and if the phagocyte cannot elaborate large amounts of nitric oxide (63). However there are other mouse strains which fail to either stop bacterial growth or are unable to maintain control of the infection. The cause of increased susceptibility is variable depending on strain (25, 64–68). The variability seen in the mouse models illustrates how complex the control of this Mtb infection is and provides tools to investigate the mechanisms involved. Specifically, a potentially novel function of infected phagocytes has recently been suggested by the use of linkage analysis. By crossing C3HeB/J mice, which are susceptible to Mtb, with resistant C57BL/6J mice, a locus has been identified which is associated with decreased survival and increased development of necrotic lesions (sst1) (68). The candidate gene within the sst1 locus Ipr1, is induced in phagocytes upon infection with Mtb and is associated with ability to control bacteria and limit necrotic death of infected macrophages (68). Importantly the impact of the locus is highest in the lung, with resistance being associated with reduced necrosis and increased apoptosis in the lung lesions. The development of T cell responses is not different between resistant and susceptible mice however vaccination is less effective in the susceptible mice suggesting that defective phagocyte responses can limit the protective effect of vaccination (69).
Unlike most animal models, the data from humans regarding the role of specific cellular pathways in controlling Mtb is less clear. It is apparent that although most humans who are exposed do not become diseased, exposure to the pathogen does result in the development of an acquired delayed type hypersensitivity (8). The mechanism mediating control of Mtb in people exposed but not developing disease is currently unknown. However the prolonged expression of an antigen-specific T cell response and the increased risk of disease in AIDS patients and in those lacking elements of the IFN-γ/IL-12 pathway suggests that acquired cellular immunity is induced upon infection and provides protection in the majority of those exposed.
Recent studies have focused on the unique abilities of human phagocytes to control Mtb. In particular, mechanistic analysis of anti-mycobacterial activity in humans has resulted in potential explanations for the increased susceptibility to disease exhibited by those with Vitamin D deficiency. Early work implicated vitamin D in the ability of human macrophages to control Mtb proliferation (70, 71). More recently it has been shown that addition of 1α,25 dihydroxyvitamin D3 (the biologically active metabolite of Vitamin D) reduces growth of mycobacteria in human peripheral blood mononuclear cells in a dose dependent manner. This action is mediated via the nuclear vitamin D receptors and is associated with induction of the cathelicidin hCAP-18 gene. Addition of the active peptide LL-37, which is cleaved from cathelicidin, reduces growth of Mtb in culture (72). Toll like receptor (TLR) activation of human macrophages and monocytes (but not dendritic cells) results in upregulation of the vitamin D receptor and the vitamin-D-1-hydroxylase genes and leads to induction of the antimicrobial peptide cathelicidin and killing of Mtb (73). The link between vitamin D-triggered anti-mycobacterial activity in monocytes and cathelicidin has been confirmed using siRNA inhibition of 1α,25 dihydroxyvitamin D3-induced cathelicidin protein production which resulted in increased mycobacterial growth (74).
A second macrophage function that has come to the fore with regard to anti-mycobacterial activity in recent years is autophagy. Cells perform autophagy to ‘clean house’ by sequestering their own cytoplasm into an autophagosome that is then delivered to the lysosome (75). It was recently shown that IFN-γ induces autophagy and that inhibition of autophagy increases the viability of intracellular mycobacteria in mice and humans (76, 77). Interestingly, this activity has been linked to ‘immunity-related p47 guanosine triphosphatases’ one of which, lrgm1 (LRG-47) has been previously shown to be essential for IFN-γ-mediated control of mycobacterial growth in mice (78); the human ortholog of this gene IRGM also plays a role in autophagy and the control of mycobacterial burden (77). The autophagic response to Mtb is abrogated by IL-4 and IL-13 (79) and enhanced by TLR4 ligation (80).
Careful analysis and identification of novel effector functions capable of limiting bacterial growth in both animal models and humans will provide increased insight into potential pathways that can be used to augment immunity both in intact and in immunocompromised individuals.
TLR and pattern recognition receptors
Both the innate and acquired response to Mtb infection depends to a large degree on recognition of Mtb as a pathogen by the pattern recognition receptors. The role of these receptors in mycobacterial infection has been extensively investigated recently. Of recent primary interest have been the TLR as well as the common adapter molecule MyD88, which mediates many of the intracellular signaling events subsequent to pattern recognition receptor ligation. MyD88 is required for survival upon mycobacterial infection (81–84) and the disease phenotype exhibited as a result of this deficiency is interesting. MyD88 deficient mice are able to generate high levels of cytokine in the lung and ex vivo stimulation of cells with mycobacterial antigen results in an IFN-γ response; however bacterial growth is not regulated and the lung develops an acute necrotic pneumonia. Analysis of the phagocytes in these mice reveals that they exhibit reduced responsiveness to the activating signals produced by the acquired response (82–84). It appears therefore that, in Myd88-deficient mice the induction of some aspects of acquired immunity remain intact while the effector function of the phagocytes is lost. It was initially thought that loss of TLR2, TLR4 and/or TLR9-induced phagocyte activation was the defining element of MyD88 deficiency phenotype and indeed it has been shown that deficiency of TLR2 and TLR9 results in increased susceptibility to Mtb infection (85). However, in direct comparison between TLR2/4/9 and MyD88 deficiency it is clear that deficiency in these TLR does not account for the susceptibility seen in the MyD88 mice (84). In other work, the function of MyD88 as an adapter of IL-1 and IL-18 receptor signaling was investigated and it was found that the absence of IL-1R but not the absence of IL-18R or the Toll-IL-1R domain-containing adapter protein (TIRAP) recapitulated the MyD88 phenotype; specifically, increased susceptibility to disease, induction of cellular responses with the absence of phagocyte activation and the development of necrotic pneumonia (86).
While MyD88 and IL-1R appear essential for induction of phagocyte activation and survival of acute infection, deficiencies in TLR are associated with reduced induction of acquired immunity and increased susceptibility to chronic or high dose infection. For example while the absence of TLR4 or TLR6 results in little impact on infection or disease in mice, the absence of TLR2 results in slightly increased bacterial growth in low dose aerosol and greater susceptibility to high dose aerosol (85, 87–89). TLR2 has been implicated in recognition of mycobacterial antigens and modulation of phagocyte function. In particular, lipoproteins from Mtb that are recognized by TLR2 can limit the ability of macrophages to upregulate class II MHC in response to IFN-γ; this effect is seen after prolonged exposure and is associated with reduced antigen-presentation (90–95). Mycobacterial peptidoglycan can also inhibit IFN-γ-mediated induction of the class II transactivator (CIITA) by a TLR2, Myd-88 independent pathway (96, 97). Despite the strong in vitro data, it has been difficult to address the impact of the bacteria on phagocyte function in vivo, however this was attempted recently. By infecting a chimeric mouse containing congenically marked phagocytes that were either TLR2 sufficient or deficient, the relative importance of TLR2 dependent and independent pathways on class II MHC expression in vivo in response to Mtb was assessed. While it was demonstrated that the regulation of class II MHC in infected lungs is dependent upon the presence of the IFN-γ-R, the presence of TLR2 does not impact the level of class II MHC on either infected or uninfected phagocytes within the infected lung (98). This could mean that either both TLR2-dependent and TLR2-independent pathways are involved in regulating class II MHC in vivo or that Mtb does not regulate class II MHC in vivo. There is some evidence that Mtb can alter T cell activation without altering class II expression (35), but this still needs to be demonstrated in vivo. In other studies, it is clear that when macrophages enter infected lung tissue they upregulate class II MHC in an IFN-γ dependent manner (99).
The fact that reduced acquired immunity and increased immunopathologic consequences seen in TLR2 and TLR9 mice (85, 88) only modestly impacts the control of bacterial growth indicates that the low dose aerosol model may not stress the immune response to a sufficient degree. This may explain why the profound differences in cellular responses of TLR deficient cells seen in vitro are not recapitulated in vivo.
With regard to induction of the acquired response it is clear that dendritic cells and macrophages have different responses to Mtb based on the expression of specific TLR (85, 100). The level and activity of TLR mediated signaling by the dendritic cells encountering the bacteria will influence the outcome of the cellular response but in the low dose aerosol infection any differences may result in subtle outcomes early in infection and may only be seen by careful analysis. For example, the relative levels of IFN-γ and IL-17-producing cells during mycobacterial infection in both mouse and human will depend on the level of specific cytokines present during and subsequent to T cell activation (101). In a recent analysis of the response of human dendritic cells to Mtb it was found that IL-23 was preferentially expressed, likely in a TLR2-dependent manner, compared to IL-12p70 in the absence of IFN-γ; IL-10 was also induced. Following IFN-γ activation, the dendritic cell responded to Mtb by producing both IL-23 and IL-12p70 and reducing the level of IL-10 (102). These data suggest that in humans prior to expression of IFN-γ, the preferential T cell effector type would likely be an IL-17 producer but that thereafter both the IFN-γ and IL-17 response would be promoted. In the murine model, Mtb-infected dendritic cells generate IL-12 and IL-23 and promote both IFN-γ and IL-17 populations in CD4 T cells (54) (Figure 1).
The C-type lectins have also recently been implicated in modulation of phagocyte function in response to mycobacteria (103, 104). Mycobacteria are able to impact dendritic cell function via ligation of the C-type lectin DC-SIGN on immature human dendritic cell and this interferes with LPS induced maturation while promoting IL-10 production (105). This interference appears to be mediated by the acetylation of the p65 subunit of NF-kB that prolongs the transcription rate of the IL10 gene (106). The C-type lectin, Dectin-1, in conjunction with TLR2, is involved in the cytokine response of macrophages to Mtb (107) and has also been implicated in Mtb-induction of IL-12p40 in splenic dendritic cells (108). Our understanding of the role of specific cell types and receptors in the development of tuberculosis is still being developed, it is therefore important that while we may not consider a specific ligand and receptor interaction as essential to survival it could be crucial for the development of balanced acquired cellular responses. We should therefore continue to determine the role of these molecules in both protection and development of pathologic consequences.
Phagocyte phenotypes in the infected lung
To understand the dynamic environment within which the acquired cellular response must act to mediate protection it is necessary to define the cellular components of the inflammatory response to Mtb infection. It has long been assumed that the phagocytic mononuclear cells that accumulate at the inflammatory site in the Mtb-infected lung are monocyte-derived macrophages. It is clear from recent data however that even in the naive lung the myeloid cell population is a complex mixture of phenotypes (28). Indeed, investigation of the cell phenotypes accumulating in the lesion following infection using flow cytometry and immunohistochemistry have challenged the conventional view that macrophages are the principal host cell for Mtb. Specifically, early observations that mycobacteria could infect cells of dendritic cell phenotype in vivo (109) have been followed up to demonstrate that Mtb infects a variety of cells within the lung as the lesion develops (35, 110). The potential importance of the dendritic cell in the lung lesion was suggested when the DEC-205 (CD205) dendritic cell marker was used to determine that the majority of “foamy macrophages” within the inflammatory lesion were in fact positive for this dendritic cell marker (111) as well as being CD11b and CD11c high. A similar subset was recently determined to be a primary host for Mtb within the infected lung (35). The origin of these cells and their function in controlling infection is still under investigation, however a recent elegant study using cell transfer techniques has shown that circulating monocytes can become one of five different types of cells when entering the Mtb infected lung. In these studies it was shown that when circulating monocytes are recruited to the lung in response to mycobacterial infection they express high levels of CD11c, MAC-3 and class II MHC (112). In an important analysis demonstrating the phenotype of the protective effector cell, a correlation between CD205 and iNOS expression was shown to occur in the CD11b+CD11c+ monocyte derived cells that accumulated in the lung (112).
It is clear that the cellular environment within the lung becomes more complex as infection develops into disease. Determining the nature of the infected cells and indeed the overall environment within the lesion is crucial to rational design of vaccines as we need to know whether cells induced by vaccination are capable of functioning within the inflammatory environment generated by the bacteria. In this regard, it has recently been demonstrated that the inflammatory lesion in several animal models (but not the intact mouse (113)) are locally hypoxic environments (114). Whether this environment could impact cellular immunity is unclear as these hypoxic sites are highly disrupted; the possibility should however be considered. One final consideration of an indirect impact of mycobacterial infection on the T cell response is the fact that several mycobacterial species are known to infect the thymus even when they have entered the body though the lung. Although this infection does not result in granulomatous responses it is likely that the normal function of the thymus may be impacted by this infection (115).
Effector B lymphocytes
The ability of acquired immune responses to limit bacterial growth is key to survival upon Mtb infection, however the fact that this immunity is insufficient to eliminate the bacteria suggests several potential failures in the response. The first is that the phagocyte is not capable of eliminating bacteria regardless of activated state. The second is that macrophage activation is optimal but that it is limited by the bacterium. The third is that activation of the infected phagocyte by T cells is compromised within the inflammatory environment. This latter failure could be addressed and potentially remedied by generating either T cells more able to express function in the inflammatory site or by modulating the inflammatory site. The importance of CD4 and CD8 T lymphocytes in controlling Mtb in the lung has been reviewed extensively elsewhere (2, 3) and it is these cells that are the focus of most current vaccine development. In this section the potential for B cells to impact the cellular control of Mtb infection will be proposed.
While the location of antigen-specific T cells within the granuloma has not been defined, it is clear that CD3+, CD4+ and CD8+ T cells are present within the inflammatory lesion. The largest population of lymphocytes within the lesions are however the B220+ cells, which are likely B cells (116). These cells accumulate in follicles that look like nascent lymphoid tissue, are dependent upon specific chemokines and may play a role in regulating immunity within the lesional site (117) (Figure 2). Further, while low dose aerosol infection of B cell deficient mice does not result in increased susceptibility to bacterial growth it does alter lesion development (118–120) and when B cell deficient mice are challenged intravenously with a large dose of bacteria they are more susceptible to disease (121). The role of B cells in immunity to tuberculosis may therefore need reassessment. Recently, higher dose aerosol infection has supported a role for B cells in immunity against lung disease (122, 123). In these recent papers, the nascent follicles in the lung are proposed to be germinal centers and are shown to contain PNA and GL7 positive cells as well as T helper follicular cells expressing CXCR5 and which respond to CXCL13. In the absence of B cells, mice exposed to these higher dose aerosols have exacerbated immunopathologic consequences with increased neutrophil accumulation; these changes in the cellular response correlate with an increase in bacterial burden (122). In an extension of this work it has recently been shown that the nature of the Fcγ receptor expressed in the lung correlates with susceptibility, thus while activating receptors aid protection, inhibiting receptors limit protection (123). The expression of immunity correlates with IL-12p40, CD4+IFNγ+ cells and class II MHC expression. These data suggest that either the B cells and their products or the follicles themselves are able to modulate both the inflammatory and the cytokine response. In this regard the ability of B cells to impact the inflammatory site has been demonstrated in a post-exposure vaccine model wherein the absence of B cells resulted a severe increase in pathologic consequences (120). Based on the high frequency and potential of B cells to impact both immunity and immunopathologic consequences at the site, it is difficult to ignore the potential of these cells. Whether it will be possible to harness the power of B cells by vaccination will depend upon our understanding of the degree of antigen-specificity and of the specific functions expressed by these cells. An element of caution should be raised here however as guinea pigs, which are highly susceptible to death from Mtb infection, elaborate an early T cell response in lung lesions which is subsequently replaced by a predominantly B cell and granulocyte response (124). Whether the B cells are causative of the susceptibility of guinea pigs will have to be determined and the function of B cells within protected and susceptible species may provide insight into what represents a useful B cell.
Figure 2.
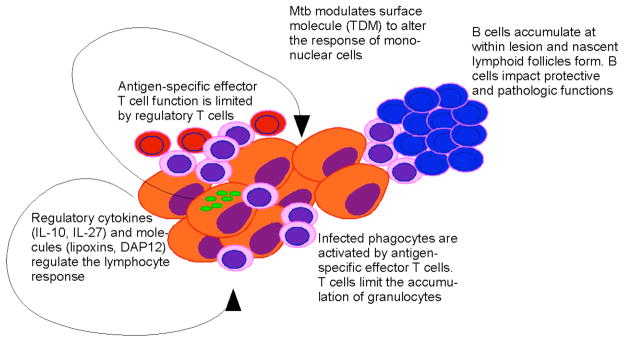
The inflammatory lesion within the lung is a dynamic environment containing a variety of protective and regulatory cells. Effector T lymphocytes (pink) mediate control of bacterial growth and the mononuclear composition of the granuloma. Regulatory T lymphocytes (red) also accumulate in the lesion and limit the ability of the acquired response to stop bacterial growth. Infected phagocytes elaborate cytokines and effector molecules that limit the activity of the lymphocyte response. B cells (blue) accumulate within the lesion in the form of nascent lymphoid follicles; these cells can impact bacterial control and the immunopathologic consequences of infection. Mtb (green) can modulate the inflammatory response via the modification of TDM expressed at the bacterial surface.
Control of T cell responses
Despite the potential of B cells, it is the T cell response that must mediate protection once Mtb is within host cells. In mice, the lung, liver and spleen are infected following aerosol infection however significant disease, as measured by the level of inflammatory involvement of parenchymal tissue, occurs primarily in the lung (125, 126). In the spleen and liver, inflammatory lesions are small and contained whereas in the lung, these lesions continue to develop and become destructive at rates depending upon the strain of mouse or species of host (60). In order to control tuberculosis we need to understand what is regulating the cellular response in the lung and also whether removal of this regulation will allow for efficient clearance of the bacteria or result in overwhelming inflammation, which could kill the host. There is mounting evidence that the immune response to Mtb infection is regulated.
With the increased interest in regulatory T cells the potential of these cells to modulate the immune response to tuberculosis has been the focus of several investigations. When a CD4 T cell population is depleted of regulatory T cells (based on surface phenotype) and transferred into Rag mice this population can mediate efficient control of bacteria; the regulatory activity is not dependent upon IL-10 (127). Further, in a direct aerosol infection it has been demonstrated that T cells with a regulatory cell phenotype accumulate both in the draining lymph nodes of the lung and within the lung itself at a rate similar to effector T cells (128). These regulatory T cells are also located within the inflammatory lesion (Figure 2). Using a chimera model wherein cells sufficient for the transcription factor required for regulatory T cell development were congenically marked, it was possible to deplete regulatory T cells from infected mice and demonstrate that depletion of regulatory T cells results in a modest reduction in bacterial number (128). Together these data suggest that regulatory T cells do impact the ability of the effector T cell response to mediate immunity however it is unclear what the long-term consequences of depletion of this regulatory activity are.
The potential role of IL-10 to regulate the protective cellular response to Mtb has been investigated extensively in recent years. The human data is of interest as there are reports of an association between the risk of developing tuberculosis and the presence of specific IL-10 polymorphisms. One example is the increased tendency toward development of primary progressive tuberculosis in humans with increased innate IL-10 responses (129). This association is not seen in all populations and a recent meta-analysis suggests that while a tendency towards increased susceptibility and IL-10 polymorphisms can be seen it is not strong (130). In the mouse model, CBA mice have a strong IL-10 response to Mtb as expressed in the lung phagocytes and this strain develops progressive tuberculosis, which is poorly controlled by the cellular response (65). If this excess IL-10 is modulated by anti-IL-10 treatment the susceptibility of this strain is reduced (131). If IL-10 is over-expressed in the lungs of Mtb-infected B6 mice, then bacterial growth is poorly controlled compared to wild type mice (65). In the B6 model, IL-10 is not expressed at high levels in lung phagocytes (65) and in its absence there is little impact on control of bacterial growth (132) (Figure 2). These data suggest that IL-10 can impact cellular immunity but that its involvement is dependent upon genetic factors impacting its expression in response to infection; this may explain the variable nature of the impact of IL-10 polymorphisms on the progression of human disease.
In several other mouse models, the loss of one gene can result in improved bacterial control (Figure 2). For example, in the absence of IL-27R activity, bacterial burden is decreased while there is an increase in the amount of inflammation within the parenchyma (133, 134). Despite this improved bacterial control the gene-deficient mice succumb to infection earlier than do wild type mice (134). In a different model, the enzyme required for the generation of the immunoregulatory lipoxins, 5-lipoxygenase, is detrimental to reduction of bacterial numbers as mice deficient for this enzyme elaborate greater Type 1 immunity to mycobacterial infection (135). While the impact of this deficiency on survival has not been reported, the absence of this enzyme is detrimental to the survival of mice infected with Toxoplasma gondii due to excessive Type 1-induced damage (136). In a further model, the absence of the chemokine receptor CXCR3 also results in improved control of bacterial burden in the chronic phase of disease, the extent of which is mouse strain dependent. In this model there is no apparent impact on the inflammatory response and it appears that CXCR3 expression regulates the size of the antigen-specific T cell population (137). Finally comparison of the phagocyte response to infection between DAP12 deficient and wild type mice, demonstrated that the DAP12-deficient cells had a much greater NFκB activation and an increased cytokine response to infection than did wild type cells. This resulted in an enhanced inflammatory response and increased numbers of IFN-γ-producing antigen-specific cells in the tissue. This increase in response resulted in reduced bacterial burden within 25 days of infection, which was combined with an accelerated and increased granulomatous response within the lung (138). These data suggest that DAP12, which is an immunoreceptor tyrosine-based activation motif (ITAM) containing co-receptor, regulates the cellular response following mycobacterial infection likely through its ability to regulate the phagocyte response to the pathogen.
The regulatory nature of the inflammatory lesion that occurs in response to Mtb means that acquired cellular immunity should be expressed as rapidly as possible in order to control bacterial growth before any regulatory activity is expressed. One way to do this is via vaccine-induced generation of immunological memory.
Memory T cell responses
As discussed above, following infection via the aerosol route there is a significant delay in the induction of acquired cellular responses. It is likely that this delay contributes to the establishment of a productive infection and progression to disease in susceptible individuals. The best way to accelerate the kinetics of acquired cellular responses is to expose the adaptive cells to specific antigens of the pathogen in an environment that will generate long lived antigen-specific cells capable of ‘remembering’ the pathogens upon subsequent challenge; this is of course the basis of vaccination. While we know that vaccination is an excellent way to protect against some diseases (primarily those controlled by antibody) vaccination against tuberculosis is not generally protective against pulmonary disease in adults (23) (Figure 3). In contrast, vaccination can significantly limit the dissemination of disease to other organs and does protect children from disease sequellae such as tuberculous meningitis (22).
Figure 3.

(A) The host with previous antigenic experience has an increased frequency of memory lymphocytes either in the circulation (pink) or within the lung (green). Cells within the lung can respond to infection before bacteria migrate to the lymph node and can therefore activate infected phagocytes and stop bacteria growth. Cells within the lung can also recognize infection and act to rapidly to recruit circulating memory cells that may require dissemination of bacteria to become activated. As effector cells arrive in the lung they activate phagocytes and stop bacterial growth. (B) The cessation of bacterial growth by memory cells can occur 5 days sooner than is seen for naive cells. This acceleration though significant does not stop substantial bacterial growth and thus effector cells must act within a site that has been initiated and modulated by Mtb. If the memory response can be improved such that the response occurs within 10 days as opposed to 15 then the T cell response may be expressed more efficiently and thereby limit the development of disease.
To improve vaccination strategy we must understand why the current strategy is ineffective. One potential issue with current models is that while sizeable populations of circulating antigen-specific memory cells are induced, the kinetics of the protective cellular memory response to aerosol challenge is only accelerated by a few days (45, 46, 139, 140). This modest acceleration allows for earlier expression of anti-mycobacterial activity within the lung but as there is still a substantial delay, a significant bacterial burden becomes established prior to expression of the memory response. The memory cells then have to act within an environment that the Mtb has had an opportunity to modulate (Figure 3). As dissemination occurs at the same time as activation of T cell responses it is likely that disseminated bacteria encounter the acquired response very shortly after arrival at a site and are therefore less able to modulate the environment. This may be why BCG is only modestly protective against lung disease but it is very effective against disseminated disease (22). It also explains why measuring the circulating population of antigen-specific IFN-γ-producing cells does not provide a good correlate of protection (141). Specifically, while memory cells may be present in the circulation it is the ability of these cells either to populate the lung or to get to the lungs quickly upon infection that is important; this ability is not measured by determining frequency of antigen-specific effector cells within the peripheral blood.
In order to overcome the low frequency of antigen-specific cells within the lung, investigators have employed in situ boosting regimens. Using BCG priming in the lung followed by boosting with either BCG or modified vaccinia virus Ankara (MVA) expressing antigen85 (Ag85) it has been found that extremely good protection against aerosol challenge could be obtained and that this correlated with induction of Ag85 specific cells within the lung draining lymph node (142). In a separate study using intramuscular DNA vaccination, it was found that very few Mtb reactive CD8 T cells were found in the airway despite a substantial systemic population. Unlike BCG vaccination wherein a predominantly systemic population of cells can mediate a modest level of protection in the murine model, this systemic CD8 population was not protective. However if the cells were recruited to the lung lumen by low dose antigen exposure these cells could mediate protection from challenge (143) (Figure 3). The authors of this work took these data to demonstrate that the protective activity of antigen-specific memory T cells is defined by the location of the cells. In an effort to induce mucosally located antigen-specific T cells, these authors have delivered dendritic cells transduced to express Ag85 intranasally to mice. These dendritic cells migrate to the lung mucosa and the draining lymph nodes in an IL-12p40-dependent manner and generate antigen-specific effector cells in the airway lumen in an IL-12-independent manner. This route of vaccination resulted in a modest level of protection (36). Further data on the longevity and any potential immunopathogic role of lung resident effector cells should be obtained prior to using pulmonary vaccination as a tool to control tuberculosis.
Prime-boost regimens have also been proposed in order to augment the routine BCG vaccination given either to newborns or adolescents, the rationale for this is that the response to BCG wanes; this has been demonstrated recently in a study from the UK (144). The MVA expressing Ag85, shown to be protective in mice (142), has been used to boost BCG induced responses in people and has been found to be highly immunogenic (145). It has also been shown that boosting with this vaccine results in populations of polyfunctional cells that retain proliferative potential (146). While it is difficult to test the protective efficacy of these prime-boost regimens in humans the data support the use of these regimens to maintain cellular immune responses.
One concern with repeated boosting is the potential to induce pathologic consequences. As discussed above, the repeated delivery of mycobacterial antigen to already infected mice can result in augmentation of the pathologic response in the lung (57, 58, 120). This is reminiscent of the work by Koch wherein he demonstrated that delivery of live mycobacteria or mycobacterial antigen to tuberculous guinea pigs could result in necrosis and sloughing of the skin. This effect was dependent upon dose of antigen and at lower doses hypersensitivity is seen without necrosis (1). While there has been little evidence of this “Koch phenomenon” during the currently performed prime-boost models, it is likely that if we can identify what mediates any damaging response and isolate it from the protective response we may be able to boost the protective response to a greater degree. In this regard we have determined that IL-23 and IL-17 appear to be required for the enhanced pathologic responses seen in repeatedly exposed Mtb-infected mice (Cruz pers comm).
While it is possible that IL-17-producing cells are detrimental during chronic infection we have identified a function for these cells during the recall of antigen-specific IFN-γ-producing memory CD4 T cells. In a model of vaccine-induced protection using a sub-unit vaccine containing a peptide from an early expressed Mtb antigen (ESAT-6) delivered in an adjuvant of monophosphoryl lipid A (MPL), trehalose dicorynomycolate (TDM) and dimethyldioctadecylammonium (DDA), we have shown that a population of antigen-dependent cells responsible for IL-17 production is induced in the lung (46). These cells respond more rapidly to aerosol challenge and are required for early expression of chemokines within the lung. In their absence neither an accelerated IFN-γ response nor vaccine-induced protection is seen in the lung (46). These data suggest that surveillance cells can be induced in the lung by subcutaneous vaccination and that vaccination may be rationally targeted to these cells. The caveat to this is of course that these cells could also mediate destructive immunopathologic consequences if not sufficiently regulated.
The increase in our ability to modulate mycobacteria genetically has allowed the design and testing of genetically modified mycobacteria as vaccine candidates. This is a rational method of generating candidates as long as we can augment immunogenicity while reducing virulence. Two recent studies highlight the practicality of this approach. In one, the expression of two genes in BCG that promote antigen translocation to the cytoplasm of infected cells results in improved protection compared to the parental BCG and this is thought to be due to cross-priming (147). In a more recent study, vaccination with an Mtb mutant lacking the ability to inhibit apoptosis of infected macrophages resulted in enhanced CD8 T cell responses and excellent early protection against protection both in mice and guinea pigs (148).
It is by increasing our understanding of the nature and functionality of vaccine-induced cells that we will provide a rational basis for new vaccine design. Key elements of this understanding include the location of the vaccine-induced cells, their ability to respond quickly and their ability to respond efficiently within a highly regulated environment. We can achieve this using basic animal models, however we also need to improve our analysis of humans to vaccination.
Human cellular responses
Recent technological advances have allowed for improved analysis of the cellular responses occurring in vaccinated, Mtb-exposed, and tuberculosis patients. The most dramatic improvement in data generation has been in analysis of antigen-specific responses and in the ability to identify several parameters of responding cells using flow cytometry. Analysis of cellular responses has shown that infected and diseased individuals express a high frequency of multifunctional cells (49). In recent studies of a European population there is a limited Mtb-specific IL-17-specific response whereas there is a strong IL-17 response to fungal antigens (149). In contrast, when South African populations are examined, Mtb-specific IL-17 producing and IL-22-producing T cells can clearly be identified both in exposed and disease individuals although the frequency of such cells in the periphery is lower in the diseased patients (50). Despite low frequency of these cells peripherally, IL-22 can be detected in the BAL of patients suggesting that IL-22-producing cells are active in the lung of patients (50). This compartmentalization of the response has also been seen in a recent study using an Ag85/HLA-A*0201 pentamer to assess frequency of antigen-specific cells in tuberculous children pre and post drug treatment. In this study it was found that patients have a low frequency of antigen-specific CD8 T cells in the blood but that this frequency increases after treatment (150). This could be due to the treatment reducing inhibition of T cell activation by the bacteria or because lung lesions were resolving and thus recruitment of antigen-specific cells from the blood to the lesional site was reduced. Interestingly the circulating antigen-specific cells had low cytokine and cytolytic activity pre treatment whereas more activated cells were present in the lung pre-treatment (150). In a separate study, it was found that at the beginning of treatment the frequency of antigen-specific IFN-γ-producing cells is higher than that for IL-2-producing antigen-specific cells but that as treatment progressed over 28 months the dominance of the IFN-γ response is lost and the majority of responding cells are of the IL-2 producing phenotype (151). Further detailed studies will allow for greater understanding of the kinetics and nature of the antigen-specific cellular response in humans.
The high level of BCG vaccination occurring in newborns in South Africa has allowed for an unprecedented screen of the ability of BCG to induce specific cell types in this population. In a recent study, cells from BCG vaccinated newborns were restimulated in vitro with BCG and the cellular response analyzed by flow cytometry. Importantly while IL-4 and IL-10 producing CD4 T cells occurred at low frequency, IFN-γ, IL-2, and TNF were all produced either singly or in combination by activated CD4 T cells. Activated CD8 T cells were less frequent and when present were predominantly IL-2 and/or IFN-γ positive. In a detail which is telling, it was noted that many of the responding T cells were not positive for IFN-γ thus the effector phenotype of many of the responding cells is still to be determined. The majority of the responding, IFN-γ-producing cells had an effector cell surface phenotype whereas those expressing IL-2 alone were of the central memory phenotype (51, 152).
One aspect of the human immune response that has received increased attention recently is the identification of antigens recognized by exposed individuals. In a study using synthetic peptide arrays of known immunodominant antigens, the antigen-specificity of a large number of CD8 T cells clones was obtained and new epitopes identified (153). In a separate study, the regions of difference (RD) that have been identified between Mtb and BCG were utilized to compare the peptide specific responses of patients with tuberculosis and healthy individuals vaccinated with BCG. In this study both groups responded to peptides from the RD1 region (perhaps reflecting a high degree of exposure to Mtb) however peptides from RD12, RD13 and RD15 peptides elicited an IL-10 response from peripheral blood mononuclear cells suggesting that antigens within the different RD are associated with distinct cellular responses (154). These kinds of studies are being used to fill the gaps in our understanding of the specificity of the T cell response to Mtb in humans (155).
Although acquired cellular immunity is the focus of many studies, the study of the innate response of humans to infection has been gaining momentum recently. Neutrophils have been implicated in anti-mycobacterial immunity due to their ability to provide antibacterial activity to infected macrophages as a result of being phagocytosed (156). This was first shown in the murine model with M. avium and M. microti experiments (157) and has recently been confirmed using human cells and virulent Mtb (158). It has been shown that both apoptotic neutrophils and purified neutrophil granules are able to reduce the viability of extracellular bacteria and augment the ability of infected macrophages to reduce growth of bacteria. The granules traffic to the early endosomes and colocalize with the bacteria (158). In an exciting extension of this work, the potential importance of neutrophils in protection against infection was assessed in 187 contacts of newly diagnosed Mtb patients (159). In this study, a whole blood assay was used to demonstrate that when neutrophils were depleted from whole blood the release of anti-microbial peptides in response to mycobacterial exposure was dramatically reduced; the same peptides were also shown to be anti-mycobacterial. Also when neutrophils were depleted from the whole blood the ability of the blood to limit mycobacterial viability was reduced. Importantly, an inverse relationship between the number of peripheral neutrophils and the risk of Mtb infection in contacts of pulmonary tuberculosis patients was observed suggesting that neutrophils may play a protective role very early in infection (159). Human NK cells have also been investigated for their ability to mediate anti-mycobacterial activity. When macrophages are infected with Mtb they upregulate vimentin and NK cells are able to lyse infected mycobacteria via ligation of the vimentin by the NKp46 molecule on NK cells (160–162). This ability of NK cells to lyse infected cells is seen with infected human alveolar macrophages suggesting that this interaction could play a role in the earliest responses to infection (160). In an extension of this work, it has been demonstrated that human NK cells can lyse expanded T cells that expres a regulatory phenotype (CD25+FoxP3+) (163). How this ability of NK cells to regulate the acquired response impacts disease will be intriguing to determine. Indeed, while the impact of NK activity on disease is not clear NK-mediated cell lysis is reduced in patients with advanced disease (162). In the murine model, IL-12p70 dependent NK cell activity can control bacterial growth in the absence of acquired immunity and also limits accumulation of granulocytes at lesional sites (164).
The study of human immunity is being improved by the availability of tools that allow for the visualization of the cellular response both immediately ex vivo and even in vivo (24). Despite these advances it is still difficult to experiment on humans directly and it is therefore imperative that we maintain an active dialog between those studying mycobacterial infection in the variety of animal models and those studying the disease in humans. Comparative pathology is an excellent tool to improve our knowledge of this complex disease.
Mtb modulates its environment
The chronicity of Mtb infection results in an inflammatory and dynamic environment within which the acquired cellular response must act. In this regard the impact of the mycobacterium on cellular inflammation is a key element. The different mycobacterial species such as M. leprae and M. ulcerans induce profoundly different consequences upon infection compared to Mtb. However it is only recently that the genetic variability and strain differences within Mtb have been appreciated to be extensive and to contribute to the pathogenesis of disease (165). The impact of this genetic variability is now well established. As more strains are identified and their ability to modulate immunity has been investigated it is clear that Mtb has a variety of tools with which to modulate the inflammatory response to infection. This is not surprising as the bacterium depends upon the inflammatory response to mediate dissemination of disease. It is also not surprising as the surface of the bacteria contains a variety of highly stimulatory molecules the structures of which are finely tuned to interact with the immune response (Figure 2).
The impact of very small changes in the molecules that comprise the cell wall of mycobacteria can be dramatic. For example, an Mtb mutant that lacks the pcaA gene (Rv0470c) and thus is unable to cyclopropanate alpha mycolates, is unable to persist in the mouse model despite reaching initially equivalent levels of infection in the lung (166). The reduced virulence of this mutant is not apparent in TNF-deficient mice suggesting that it is the induction of TNF by the altered molecule that is associated with the decreased virulence of this mutant. It appears that it is the defective structure of trehalose dimycolate (TDM) in this mutant that results in the hypoinflammatory response. Indeed, while systemic delivery of wild type TDM results in long-lived granulomatous inflammation in the lung, the mutant TDM is much less able to induce this response. It also fails to induce an early TNF response in macrophages in vitro (167).
In an interesting contrast to the impact of the pcaA gene, a mutant lacking cmaA2, which is required to trans-cycloproponate methoxy and ketomycolates but does not impact alpha-mycolate, exhibits normal bacterial growth in vivo but is more virulent than wild type in mice following aerosol infection. This virulence correlates with increased inflammatory involvement in infected organs where larger but lymphopenic lesions are seen. The mutant is hyperinflammatory and the cmaA2-dependent lipid fraction from wild type bacteria inhibits this hyperinflammatory response. As was seen for the pcaA mutant, the defect in the cmaA2 mutant is associated with the TDM fraction and the wild type TDM inhibits the ability of the cmaA2 mutant TDM to induce a TNF response (168).
Modification of the TDM is also involved in induction of the IL-12p40 gene. An Mtb mutant lacking the mmaA4 gene, which is required for oxygen-containing modifications of cell wall mycolic acids, is less virulent in vivo and this reduced virulence is not observed in IL-12p40 deficient mice. Both the mutant bacteria and its TDM induce more IL-12p40 than the wild type Mtb and the wild type TDM is inhibitory (169).
In a further example of the role of TNF induction in the virulence of mycobacteria it appears that some isolates of the highly successful W-Beijing strains are able to modulate their induction of TNF. These strains are present throughout the world and are associated with acute outbreaks (170). It appears that several virulent isolates of the W-Beijing strains do not have the frameshift mutation, present in many strains of Mtb, that prevents generation of phenolic glycolipid (PGL) and thus they produce this lipid. In the mouse model, there is little impact on the bacterial burden as a result of the generation of PGL however infected mice are subject to greater mortality. When the ability to make this lipid is disrupted by mutation, the hypervirulence is lost and there is an increase in induction of TNF, IL-6 and IL-12. Over expression of the PGL by mycobacteria or treatment with free PGL results in decreased induction of these cytokines (171). Virulence of the W-Beijing strains in a rabbit model of tuberculosis meningitis is also dependent upon the production of PGL (172). Further analysis of the role of the PGL suggests that its ability to impact virulence may depend on other as yet undefined aspects of the bacteria, as simple expression of the PGL in the laboratory strain H37Rv does not result in hypervirulence but does inhibit cytokine induction (173). It also appears that when the acquired cellular response to one of the hypervirulent W-Beijing strains is examined, a significant Th1 type immune response is induced but is lost as a regulatory T cell population expands (174). Thus while the W-Beijing strains are clearly altered in their ability to modulate the immune response to infection and it is clear that the expression of PGL is essential to this process, the exact mechanism by which this hypervirulence occurs is still to be definitively determined.
Together the above evidence suggest that Mtb devotes considerable energy to directing the induction of the cellular response to infection and that the ability of strains and mutants to modulate the kinetics and extent of cytokine induction plays an important role not in the control of bacteria but in the nature of the inflammatory response induced.
Conclusions
Our knowledge of the cellular response to Mtb infection has improved dramatically over the years. We are have not however been able to define the type of acquired cellular response that will mediate immunity and which should therefore be induced by vaccination; this severely limits our ability to develop novel vaccines. We are also at a loss to test new vaccines in humans, as we do not have a definitive correlate of protection. Improvements in locally available technology at sites of high disease incidence are improving our ability to define the human cellular response to both infection and disease. These improvements combined with the much greater communication between groups that is being facilitated by funding of large consortia should dramatically improve our understanding of human disease. Combining this greater understanding with definitive experimentation within the animal models will allow for the determination of the protective and pathologic function of specific cellular responses as well as how best to induce each type of response.
Summary Points.
The induction of naive T cell activation occurs more than a week after delivery of bacteria to the lung. Bacterial dissemination to the draining lymph node correlates with initiation of T cell responses in the draining lymph node
A variety of effector lymphocyte subsets are induced by infection including multifunctional and cytolytic cells. Not all responding cells make IFN-γ.
Phagocyte effector function is key to the ability of the host to express immunity.
The MyD88 mediated phagocyte response required for control of Mtb is not a TLR dependent event but likely a result of IL-1R activity. TLR ligation is involved in efficient induction of lymphocyte effector subsets and may be important in the balance between protection and pathologic consequences.
The phagocytes that accumulate in the lung in response to Mtb infection represent a complex population that includes cells that express both macrophage and dendritic cell markers.
The inflammatory lesion is a dynamic environment that is regulated both by the bacteria and by the host. TDM is a key bacterial modulator of the immune response while B lymphocytes, T lymphocytes and phagocytes are key host regulators of the inflammatory site.
Vaccine-induced memory cells need to be either in the lung or capable of being rapidly recruited to the lung in order to be effective in limiting bacterial growth following aerosol infection.
Detailed investigation of the human response to infection is highlighting the complexity of the response. Demonstration of novel lymphocyte subsets as well as novel phagocyte functions highlights the need to maintain an open view as to what constitutes a protective response in humans.
Future Issues.
In order to improve current vaccine strategies the field should:
Determine where bacteria are deposited in the lung during primary infection and why it takes 8–9 days for the acquired immune response to be initiated.
Determine the factors, both bacterial and host, that regulate expression of immunity in the lung
Identify effector lymphocyte subsets that can mediate protection within the context of an Mtb-induced inflammatory site.
Identify novel phagocyte functions that can be activated by vaccine-inducible antigen-specific lymphocytes.
Determine how to augment the protective activity of the acquired cellular response without increasing immune-mediated damage.
Determine the potential of B cells, which are plentiful at the lesional site, to modulate immunity and/or immunopathologic consequences. Can we (should we) harness B cells by vaccination?
Determine how to generate antigen-specific lymphocytes capable of persisting in the lung and of responding rapidly to aerosol infection.
Continue to enhance the consortium arrangements that promote communication between bacteriologists, animal modelers and those investigating the human disease.
Acknowledgments
Trudeau Institute Librarian, Ms Kelly Stanyon provided excellent Library assistance. I apologize in advance to all the Investigators who could not be cited due to space limitations. I would like to thank my colleagues who kindly provided preprints and informative discussions. AMC is supported by the Trudeau Institute, Inc, and by grants from the National Institutes of Health (AI067723, AI46530, AI069121, AG028878) and the American Lung Association (DeSouza award).
Terms and Definitions
- Immunity
Being able to stop or significantly limit the ability of a pathogen to cause disease
- Acquired cellular immune response
The adaptive response of the vertebrate immune system resulting in the development of antigen-specific lymphocytes and thereby the enhanced specificity of the effector mechanisms of the system
- Inflammation
The physiological process by which the body responds (in this case) to an invading and persistent pathogen. This includes the acute inflammation associated with activation of infected tissue macrophages as well as the chronic inflammation associated with mononuclear cell accumulation
Literature Cited
- 1.Rich A. The pathogenesis of tuberculosis. Baltimore: Chales C Thomas; 1944. [Google Scholar]
- 2.North R, Jung Y. Immunity to tuberculosis. Ann Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 3.Flynn J, Chan J. Immunology of tuberculosis. Ann Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 4.Waterston R, Lindblad-Toh K, Birney E, Rogers J, et al. Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 5.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 6.Lee M, Pascopella L, Jacobs WJ, Hatfull G. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guérin. Proc Natl Acad Sci USA. 1991;88:3111–5. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelicic V, Jackson M, Reyrat J, Jacobs WJ, Gicquel B, Guilhot C. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 1997;94:10955–60. doi: 10.1073/pnas.94.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dye C, Scheele S, Dolin P, Pathania V, Raviglione M. Global Burden of Tuberculosis. Estimated Incidence, Prevalence, and Mortality by Country. JAMA. 1999;282:677–86. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 9.Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, et al. The Growing Burden of Tuberculosis: Global Trends and Interactions With the HIV Epidemic. J Arch Intern Med. 2003;163:1009–21. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]
- 10.Koenig R. Drug-resistant tuberculosis. In South Africa, XDR TB and HIV prove a deadly combination. Science. 2008;319:894–7. doi: 10.1126/science.319.5865.894. [DOI] [PubMed] [Google Scholar]
- 11.Havlir D, Barnes P. Tuberculosis patients with human immunodeficiency virus infection. N Eng J Med. 1999;340:367–73. doi: 10.1056/NEJM199902043400507. [DOI] [PubMed] [Google Scholar]
- 12.de Noronha A, Báfica A, Nogueira L, Barral A, Barral-Netto M. Lung granulomas from Mycobacterium tuberculosis/HIV-1 co-infected patients display decreased in situ TNF production. Pathology - Research and Practice. 2008;204:155–61. doi: 10.1016/j.prp.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Saunders B, Frank A, Orme I, Cooper A. CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell Immunol. 2002;216:65–71. doi: 10.1016/s0008-8749(02)00510-5. [DOI] [PubMed] [Google Scholar]
- 14.North RJ. Importance of thymocyte-derived lymphocytes in cell-mediated immunity to infection. Cell Immunol. 1973;7:166–76. doi: 10.1016/0008-8749(73)90193-7. [DOI] [PubMed] [Google Scholar]
- 15.Lefford MJ. Transfer of adoptive immunity to tuberculosis in mice. Infect Immun. 1975;11:1174–81. doi: 10.1128/iai.11.6.1174-1181.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orme I, Collins F. Protection against Mycobacterium tuberculosis infection by adoptive immunotherapy. Requirement for T cell-deficient recipients. J Exp Med. 1983;158:74–83. doi: 10.1084/jem.158.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orme IM. The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. J Immunol. 1987;138:293–8. [PubMed] [Google Scholar]
- 18.Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2001;193:271–80. doi: 10.1084/jem.193.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–7. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–54. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ottenhof T, Kumararante D, Casanova J. Novel human immunodeficiencies reveal the essential role of type-1 cytokines in immunity to intracellular bacteria. Immunol Today. 1998;19:491–4. doi: 10.1016/s0167-5699(98)01321-8. [DOI] [PubMed] [Google Scholar]
- 22.Trunz B, Fine P, Dye C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet. 2006;367:1173–80. doi: 10.1016/S0140-6736(06)68507-3. [DOI] [PubMed] [Google Scholar]
- 23.Colditz G, Brewer T, Berkey C, Wilson M, Burdick E, et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA. 1994;271:698–702. [PubMed] [Google Scholar]
- 24.Young D, Perkins M, Duncan K, Barry Cr. Confronting the scientific obstacles to global control of tuberculosis. J Clin Invest. 2008;118:1255–65. doi: 10.1172/JCI34614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chackerian A, Alt J, Perera T, Dascher C, SMB Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70:4501–9. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf A, Desvignes L, Linas B, Banaiee N, Tamura T, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–15. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiley W, Calayag M, Wittmer S, Huntington J, Pearl J, et al. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in mediastinal lymph nodes. Proc Natl Acad Sci USA. 2008 doi: 10.1073/pnas.0801496105. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Garnier C, Filgueira L, Wikstrom M, Smith M, Thomas J, et al. Anatomical location determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J Immunol. 2005;175:1609–18. doi: 10.4049/jimmunol.175.3.1609. [DOI] [PubMed] [Google Scholar]
- 29.Demangel C, Bertolino P, Britton WJ. Autocrine IL-10 impairs dendritic cell(DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. Eur J Immunol. 2002;32:994–1002. doi: 10.1002/1521-4141(200204)32:4<994::AID-IMMU994>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Bhatt K, Hickman SP, Salgame P. Cutting Edge: A new approach to modeling early lung immunity in murine tuberculosis. J Immunol. 2004;172:2748–51. doi: 10.4049/jimmunol.172.5.2748. [DOI] [PubMed] [Google Scholar]
- 31.Khader S, Partida-Sanchez S, Bell G, Jelley-Gibbs D, Swain S, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203:1805–15. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia W, Pinto C, Kradin R. The antigen-presenting activities of Ia+ dendritic cells shift dynamically from lung to lymph node after an airway challenge with soluble antigen. J Exp Med. 1995;181:1275–83. doi: 10.1084/jem.181.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holt P, Haining S, Nelson D, Sedgwick J. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J Immunol. 1994;153:256–61. [PubMed] [Google Scholar]
- 34.Inaba K, Inaba M, Naito M, Steinman R. Dendritic cell progenitors phagocytose particulates, including Bacillus Calmatte-Guerin organisms, and sensitize mice to mycobacterial antigens in vivo. J Exp Med. 1993;178:479–88. doi: 10.1084/jem.178.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. 2007;179:2509–19. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 36.McCormick S, Santosuosso M, Small C-L, Shaler C, Zhang X, et al. Mucosally delivered dendritic cells activate T cells independently of IL-12 and endogenous APCs. J Immunol. 2008 doi: 10.4049/jimmunol.181.4.2356. in press. [DOI] [PubMed] [Google Scholar]
- 37.Mueller S, Hosiawa-Meagher K, Konieczny B, Sullivan B, Bachmann M, et al. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science. 2007;317:670–4. doi: 10.1126/science.1144830. [DOI] [PubMed] [Google Scholar]
- 38.Mueller S, Matloubian M, Clemens D, Sharpe A, Freeman G, et al. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci USA. 2007;104:15430–5. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egen J, Rothfuchs A, Feng C, Winter N, Sher A, Germain R. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–84. doi: 10.1016/j.immuni.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cosma C, Humbert O, Ramakrishnan L. Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol. 2004;5:828–35. doi: 10.1038/ni1091. [DOI] [PubMed] [Google Scholar]
- 41.Davis J, Clay H, Lewis J, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. 2002;17:693–702. doi: 10.1016/s1074-7613(02)00475-2. [DOI] [PubMed] [Google Scholar]
- 42.Meijer A, van der Sar A, Cunha C, Lamers G, Laplante M, et al. Identification and real-time imaging of a myc-expressing neutrophil population involved in inflammation and mycobacterial granuloma formation in zebrafish. Dev Comp Immunol. 2008;32:36–49. doi: 10.1016/j.dci.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Orme IM, Miller ES, Roberts AD, Furney SK, Griffin JP, et al. T lymphocytes mediating protection and cellular cytolysis during the course of Mycobacterium tuberculosis infection. Evidence for different kinetics and recognition of a wide spectrum of protein antigens. J Immunol. 1992;148:189–96. [PubMed] [Google Scholar]
- 44.Chackerian A, Perera T, Behar S. Gamma interferon-producing CD4+ T lymphocytes in the lung correlate with resistance to infection with Mycobacterium tuberculosis. Infect Immun. 2001;69:2666–74. doi: 10.1128/IAI.69.4.2666-2674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung Y, Ryan L, Lacourse R, North R. Properties and protective value of the secondary versus primary T helper type 1 response to airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2005;201:1915–24. doi: 10.1084/jem.20050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khader S, Bell G, Pearl J, Fountain J, Rangel-Moreno J, et al. IL-23 and IL-17 in establishment of protective pulmonary CD4+ T cell responses upon vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 47.Darrah P, Patel D, De Luca P, Lindsay R, Davey D, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–50. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 48.Klucar P, Barnes P, Kong Y, Samten B, Tvinnereim A, et al. Characterization of effector functions of human peptide-specific CD4+ T-cell clones for an intracellular pathogen. Hum Immunol. 2008 doi: 10.1016/j.humimm.2008.05.008. epub. [DOI] [PubMed] [Google Scholar]
- 49.Winkler S, Necek M, Winkler H, Adegnika A, Perkmann T, et al. Increased specific T cell cytokine responses in patients with active pulmonary tuberculosis from Central Africa. Microbes Infect. 2005;7:1161–9. doi: 10.1016/j.micinf.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 50.Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol. 2008;180:1962–70. doi: 10.4049/jimmunol.180.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soares AP, Scriba TJ, Joseph S, Harbacheuski R, Murray RA, et al. Bacillus calmette-guerin vaccination of human newborns induces T cells with complex cytokine and phenotypic profiles. J Immunol. 2008;180:3569–77. doi: 10.4049/jimmunol.180.5.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooper A, Solache A, Khader S. Interleukin-12 and tuberculosis: An old story revisited. Curr Op Immunol. 2007;19:441–7. doi: 10.1016/j.coi.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khader S, Cooper A. IL-23 and IL-17 in tuberculosis. Cytokine. 2008;41:79–83. doi: 10.1016/j.cyto.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khader S, Pearl J, Sakamoto K, Gilmartin L, Bell G, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol. 2005;175:788–95. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 55.Umemura M, Yahagi A, Hamada S, Begum M, Watanabe H, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis Bacille Calmette-Guerin infection. J Immunol. 2007;178:3786–96. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 56.Lockhart E, Green A, Flynn J. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 57.Taylor J, Turner O, Basaraba R, Belisle J, Huygen K, Orme I. Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect Immun. 2003;71:2192–8. doi: 10.1128/IAI.71.4.2192-2198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner J, Rhoades E, Keen M, Belisle J, Frank A, Orme I. Effective preexposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect Immun. 2000;68:1706–9. doi: 10.1128/iai.68.3.1706-1709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper AM, Adams LB, Dalton DK, Appelberg R, Ehlers S. IFN-γ and NO in mycobacterial disease: new jobs for old hands. Trends Microbiol. 2002;10:221–6. doi: 10.1016/s0966-842x(02)02344-2. [DOI] [PubMed] [Google Scholar]
- 60.Dannenberg AJ, Collins F. Progressive pulmonary tuberculosis is not due to increasing numbers of viable bacilli in rabbits, mice and guinea pigs, but is due to a continuous host response to mycobacterial products. Tuberculosis. 2001;81:229–42. doi: 10.1054/tube.2001.0287. [DOI] [PubMed] [Google Scholar]
- 61.Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuberc Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 62.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–72. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 63.MacMicking JD, North RJ, LaCourse R, Mudgett J, Shah SK, Nathan CF. Identification of NOS2 as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–8. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marquis J, Nantel A, LaCourse R, Ryan L, North R, Gros P. Fibrotic response as a distinguishing feature of resistance and susceptibility to pulmonary infection with Mycobacterium tuberculosis in mice. Infect Immun. 2008;76:78–88. doi: 10.1128/IAI.00369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turner J, Gonzalez-Juarrero M, Ellis D, Basaraba R, Kipnis A, et al. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J Immunol. 2002;169:6343–51. doi: 10.4049/jimmunol.169.11.6343. [DOI] [PubMed] [Google Scholar]
- 66.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun. 2006;74:4295–309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lyadova I, Eruslanov E, Khaidukov S, Yeremeev V, Majorov K, et al. Comparative analysis of T lymphocytes recovered from the lungs of mice genetically susceptible, resistant, and hyperresistant to Mycobacterium tuberculosis-triggered disease. J Immunol. 2000;165:5921–31. doi: 10.4049/jimmunol.165.10.5921. [DOI] [PubMed] [Google Scholar]
- 68.Pan H, Yan B, Rojas M, Shebzukhov Y, Zhou H, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434:767–72. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan BS, Pichugin AV, Jobe O, Helming L, Eruslanov EB, et al. Progression of pulmonary tuberculosis and efficiency of bacillus Calmette-Guerin vaccination are genetically controlled via a common sst1-mediated mechanism of innate immunity. J Immunol. 2007;179:6919–32. doi: 10.4049/jimmunol.179.10.6919. [DOI] [PubMed] [Google Scholar]
- 70.Crowle A, Ross E, May M. Inhibition by 1,25(OH)2-vitamin D3 of the multiplication of virulent tubercle bacilli in cultured human macrophages. Infect Immun. 1987;55:2945–50. doi: 10.1128/iai.55.12.2945-2950.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rook GAW, Steele J, Fraher L, Barker S, Karmali R, O’Riordan J. Vitamin D3, gamma interferon, and control of proliferation of Mycobacterium tuberculosis by human monocytes. Immunology. 1986;57:159–63. [PMC free article] [PubMed] [Google Scholar]
- 72.Martineau AR, Wilkinson KA, Newton SM, Floto RA, Norman AW, et al. IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J Immunol. 2007;178:7190–8. doi: 10.4049/jimmunol.178.11.7190. [DOI] [PubMed] [Google Scholar]
- 73.Liu P, Stenger S, Li H, Wenzel L, Tan B, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–3. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 74.Liu PT, Stenger S, Tang DH, Modlin RL. Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol. 2007;179:2060–3. doi: 10.4049/jimmunol.179.4.2060. [DOI] [PubMed] [Google Scholar]
- 75.Levine B, Klionsky D. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 76.Gutierrez M, Master S, Singh S, Taylor G, Colombo M, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 77.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–41. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 78.MacMicking J, Taylor G, McKinney J. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science. 2003;302:654–9. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 79.Harris J, De Haro SA, Master SS, Keane J, Roberts EA, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–17. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 80.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feng C, Scanga C, Collazo-Custodio C, Cheever A, Hieny S, et al. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J Immunol. 2003;171:4758–64. doi: 10.4049/jimmunol.171.9.4758. [DOI] [PubMed] [Google Scholar]
- 82.Fremond C, Yeremeev V, Nicolle D, Jacobs M, Quesniaux V, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114:1790–9. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scanga C, Bafica A, Feng C, Cheever A, Hieny S, Sher A. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun. 2004;72:2400–4. doi: 10.1128/IAI.72.4.2400-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hölscher C, Reiling N, Schaible U, Hölscher A, Bathmann C, et al. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol. 2008;38:680–94. doi: 10.1002/eji.200736458. [DOI] [PubMed] [Google Scholar]
- 85.Bafica A, Scanga C, Feng C, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–24. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179:1178–89. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 87.Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning C, et al. Cutting Edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–4. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 88.Sugawara I, Yamada H, Li C, Mizuno S, Takeuchi O, Akira S. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol Immunol. 2003;47:327–36. doi: 10.1111/j.1348-0421.2003.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 89.Heldwein KA, Liang MD, Andresen TK, Thomas KE, Marty AM, et al. TL2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J Leukocyte Biol. 2003;74:277–86. doi: 10.1189/jlb.0103026. [DOI] [PubMed] [Google Scholar]
- 90.Noss E, Pai R, Sellati T, Radolf J, Belisle J, et al. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J Immunol. 2001;167:910–8. doi: 10.4049/jimmunol.167.2.910. [DOI] [PubMed] [Google Scholar]
- 91.Pai R, Convery M, Hamilton T, Boom W, Harding C. Inhibition of IFN-gamma-induced class II transactivator expression by a 19-kDa lipoprotein from Mycobacterium tuberculosis: a potential mechanism for immune evasion. J Immunol. 2003;171:175–84. doi: 10.4049/jimmunol.171.1.175. [DOI] [PubMed] [Google Scholar]
- 92.Fulton S, Reba S, Pai R, Pennini M, Torres M, et al. Inhibition of major histocompatibility complex II expression and antigen processing in murine alveolar macrophages by Mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect Immun. 2004;72:2101–10. doi: 10.1128/IAI.72.4.2101-2110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pai R, Pennini M, Tobian A, Canaday D, Boom W, Harding C. Prolonged toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect Immun. 2004;72:6603–14. doi: 10.1128/IAI.72.11.6603-6614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gehring A, Rojas R, Canaday D, Lakey D, Harding C, Boom W. The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and Fc gamma R1 on human macrophages through Toll-like receptor 2. Infect Immun. 2003;71:4487–97. doi: 10.1128/IAI.71.8.4487-4497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, Curry H, Zwilling B, Lafuse W. Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J Immunol. 2005;174:5687–94. doi: 10.4049/jimmunol.174.9.5687. [DOI] [PubMed] [Google Scholar]
- 96.Fortune S, Solache A, Jaeger A, Hill P, Belisle J, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J Immunol. 2004;172:6272–80. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- 97.Banaiee N, Kincaid E, Buchwald U, Jacobs WJ, Ernst J. Potent inhibition of macrophage responses to IFN-gamma by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. J Immunol. 2006;176:3019–27. doi: 10.4049/jimmunol.176.5.3019. [DOI] [PubMed] [Google Scholar]
- 98.Kincaid EZ, Wolf AJ, Desvignes L, Mahapatra S, Crick DC, et al. Codominance of TLR2-dependent and TLR2-independent modulation of MHC class II in Mycobacterium tuberculosis infection in vivo. J Immunol. 2007;179:3187–95. doi: 10.4049/jimmunol.179.5.3187. [DOI] [PubMed] [Google Scholar]
- 99.Sköld M, Xiong X, Illarionov P, Besra G, Behar S. Interplay of cytokines and microbial signals in regulation of CD1d expression and NKT cell activation. J Immunol. 2005;175:3584–93. doi: 10.4049/jimmunol.175.6.3584. [DOI] [PubMed] [Google Scholar]
- 100.Pompei L, Jang S, Zamlynny B, Ravikumar S, McBride A, et al. Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J Immunol. 2007;178:5192–9. doi: 10.4049/jimmunol.178.8.5192. [DOI] [PubMed] [Google Scholar]
- 101.O’Garra A, Stockinger B, Veldhoen M. Differentiation of human T(H)-17 cells does require TGF-beta! Nat Immunol. 2008;9:588–90. doi: 10.1038/ni0608-588. [DOI] [PubMed] [Google Scholar]
- 102.Gerosa F, Baldani-Guerra B, Lyakh L, Batoni G, Esin S, et al. Differential regulation of interleukin 12 and interleukin 23 production in human dendritic cells. J Exp Med. 2008;205:1447–61. doi: 10.1084/jem.20071450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jo E. Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Curr Op Infect Dis. 2008;21:279–86. doi: 10.1097/QCO.0b013e3282f88b5d. [DOI] [PubMed] [Google Scholar]
- 104.Torrelles J, Azad A, Henning L, Carlson T, Schlesinger L. Role of C-type lectins in mycobacterial infections. Curr Drug Targets. 2008;9:102–12. doi: 10.2174/138945008783502467. [DOI] [PubMed] [Google Scholar]
- 105.Geijtenbeek T, Van Vliet S, Koppel E, Sanchez-Hernandez M, Vandenbroucke-Grauls C, et al. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity. 2007;26:605–16. doi: 10.1016/j.immuni.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 107.Yadav M, Schorey J. The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood. 2006;108:3168–75. doi: 10.1182/blood-2006-05-024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rothfuchs A, Bafica A, Feng C, Egen J, Williams D, et al. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol. 2007;179:3463–71. doi: 10.4049/jimmunol.179.6.3463. [DOI] [PubMed] [Google Scholar]
- 109.Jiao X, Lo-Man R, Guermonprez P, Fiette L, Deriaud E, et al. Dendritic cells are host cells for mycobacteria in vivo that trigger innate and acquired immunity. J Immunol. 2002;168:1294–301. doi: 10.4049/jimmunol.168.3.1294. [DOI] [PubMed] [Google Scholar]
- 110.Gonzalez-Juarrero M, Orme I. Characterization of murine lung dendritic cells infected with Mycobacterium tuberculosis. Infect Immun. 2001;69:1127–33. doi: 10.1128/IAI.69.2.1127-1133.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ordway D, Henao-Tamayo M, Orme I, Gonzalez-Juarrero M. Foamy macrophages within lung granulomas of mice infected with Mycobacterium tuberculosis express molecules characteristic of dendritic cells and antiapoptotic markers of the TNF receptor-associated factor family. J Immunol. 2005;175:3873–81. doi: 10.4049/jimmunol.175.6.3873. [DOI] [PubMed] [Google Scholar]
- 112.Skold M, Behar SM. Tuberculosis triggers a tissue dependent program of differentiation and acquisition of effector functions by circulating monocytes. J Immunol. 2008 doi: 10.4049/jimmunol.181.9.6349. in press. [DOI] [PubMed] [Google Scholar]
- 113.Aly S, Wagner K, Keller C, Malm S, Malzan A, et al. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J Pathol. 2006;210:298–305. doi: 10.1002/path.2055. [DOI] [PubMed] [Google Scholar]
- 114.Via L, Lin P, Ray S, Carrillo J, Allen S, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun. 2008;76:2333–40. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nobrega C, Cardona P, Roque S, Pinto do OP, Appelberg R, Correia-Neves M. The thymus as a target for mycobacterial infections. Microbes Infect. 2007;9:1521–9. doi: 10.1016/j.micinf.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 116.Gonzalez-Juarrero M, Turner O, Turner J, Marietta P, Brooks J, Orme I. Temporal and spatial arrangement of lymphocytes within lung granulomas induced by aerosol infection with Mycobacterium tuberculosis. Infect Immun. 2001;69:1722–8. doi: 10.1128/IAI.69.3.1722-1728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kahnert A, Höpken U, Stein M, Bandermann S, Lipp M, Kaufmann S. Mycobacterium tuberculosis triggers formation of lymphoid structure in murine lungs. J Inf Dis. 2007;195:46–54. doi: 10.1086/508894. [DOI] [PubMed] [Google Scholar]
- 118.Johnson CM, Cooper AM, Frank AA, Bonorino CBC, Wysoki LJ, Orme IM. Mycobacterium tuberculosis aerogenic rechallenge infections in B-cell deficient mice. Tuberc Lung Dis. 1997;78:257–61. doi: 10.1016/s0962-8479(97)90006-x. [DOI] [PubMed] [Google Scholar]
- 119.Bosio C, Gardner D, Elkins K. Infection of B cell-deficient mice with CDC 1551, a clinical isolate of Mycobacterium tuberculosis: delay in dissemination and development of lung pathology. J Immunol. 2000;164:6417–25. doi: 10.4049/jimmunol.164.12.6417. [DOI] [PubMed] [Google Scholar]
- 120.Taylor J, Ordway D, Troudt J, Gonzalez-Juarrero M, Basaraba R, Orme I. Factors associated with severe granulomatous pneumonia in Mycobacterium tuberculosis-infected mice vaccinated therapeutically with hsp65 DNA. Infect Immun. 2005;73:5189–93. doi: 10.1128/IAI.73.8.5189-5193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vordermeier HM, Venkatprasad N, Harris DP, Ivanyi J. Increase of tuberculosis infection in the organs of B-cell deficient mice. Clin Exp Immunol. 1996;106:312–6. doi: 10.1046/j.1365-2249.1996.d01-845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maglione P, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol. 2007;178:7222–34. doi: 10.4049/jimmunol.178.11.7222. [DOI] [PubMed] [Google Scholar]
- 123.Maglione P, Xu J, Casadevall A, Chan J. Fc{gamma} Receptors Regulate Immune Activation and Susceptibility during Mycobacterium tuberculosis Infection. J Immunol. 2008;180:3329–38. doi: 10.4049/jimmunol.180.5.3329. [DOI] [PubMed] [Google Scholar]
- 124.Ordway D, Palanisamy G, Henao-Tamayo M, Smith EE, Shanley C, et al. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J Immunol. 2007;179:2532–41. doi: 10.4049/jimmunol.179.4.2532. [DOI] [PubMed] [Google Scholar]
- 125.North JN. Mycobacterium tuberculosis is strikingly more virulent for mice when given via the respiratory than the intravenous route. J Inf Dis. 1995;172:1550–3. doi: 10.1093/infdis/172.6.1550. [DOI] [PubMed] [Google Scholar]
- 126.Cardona P-J, Cooper AM, Luquin M, Ariza A, Filipo F, et al. The intravenous model of murine tuberculosis is less pathogenic than the aerosol model owing to a more rapid induction of systemic immunity. Scand J Immunol. 1999;49:362–6. doi: 10.1046/j.1365-3083.1999.00522.x. [DOI] [PubMed] [Google Scholar]
- 127.Kursar M, Koch M, Mittrücker H, Nouailles G, Bonhagen K, et al. Cutting Edge: Regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol. 2007;178:2661–5. doi: 10.4049/jimmunol.178.5.2661. [DOI] [PubMed] [Google Scholar]
- 128.Scott-Browne J, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot J, et al. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med. 2007;204:2159–69. doi: 10.1084/jem.20062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Awomoyi A, Marchant A, Howson J, McAdam K, Blackwell J, Newport M. Interleukin-10, polymorphism in SLC11A1 (formerly NRAMP1), and susceptibility to tuberculosis. J Inf Dis. 2002;186:1808–14. doi: 10.1086/345920. [DOI] [PubMed] [Google Scholar]
- 130.Pacheco A, Cardoso C, Moraes M. IFNG +874T/A, IL10 -1082G/A and TNF -308G/A polymorphisms in association with tuberculosis susceptibility: a meta-analysis study. Hum Genet. 2008;123:477–84. doi: 10.1007/s00439-008-0497-5. [DOI] [PubMed] [Google Scholar]
- 131.Beamer G, Flaherty D, Assogba B, Stromberg P, Gonzalez-Juarrero M, et al. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J Immunol. 2008 doi: 10.4049/jimmunol.181.8.5545. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jung Y, Ryan L, LaCourse R, North R. Increased interleukin-10 expression is not responsible for failure of T helper 1 immunity to resolve airborne Mycobacterium tuberculosis infection in mice. Immunology. 2003;109:295–9. doi: 10.1046/j.1365-2567.2003.01645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pearl JE, Shabaana AK, Solache A, Gilmartin L, Ghilardi N, et al. IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J Immunol. 2004;173:7490–6. doi: 10.4049/jimmunol.173.12.7490. [DOI] [PubMed] [Google Scholar]
- 134.Holscher C, Holscher A, Ruckerl D, Yoshimoto T, Yoshida H, et al. The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J Immunol. 2005;174:3534–44. doi: 10.4049/jimmunol.174.6.3534. [DOI] [PubMed] [Google Scholar]
- 135.Bafica A, Scanga C, Serhan C, Machado F, White S, et al. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest. 2005;115:1601–6. doi: 10.1172/JCI23949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aliberti J, Serhan C, Sher A. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J Exp Med. 2002;196:1253–62. doi: 10.1084/jem.20021183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chakravarty SD, Xu J, Lu B, Gerard C, Flynn J, Chan J. The chemokine receptor CXCR3 attenuates the control of chronic Mycobacterium tuberculosis infection in BALB/c mice. J Immunol. 2007;178:1723–35. doi: 10.4049/jimmunol.178.3.1723. [DOI] [PubMed] [Google Scholar]
- 138.Divangahi M, Yang T, Kugathasan K, McCormick S, Takenaka S, et al. Critical negative regulation of type 1 T cell immunity and immunopathology by signaling adaptor DAP12 during intracellular infection. J Immunol. 2007;179:4015–26. doi: 10.4049/jimmunol.179.6.4015. [DOI] [PubMed] [Google Scholar]
- 139.Cooper AM, Callahan JE, Keen M, Belisle JT, Orme IM. Expression of memory immunity in the lung following re-exposure to Mycobacterium tuberculosis. Tuberc Lung Dis. 1997;78:67–73. doi: 10.1016/s0962-8479(97)90017-4. [DOI] [PubMed] [Google Scholar]
- 140.Serbina N, Flynn J. CD8(+) T cells participate in the memory immune response to Mycobacterium tuberculosis. Infect Immun. 2001;69:4320–8. doi: 10.1128/IAI.69.7.4320-4328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Goldsack L, Kirman J. Half-truths and selective memory: Interferon gamma, CD4(+) T cells and protective memory against tuberculosis. Tuberculosis. 2007;87:465–73. doi: 10.1016/j.tube.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 142.Goonetilleke N, McShane H, Hannan C, Anderson R, Brookes R, Hill A. Enhanced Immunogenicity and Protective Efficacy Against Mycobacterium tuberculosis of Bacille Calmette-Guerin Vaccine Using Mucosal Administration and Boosting with a Recombinant Modified Vaccinia Virus Ankara. J Immunol. 2003;171:1602–9. doi: 10.4049/jimmunol.171.3.1602. [DOI] [PubMed] [Google Scholar]
- 143.Santosuosso M, McCormick S, Roediger E, Zhang X, Zganiacz A, et al. Mucosal luminal manipulation of T cell geography switches on protective efficacy by otherwise ineffective parenteral genetic immunization. J Immunol. 2007;178:2387–95. doi: 10.4049/jimmunol.178.4.2387. [DOI] [PubMed] [Google Scholar]
- 144.Weir R, Gorak-Stolinska P, Floyd S, Lalor M, Stenson S, et al. Persistence of the immune response induced by BCG vaccination. BMC Infect Dis. 2008;8:9. doi: 10.1186/1471-2334-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.McShane H, Pathan A, Sander C, Keating S, Gilbert S, et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med. 2004;10:1240–4. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 146.Beveridge N, Price D, Casazza J, Pathan A, Sander C, et al. Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur J Immunol. 2007;37:3089–100. doi: 10.1002/eji.200737504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guérin mutants that secrete listeriolysin. J Clin Invest. 2005;115:24722479. doi: 10.1172/JCI24617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hinchey J, Lee S, Jeon B, Basaraba R, Venkataswamy M, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest. 2007;117:2279–88. doi: 10.1172/JCI31947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Acosta-Rodriguez E, Rivino L, Geginat J, Jarrossay D, Gattorno M, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 150.Caccamo N, Meraviglia S, La Mendola C, Guggino G, Dieli F, Salerno A. Phenotypical and functional analysis of memory and effector human CD8 T cells specific for mycobacterial antigens. J Immunol. 2006;177:1780–5. doi: 10.4049/jimmunol.177.3.1780. [DOI] [PubMed] [Google Scholar]
- 151.Millington KA, Innes JA, Hackforth S, Hinks TS, Deeks JJ, et al. Dynamic relationship between IFN-gamma and IL-2 profile of Mycobacterium tuberculosis-specific T cells and antigen load. J Immunol. 2007;178:5217–26. doi: 10.4049/jimmunol.178.8.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Murray RA, Mansoor N, Harbacheuski R, Soler J, Davids V, et al. Bacillus Calmette Guerin vaccination of human newborns induces a specific, functional CD8+ T cell response. J Immunol. 2006;177:5647–51. doi: 10.4049/jimmunol.177.8.5647. [DOI] [PubMed] [Google Scholar]
- 153.Lewinsohn D, Winata E, Swarbrick G, Tanner K, Cook M, et al. Immunodominant tuberculosis CD8 antigens preferentially restricted by HLA-B. PLoS Pathog. 2007;3:1240–9. doi: 10.1371/journal.ppat.0030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Al-Attiyah R, Mustafa A. Characterization of human cellular immune responses to novel Mycobacterium tuberculosis antigens encoded by genomic regions absent in Mycobacterium bovis BCG. Infect Immun. 2008 doi: 10.1128/IAI.00199-08. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Blythe M, Zhang Q, Vaughan K, de Castro RJ, Salimi N, et al. An analysis of the epitope knowledge related to Mycobacteria. Immunome Res. 2007;3:10. doi: 10.1186/1745-7580-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Appelberg R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing. Trends Microbiol. 2007;15:87–92. doi: 10.1016/j.tim.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 157.Silva M, Silva M, Appelberg R. Neutrophil-macrophage cooperation in the host defence against mycobacterial infections. Microb Pathogen. 1989;6:369–80. doi: 10.1016/0882-4010(89)90079-x. [DOI] [PubMed] [Google Scholar]
- 158.Tan BH, Meinken C, Bastian M, Bruns H, Legaspi A, et al. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J Immunol. 2006;177:1864–71. doi: 10.4049/jimmunol.177.3.1864. [DOI] [PubMed] [Google Scholar]
- 159.Martineau A, Newton S, Wilkinson K, Kampmann B, Hall B, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117:1988–94. doi: 10.1172/JCI31097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Garg A, Barnes PF, Porgador A, Roy S, Wu S, et al. Vimentin expressed on Mycobacterium tuberculosis-infected human monocytes is involved in binding to the NKp46 receptor. J Immunol. 2006;177:6192–8. doi: 10.4049/jimmunol.177.9.6192. [DOI] [PubMed] [Google Scholar]
- 161.Vankayalapati R, Garg A, Porgador A, Griffith D, Klucar P, et al. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol. 2005;175:4611–7. doi: 10.4049/jimmunol.175.7.4611. [DOI] [PubMed] [Google Scholar]
- 162.Vankayalapati R, Wizel B, Weis S, Safi H, Lakey D, et al. The NKp46 receptor contributes to NK cell lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol. 2002;168:3451–7. doi: 10.4049/jimmunol.168.7.3451. [DOI] [PubMed] [Google Scholar]
- 163.Roy S, Barnes P, Garg A, Wu S, Cosman D, Vankayalapati R. NK cells lyse T regulatory cells that expand in response to an intracellular pathogen. J Immunol. 2008;180:1729–36. doi: 10.4049/jimmunol.180.3.1729. [DOI] [PubMed] [Google Scholar]
- 164.Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, et al. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol. 2006;177:7086–93. doi: 10.4049/jimmunol.177.10.7086. [DOI] [PubMed] [Google Scholar]
- 165.Fleischmann R, Alland D, Eisen J, Carpenter L, White O, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–90. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Glickman M, Cox J, Jacobs W. A novel mycolic acid synthetase is required for cording, persistence and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–27. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 167.Rao V, Fujiwara N, Porcelli S, Glickman M. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. 2005;201:535–43. doi: 10.1084/jem.20041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rao V, Gao F, Chen B, Jacobs WJ, Glickman M. Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis -induced inflammation and virulence. J Clin Invest. 2006;116:1660–7. doi: 10.1172/JCI27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dao D, Sweeney K, Hsu T, Gurcha S, Nascimento I, et al. Mycolic acid modification by the mmaA4 gene of M. tuberculosis modulates IL-12 production. PLoS Pathog. 2008;4:e1000081. doi: 10.1371/journal.ppat.1000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kremer K, Glynn J, Lillebaek T, Niemann S, Kurepina N, et al. Definition of the Beijing/W lineage of Mycobacterium tuberculosis on the basis of genetic markers. J Clin Microbiol. 2004;42:4040–9. doi: 10.1128/JCM.42.9.4040-4049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Reed M, Domenech P, Manca C, Su H, Barczak A, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–7. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 172.Tsenova L, Ellison E, Harbacheuski R, Moreira A, Kurepina N, et al. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Inf Dis. 2005;192:98–106. doi: 10.1086/430614. [DOI] [PubMed] [Google Scholar]
- 173.Sinsimer D, Huet G, Manca C, Tsenova L, Koo M, et al. The phenolic glycolipid of Mycobacterium tuberculosis differentially modulates the early host cytokine response but does not in itself confer hypervirulence. Infect Immun. 2008;76:3027–36. doi: 10.1128/IAI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol. 2007;179:522–31. doi: 10.4049/jimmunol.179.1.522. [DOI] [PubMed] [Google Scholar]