

Abstract
Group B coxsackieviruses (CVBs) are involved in triggering some cases of type 1 diabetes mellitus (T1DM). However, the molecular mechanism(s) responsible for this remain elusive. Toll-like receptor 3 (TLR3), a receptor that recognizes viral double-stranded RNA, is hypothesized to play a role in virus-induced T1DM, although this hypothesis is yet to be substantiated. The objective of this study was to directly investigate the role of TLR3 in CVB-triggered T1DM in nonobese diabetic (NOD) mice, a mouse model of human T1DM that is widely used to study both spontaneous autoimmune and viral-induced T1DM. As such, we infected female wild-type (TLR3+/+) and TLR3 knockout (TLR3−/−) NOD mice with CVB4 and compared the incidence of diabetes in CVB4-infected mice with that of uninfected counterparts. We also evaluated the islets of uninfected and CVB4-infected wild-type and TLR3 knockout NOD mice by immunohistochemistry and insulitis scoring. TLR3 knockout mice were markedly protected from CVB4-induced diabetes compared with CVB4-infected wild-type mice. CVB4-induced T-lymphocyte-mediated insulitis was also significantly less severe in TLR3 knockout mice compared with wild-type mice. No differences in insulitis were observed between uninfected animals, either wild-type or TLR3 knockout mice. These data demonstrate for the first time that TLR3 is 1) critical for CVB4-induced T1DM, and 2) modulates CVB4-induced insulitis in genetically prone NOD mice.
Type 1 diabetes mellitus (T1DM) is a disorder of glucose metabolism characterized by destruction of insulin secreting β-cells of the pancreas and is triggered by both genetic and environmental factors (eg, viruses) (1–29). Considerable evidence suggests that coxsackieviruses (group B coxsackieviruses [CVBs]), in particular, are involved in triggering T1DM in a certain subset of genetically susceptible individuals and animals (22–29), although the molecular mechanism(s) responsible for CVB induction of T1DM in these at risk individuals remains largely unidentified.
Certain members of a conserved pattern recognition receptor family, the toll-like receptors (TLRs), mediate, in part, the innate immune response to viral infections (30). Upon binding to their respective ligands, these receptors activate signaling cascades that up-regulate expression of costimulatory and adhesion molecules, chemokines, proinflammatory cytokines, and their receptors (31). TLRs also bridge the innate and adaptive immune responses by facilitating the maturation of antigen presenting cells, stimulating antibody production in B cells, and down-regulating the activity of regulatory T cells (31, 32). TLR3 is the only TLR that recognizes both viral double-stranded RNA (dsRNA) and polyinosinic-polycytidylic acid (pIC) (synthetic dsRNA that mimics host responses triggered by viral dsRNA) (30). Although TLR3 expression has primarily been characterized in leukocytes (33–36), it has also been detected and shown to be functional, in various nonimmune cells (37–42), including pancreatic β-cells (43, 44). TLR3 has also been implicated in the pathogenesis of a number of autoimmune diseases, including T1DM (43, 45–48), multiple sclerosis (reviewed in Ref. 49), and systemic lupus erythematosus (50, 51). Recent studies have suggested a possible role for TLR3 in viral-induced T1DM. However, studies linking viruses to T1DM were either associative in nature, performed in cell culture, or used synthetic dsRNA to trigger TLR3 signaling in mouse models with no known genetic risk for developing T1DM (43, 45–48). For example, 2 of these studies demonstrated that pIC stimulates activation of autoreactive T cells, TLR3 signaling in pancreatic β-cells, β-cell destruction, and subsequent diabetes in 2 different mouse models of T1DM (45, 47). Although these data suggested a role for TLR3 in the development of T1DM, it was insufficient to validate this claim, primarily because the possible contributions by other existing pathways (retinoic acid-inducible gene 1, melanoma differentiation-associated protein 5, protein kinase R) that are stimulated by pIC to this process were not examined (45, 47). Further, these studies did not address the role that TLR3 plays in environmental induction of T1DM by enterovirus infection in genetically susceptible animals, a model which more closely resembles the human condition.
The nonobese diabetic (NOD) mouse is a widely used model of T1DM, because there is a spontaneous autoimmune component that can be accelerated via environmental induction (7, 25, 26, 28, 52). Previous studies show that CVBs (ie, CVB3 and CVB4) can trigger T1DM in NOD mice once an age-dependent critical mass of spontaneous insulitis occurs (25, 27, 28), and these studies have implicated TLR3 in this process (53). However, in a study that compared the incidence of spontaneous diabetes in TLR3-deficient NOD mice in a sterile environment, there was no difference in the incidence of spontaneous diabetes between TLR3−/−, TLR3+/−, and TLR3+/+ NOD mice despite the reported impairment of pIC-induced immune responses in the TLR3-deficient mice (32). This suggested TLR3 is not the only requirement for spontaneous autoimmune diabetes in NOD mice (53). However, neither this study nor previous pIC studies addressed the role that TLR3 possibly plays in the environmental induction of T1DM by enterovirus infection in these genetically susceptible NOD mice. This report provides evidence that TLR3 is important in CVB4-triggered T1DM in NOD mice and offers the first mechanistic insight into its role in this process.
Materials and Methods
Mice, virus, and blood glucose
This work was conducted with approval from the Ohio University Institutional Animal Care and Use Committee in accord with accepted standards of humane animal care.
Eight-week-old female wild-type NOD (The Jackson Laboratory) and TLR3 knockout NOD (53) mice were infected by ip injection with 5 × 105 plaque forming units of CVB4 (kindly provided by Dr Roger Loria, Virginia Commonwealth University) (52). Eight-week-old NOD mice were used in these studies, because the preexisting critical mass of autoreactive T cells in pancreatic islets (ie, critical threshold level of insulitis) is known to be present in NOD mice by 8 weeks of age, and it has been previously shown that inoculation of NOD mice with coxsackieviruses at any age more than or equal to 8 weeks of age rapidly accelerates the onset of T1DM (25–28, 54).
The wild-type NOD and TLR3 knockout NOD mice differ only at the TLR3 locus. TLR3−/− B6 mice were backcrossed onto the NOD genetic background for over 10 generations. The purity of the NOD genetic background was examined by PCR-based microsatellite analysis with all known idd markers. There is no known diabetes susceptibility locus on mouse chromosome 8, where the TLR3 gene is located.
Blood glucose measurements were nonfasting and were consistently measured at the same time of day (ie, early morning ∼8 am). Diabetes was defined as blood glucose values more than 240 mg/dL on consecutive days and confirmed by glucosuria (Diastix, Bayer). Mice were housed in a sterile/pathogen-free facility. Weight and blood glucose were measured weekly.
The insulitis scoring, assessment of pancreatic viral titers, and histological assessments were conducted as blind studies to prevent bias. To accomplish this, all samples were randomly assigned numbers, and their identities were revealed only after the data were obtained.
Pancreatic viral titers
Whole pancreata were removed from euthanized mice, weighed, placed in sterile PBS, homogenized, frozen and thawed 3 times, and then clarified using low-speed centrifugation. Ten-fold serial dilutions of cleared lysates were prepared in PBS from pancreas of animals euthanized 3 and 4 days after infection. Two hundred-microliter aliquots were used to infect confluent HeLa cell monolayers. An overlay of Eagle's MEM (Gibco) containing 1% methylcellulose was added to monolayer and the plates were incubated for 96 hours at 37°C. After 4 days, the overlay was removed and cells were fixed with formaldehyde and stained with crystal violet. Plaques were counted and expressed as the (number of plaque forming units per mL/g of tissue)/dilution factor.
Histology/immunohistochemistry (IHC)
Histological analyses presented in Figure 3 were performed on pancreata frozen in optimal cutting temperature medium (Tissue-Tek). Frozen pancreata were sliced to 10 μm and fixed with 95% ethanol at −20°C for 15 seconds and air dried. The sections were immunostained using a rabbit polyclonal antibody against insulin (Abcam) or a rat monoclonal antibody against cluster of differentiation (CD) 4 (clone RM4–5; BD Biosciences) or CD8 (clone YTS169.4; Abcam). A rabbit polyclonal IgG and rat monoclonal IgG (Abcam) were used as isotype controls and only the rat control is presented in Figure 3. Insulin was visualized using the EXPOSE rabbit-specific horseradish peroxidase/3,3′-diaminobenzidine kit from Abcam. CD4 and CD8 immuno-staining was visualized by treating tissue sections after primary-antibody incubation with ImmPRESS Anti-Rat Ig, Mouse adsorbed (peroxidase) Polymer Detection kit (Vector Laboratories) followed by treatment with 3,3′-diaminobenzidine substrate/chromogen reagent from the EXPOSE kit (Supplemental Table 1). Sections were also counterstained with hematoxylin. Two separate individuals on independent occasions conducted analyses of histological sections, and their assessments were in agreement with one another as reported here.
Figure 3.
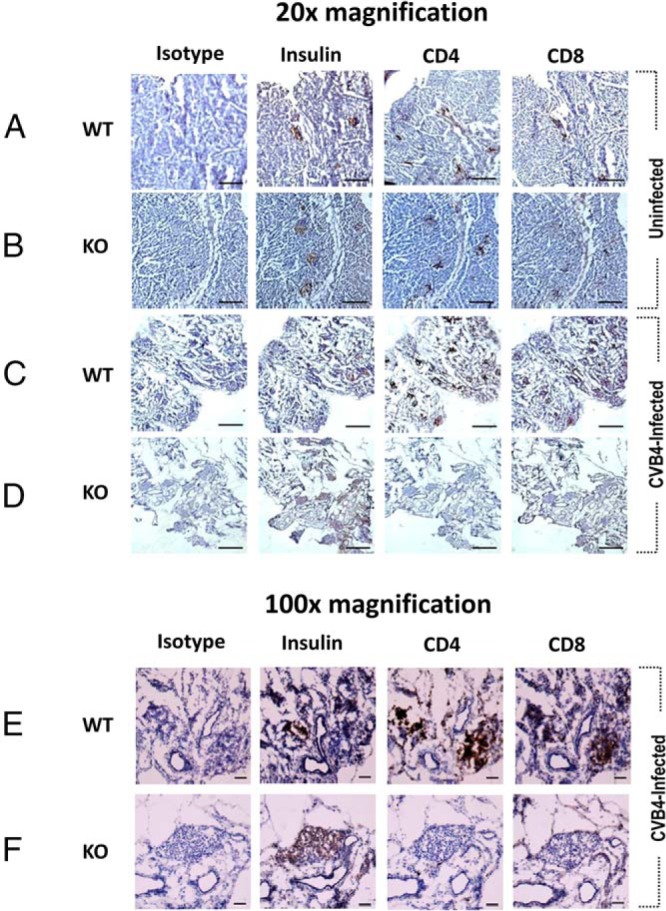
TLR3 is critical for CVB4-induced T cell-mediated insulitis in female NOD mice. Evaluation of T cell infiltration of pancreatic islets in 10-week-old uninfected and CVB4-infected female wild-type (WT) and TLR3 knockout (KO) NOD mice. Serial sections of pancreas stained for insulin and the T cell markers CD4 and CD8 revealed that T cells were abundant in CVB4-infected wild-type NOD islets but scarce in CVB4-infected TLR3 KO NOD islets. No differences in T cell infiltration of islets of uninfected wild-type and TLR3 KO NOD mice were observed. At least 4 mice per group were analyzed. Pictures were taken at ×20 (A–D) or ×100 (E and F) magnification. Scale bars in A–D indicate 500 μm, whereas scale bars in E and F indicate 50 μm. Images in this figure show isolated representative data on sections rather than summary data of all of the mice.
Insulitis scoring
Pancreata from 10-week-old mice were collected, fixed in formalin, embedded in paraffin, and sectioned. Sections of hematoxylin and eosin stained, and insulin-immunostained pancreas were used for insulitis scoring. Using ImagePro Software (Mediacybernetics), islets were photographed, and total islet area, as well as immune cell-infiltrated islet area, was measured (μm2). Percent infiltration was then calculated for each islet by dividing area of islet infiltration by total islet area. Based on percent infiltration, each islet was then assigned an insulitis score as follows: no insulitis (0% infiltration) = 0, low grade (1%–33% infiltration) = 1, moderate (34%–66% infiltration) = 2, and severe (>66% infiltration) = 3. Average insulitis scores/group were then calculated and compared. This work was conducted by a single observer.
Statistical analysis
Statistical significance was determined using a log-rank test for data in Figure 1, independent t tests were used for data in Figure 2, and a one-way ANOVA followed by a Tukey-Kramer post hoc analysis was used for data in Figure 4.
Figure 1.
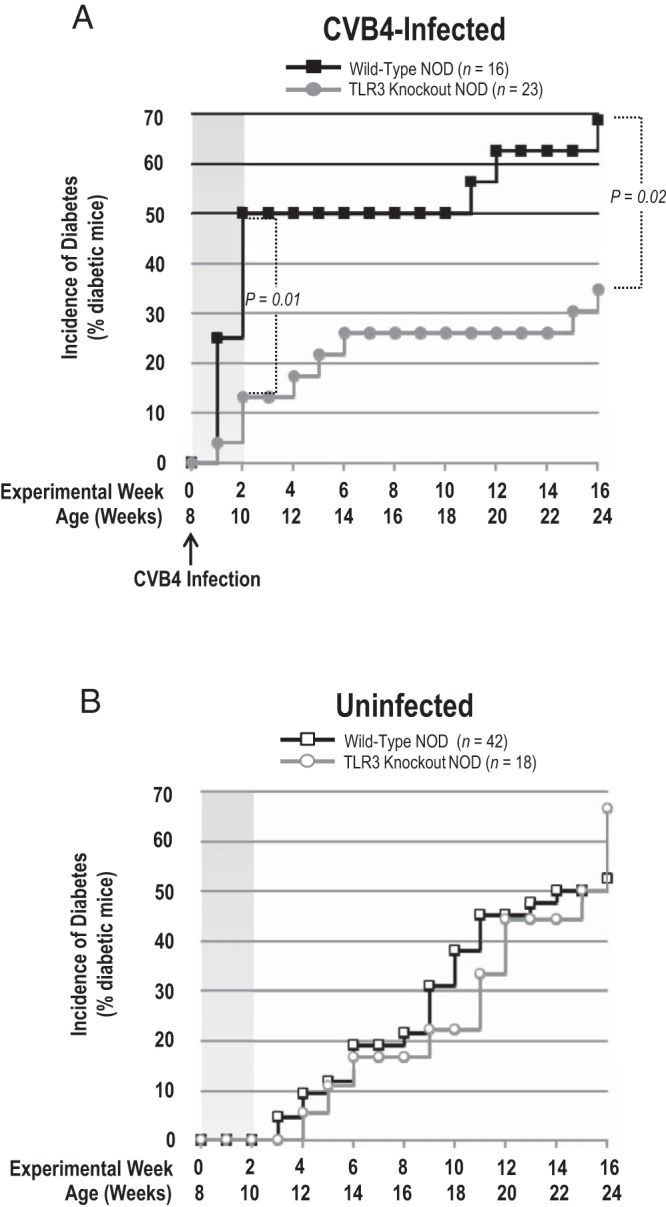
TLR3 is important for CVB4 induction of T1DM in female NOD mice. A, Incidence of diabetes in CVB4-infected wild-type and TLR3 knockout NOD mice. TLR3 knockout NOD mice were protected from CVB4 induction of T1DM. B, Incidence of diabetes in uninfected wild-type and TLR3 knockout NOD mice. There was no difference in the incidence of spontaneous autoimmune T1DM between TLR3 knockout and wild-type NOD mice. Graphs depict the percentage of mice in each group with diabetes. Statistical significance was determined using a log-rank test. Bars indicate significant differences between groups with P = .01 and P = .02, as indicated.
Figure 2.

CVB4 is present in the pancreas of CVB4-infected female wild-type and TLR3 knockout NOD mice. Pancreas was collected from euthanized mice at days 3 and 4 after CVB4 infection, and virus titers were determined. TLR3 knockout NOD mice (n = 3) had significantly (P = .01) more virus than wild-type NOD counterparts (n = 2) at 3 days after infection. This difference was gone by 4 days after infection (n = 2, for each group). Statistical significance was determined using an independent t test. Bar indicates statistical significance between groups with P = .01, as indicated.
Figure 4.

TLR3 is important for CVB4-induced insulitis in female NOD mice. A, Insulitis scoring of pancreatic islets from uninfected and CVB4-infected wild-type (WT) and TLR3 knockout (KO) NOD mice. CVB4-infected TLR3 knockout NOD mice had significantly less severe insulitis than CVB4-infected wild-type NOD mice. Statistical significance was determined using a one-way ANOVA followed by a Tukey-Kramer post hoc analysis. Each bar indicates statistical significance between groups with P < .01. B, Percentages of islets with no insulitis, low-grade insulitis, moderate insulitis, and severe insulitis for each experimental group.
Notes on experiments
Multiple studies were conducted to obtain the data presented in this manuscript. First, a large cohort of wild-type NOD mice were purchased from The Jackson Laboratory, randomly divided into appropriate groups, and a single experiment was carried out over 1 16-week period with these wild-type NOD mice. To obtain similar data with the TLR3 knockout NOD mice, the TLR3 knockout NOD mice were bred and raised in a breeding colony at Ohio University and were used in experiments that were staggered as mice became available for use (ie, 8 weeks of age). For these 16-week studies, all animals included in the studies were either followed through until diabetes development (at which time they were euthanized) or were euthanized at 24 weeks of age (ie, experimental termination). Pancreas was collected from any mouse that became diabetic at 10 weeks of age (euthanized at that time) and was used for the insulitis scoring. In a separate experiment, a second cohort of wild-type NOD mice (whose numbers were not included in the survival cohort represented in Figure 1) was obtained from The Jackson Laboratory and used in a separate 2-week study to obtain pancreatic tissue for histological analyses of T-lymphocytes in 10-week-old mice. Similarly, TLR3 knockout NOD mice were used in a separate set of staggered experiments to obtain pancreatic tissue for histological analyses of T-lymphocytes at the same age. Although separate studies were conducted for the reasons stated, the incidence of diabetes that developed in each group at 10 weeks of age was consistent with that reported here (ie, we observed consistency in incidence of diabetes between experiments). A third study was conducted to obtain pancreata from virus-infected wild-type and TLR3 knockout NOD mice for evaluation of viral titers.
Results
Diabetes development in female uninfected and CVB4-infected wild-type and TLR3 knockout NOD mice
Eight-week-old female wild-type (TLR3+/+) and TLR3 knockout (TLR3−/−) NOD mice were either infected with CVB4 (to study virus-triggered diabetes) or left uninfected (to study spontaneous diabetes), and the incidence of diabetes was assessed. Infection of 8-week-old wild-type NOD mice resulted in a rapid induction of diabetes within the first 2-weeks postvirus infection. Fifty percent of wild-type NOD mice infected with CVB4 were diabetic by 2-weeks postvirus infection, whereas only 13% of infected TLR3 knockout NOD mice were diabetic by this time (Figure 1A). None of the uninfected wild-type or TLR3 knockout NOD mice had developed diabetes by 10 weeks of age (Figure 1B). After the initial CVB4-induced spike in incidence of diabetes within the first 2 weeks, the incidence of diabetes remained at 50% for 8 weeks in wild-type NOD mice, and then at 19 weeks of age the incidence of diabetes began to slowly rise again (Figure 1A). In contrast, the incidence of diabetes postinfection in TLR3 knockout NOD mice continued to rise slowly for an additional 4 weeks, also reaching an 8-week plateau at 6 weeks after infection (14 wks of age) with a CVB4-induced incidence of diabetes of 26% at this time (Figure 1A). At 15 weeks after infection (ie, 22 wks of age), the incidence of diabetes began to slowly rise again, at a rate similar to that seen in the wild-type NOD mice (Figure 1A). By the end of the 16-week experiment, the incidence of diabetes in CVB4-infected TLR3 knockout NOD mice remained significantly lower than CVB4-infected wild-type NOD mice (35% vs 69%) (Figure 1A). The natural onset of diabetes and total incidence of diabetes between the uninfected wild-type NOD and TLR3 knockout NOD mice was not statistically different throughout the duration of the experiments (Figure 1B). Interestingly, at the conclusion of the study (16 wks), there was approximately 15% more CVB4-infected wild-type NOD mice that developed diabetes compared with uninfected wild-type controls overall, and there was an apparent decrease in the percentage of CVB4-infected TLR3 knockout NOD mice developing diabetes (∼30%) compared with uninfected control mice (∼50%). However, these differences were not statistically significant (Figure 1).
CVB4 titers in the pancreas of female wild-type and TLR3 knockout NOD mice during the first 14 days after CVB4 infection
To confirm the presence of CVB4 infection of the pancreas, the pancreas was collected from euthanized wild-type and TLR3 knockout NOD mice up to 6 days after CVB4 infection, and virus titers were determined using plaque assays. TLR3 knockout NOD mice had significantly (P = .01) higher pancreatic CVB4 titers than wild-type NOD counterparts at 3 days after infection (Figure 2). However, these differences were not evident by 4 days after infection, when overall CVB4 titers were negligible (Figure 2). CVB4 was undetectable by day 6 after CVB4 infection (data not shown), which is consistent with previous findings (26).
Insulitis in female uninfected and CVB4-infected wild-type and TLR3 knockout NOD mice
Because a critical threshold, defined by age, and the level of T-lymphocyte infiltration of pancreatic islets (insulitis) are critical for both spontaneous and CVB-induced T1DM in NOD mice (6, 7, 25–28), differences in the severity of insulitis in 10-week old uninfected and CVB4-infected wild-type and TLR3 knockout NOD mice were evaluated using IHC. Serial sections of pancreas from mice in all 4 groups were stained for the T-lymphocyte markers CD4 and CD8 and for insulin to identify functional islets, and the T-lymphocyte markers CD4 and CD8. No differences in T-lymphocyte infiltration of islets were apparent in the uninfected wild-type or TLR3 knockout NOD mice (Figure 3, rows A and B, respectively). However, it was evident that CVB4-infected wild-type NOD islets contained more T-lymphocytes (both CD4+ and CD8+) than islets from CVB4-infected TLR3 knockout NOD mice (Figure 3, C and D and E and F). It was also apparent that islets from CVB4-infected wild-type NOD mice contained more CD4+ and CD8+ T-lymphocytes than islets from age-matched uninfected wild-type and TLR3 knockout NOD mice (Figure 3). As expected, there was also an absence of insulin staining in regions of islets where β-cells had been destroyed and replaced by T-lymphocytes (Figure 3, C and E). Moreover, CVB4 infection also caused widespread exocrine pancreatic damage (ie, pancreatitis) in both wild-type and TLR3 knockout NOD mice (Figure 3, compare A and B with C and D).
To quantify islet lymphocytic infiltration in each of these different groups, insulitis scoring was performed. Although there was significant insulitis observed in all groups, there were no significant differences in insulitis scoring between uninfected wild-type and uninfected TLR3 knockout NOD mice (Figure 4A). However, insulitis scoring in the CVB4-infected wild-type NOD mice demonstrated severe lymphocytic infiltration of islets compared with uninfected wild-type and uninfected TLR3 knockout NOD mice (Figure 4A). Moreover, CVB4-infected wild-type NOD mice demonstrated severe insulitis compared with CVB4-infected TLR3 knockout NOD mice (Figure 4A), confirming the IHC findings noted in Figure 3. The percentage of islets with no insulitis, low-grade insulitis, moderate insulitis, and severe insulitis for each experimental group is represented in Figure 4B.
Discussion
The purpose of these studies was to assess the role of TLR3 in CVB4-induced T1DM in genetically susceptible NOD mice. These data confirm recent findings that TLR3 is not critical for the spontaneous, genetically determined portion of autoimmune diabetes observed in female NOD mice (53), but do demonstrate that TLR3 is critical for CVB4 (environmental) induction of T1DM. This is confirmed by the data that shows that CVB4 infection of TLR3 knockout NOD mice exhibited a significantly lower incidence of diabetes compared with wild-type CVB4-infected NOD mice.
Although the acceleration of T1DM by CVB4 was significantly diminished in the TLR3 knockout compared with wild-type NOD mice, there was a plateau in diabetes development that occurred after the first 2-week period of acceleration, and then there was a subsequent rise in diabetes development that occurred in both wild-type and TLR3 knockout NOD mice by the end of the experiment (11 and 15 wks after infection, respectively). The acceleration of T1DM within the first 2 weeks after infection reflects the acute inflammatory effects of CVB4 infection on the pancreas; the severity was more pronounced in the wild-type NOD mice. The plateau in prevalence of diabetes that follows the 2-week acceleration represents possible recovery from the acute viral insult. The acute inflammation and incidence of T1DM was much lower in the TLR3 knockout NOD mice, reflecting the role of TLR3 in the innate immune response to CVB4 and its role in accelerating the rate of the virus-triggered T1DM. We believe the subsequent rise in T1DM in both wild-type and TLR3 knockout NOD mice after the plateau (Figure 1A) reflects the natural progression of the genetically determined portion of β-cell destruction in NOD animals, which is suggested by the similar slopes.
Female TLR3 knockout NOD mice had significantly (∼5-fold) higher viral titers than female wild-type NOD mice 3 days after CVB4 infection despite being infected with the same amount of CVB4 at 8 weeks of age. Although the virus was nearly completely cleared by both groups by 4 days after CVB4 infection, the exocrine pancreatitis was markedly less, and the incidence of diabetes was significantly less than that seen in wild-type mice. The higher viral titers seen in whole pancreas at day 3 after infection suggests increased susceptibility to viral infection or enhanced viral replication as a result of a delay in the innate immune response in the TLR3 knockout NOD mice due to the lack of a major viral recognition pathway rather than differences in viral clearance (55). CVB4 infection of pancreatic islets was not specifically evaluated in these studies, but this has been previously reported (6). Despite approximately 5-fold higher pancreatic CVB4 viral titers, female TLR3 knockout NOD mice were significantly protected from CVB4-triggered T1DM compared with female wild-type NOD mice. Together, these findings suggest that TLR3 signaling pathways are critical mediators of CVB4-triggered T1DM in NOD mice.
Despite the rapid acceleration of diabetes in CVB4-infected wild-type NOD mice, there was only an approximately 15% overall increase in the percentage of mice that developed diabetes compared with noninfected mice at the end of the study period. CVB4-infected TLR3 knockout NOD mice also demonstrated an acceleration of onset of diabetes at 10 weeks. However, despite this acceleration, there was an overall decrease in the percentage of CVB4-infected TLR3 knockout NOD mice developing diabetes (∼30%) relative to uninfected controls at termination of this study. Neither of these differences in T1DM prevalence rates at the end of the study was statistically significant. However, it suggests a possible protective effect. Regardless of the lack of statistical significance for the decrease in overall conversion of CVB4-infected TLR3 knockout mice to T1DM, the data point to a protective role for the combination of TLR3 knockout and CVB4 infection. It has been previously hypothesized that immunity to viral infection might be under the control of regulatory mechanisms that would confer protection from T1DM, in the absence of extensive damage to β-cells (56). Specifically, it has been shown that when NOD mice are infected with strains of viruses that prevent rather than trigger T1DM in NOD mice, 2 distinct control mechanisms are induced during immunity to these viruses and that they operate synergistically to efficiently block the autoimmune process leading to T1DM (56). These 2 mechanisms are up-regulation of programmed cell death-1 ligand 1 and enhancement of CD4+CD25+Foxp3+ T-regs (ie, “protective” T-regulatory lymphocytes that function to suppress CD8+ effector T-cell proliferation) that produce TGF-β (56). Thus, the protective effect of the combination of TLR3 knockout and CVB4 infection observed in this study might be explained by an up-regulation of programmed cell death-1 ligand 1 and/or enhancement of CD4+CD25+Foxp3+ T-regs. This explanation is at least partially supported by the observation that CVB4-infected TLR3 knockout NOD mice in this study had significantly less severe insulitis at 2 weeks after CVB4 infection, which indicates that there is indeed less β-cell damage occurring in the pancreas of CVB4-infected TLR3 knockout NOD mice. It is unclear at this time whether there are a larger proportion of CD4+CD25+Foxp3+ T-regs in the pancreas of the CVB4-infected TLR3 knockout NOD mice. However, this will be one of the foci of our future studies.
The presence of both CD8+ and CD4+ T-lymphocytes in the islets of NOD mice has been well documented (57–61), and it is known that both CD4+ and CD8+ T-lymphocytes play a critical role in the development of autoimmune diabetes in NOD mice (reviewed in Ref. 58). IHC staining of pancreas from 10-week-old CVB4-infected wild-type and TLR3 knockout NOD mice with antibodies specific for the T-lymphocyte markers CD4 and CD8, as well as insulin, revealed that CVB4-infected TLR3 knockout NOD mice had fewer T-lymphocytes infiltrating pancreatic islets than similarly infected wild-type NOD mice. This observation was confirmed by insulitis scoring. These findings suggest that TLR3 is important in CVB4-induced insulitis in NOD mice. In support of this notion is the observation that there were a greater number of insulin-producing β-cells remaining in the islets of CVB4-infected TLR3 knockout mice compared with age-matched CVB4-infected wild-type mice that coincided with fewer T-lymphocytes in TLR3 knockout mice. Thus, the decreased incidence of T1DM in the TLR3 knockout NOD mice may well be due to the decreased number of T-lymphocytes migrating into islets after viral infection where their presence is correlated with β-cell destruction. The fact that CVB4-induced T-lymphocyte invasion of the pancreatic islets is largely absent in TLR3-knockout NOD mice also raises new questions and sets the groundwork for future studies aimed at determining exactly how TLR3 is involved in this process. For example, although it is not yet clear why there are fewer T-lymphocytes infiltrating islets in TLR3 knockout NOD mice, a dramatic shift in the important cytokine/chemokine profile (eg, cytokines such as interferon γ and IL-4) (26) in the pancreatic microenvironment in the absence of TLR3 signaling may be responsible for these observations. It will be important in future studies to evaluate the pancreatic cytokine/chemokine profile and to characterize the T-helper lymphocyte population in TLR3 knockout NOD mice to evaluate the presence or absence of T-regs (ie, Foxp3+/CD4+/CD25+ cells) (62–64), because these correlations may be of significance.
In agreement with previous reports, we also observed wide-spread pancreatitis after CVB4 infection in female wild-type NOD mice (25, 26). However, this is the first report demonstrating that there is a similar CVB4-induced pancreatitis in TLR3 knockout NOD mice. It has been suggested that the inflammatory mediators produced in response to the CVB4 infection of the exocrine pancreas may provide an “innocent by-stander activation” effect that accelerates diabetes development in NOD mice if infection occurs after a time when a critical threshold level of insulitis exists and adequate numbers of β-cell autoreactive T-lymphocytes are present (26). The fact that TLR3 knockout NOD mice have similar levels of pancreatitis as that observed in wild-type NOD mice, yet the acceleration of T1DM by CVB4 was significantly diminished in the TLR3 knockout compared with wild-type NOD mice, suggests that TLR3 may be involved in the innocent by-stander activation effect that accelerates diabetes development in NOD mice. Otherwise stated, these findings suggest that CVB4 infection of the exocrine pancreas of wild-type NOD mice, and subsequent activation of TLR3 signaling, may enhance T-lymphocyte-mediated insulitis, β-cell destruction, and onset of diabetes. Although the exact nature of how TLR3 signaling is involved in these processes is not yet known, these findings lay the foundation for future studies to more definitively delineate the role of TLR3 in the pathogenesis of viral-induced T1DM.
Together, these data indicate that TLR3 is an important mediator of the CVB4 induction of T1DM observed in the NOD mouse model and provides new evidence for its role in the environmental induction/acceleration of this process. This conclusion is further supported by our previous report that a novel TLR3 pathway inhibitor developed in our laboratory (phenylmethimazole or compound 10) can also block CVB4-triggered T1DM in female NOD mice but has no effect on the genetically determined portion of T1DM pathogenesis in NOD mice (65). Although these studies have established the importance of TLR3 in viral induction of T1DM in NOD mice, because of the limitations of our data, the exact role of TLR3 in mediating the immune response leading to diabetes in CVB4-infected NOD mice is not yet known, and as a consequence, it is not clear if this is a direct affect or a result of changes in pathways distinct from TLR3 that are secondarily modified during the development of the mice with chronic TLR3 deficiency. As such, our future studies will be aimed at identifying the exact role(s) that TLR3 plays in this process.
These findings suggest that CVB4 activation of TLR3 signaling may contribute to the viral induction of T1DM reported in humans and that inhibition of the TLR3 signaling pathways may be a useful target for the prevention of T1DM in subsets of individuals at risk for viral-induced T1DM. Furthermore, inhibition of TLR3 signaling pathways could suppress islet inflammation and help protect and/or salvage β-cells in patients with recent onset virus-related T1DM. Some of the major hurdles to overcome for this type of TLR-specific therapy will be to identify those genotypes at risk for the viral induction of T1DM and also be able to detect viral-induced onset of disease early enough to intervene. However, we have identified at least 1 of the critical molecular pathways that mediate viral induction of T1DM in genetically susceptible animals.
Acknowledgments
We thank Dr Roger Loria (Virginia Commonwealth University) for providing the CVB4 for these studies. We also thank Aili Guo, Christopher Schwartz, Eric Dickerson, Andrew Henson, Tara Edwards, Kiet Ma, Arlesia Glaspy, Kevin Albert, Alex Campolo, Sarah Metro, and Jarrod Uhrig for technical support and/or helpful discussions on this project and Dr David Serreze, Dr Mark Atkinson, and the late Dr Leonard Kohn for their discussions and expertise provided on the NOD mouse model.
This work was supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases Grant 1R15 DK081192-01 (to K.D.M.), the J.O. Watson Endowment for Diabetes Research, and the Ohio University Heritage College of Osteopathic Medicine.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CD
- cluster of differentiation
- CVB
- group B coxsackievirus
- dsRNA
- double-stranded RNA
- IHC
- immunohistochemistry
- NOD
- nonobese diabetic
- pIC
- polyinosinic-polycytidylic acid
- T1DM
- type 1 diabetes mellitus
- TLR
- toll-like receptor.
References
- 1. Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. [DOI] [PubMed] [Google Scholar]
- 2. Bach JF. Predictive medicine in autoimmune diseases: from the identification of genetic predisposition and environmental influence to precocious immunotherapy. Clin Immunol Immunopathol. 1994;72:156–161. [DOI] [PubMed] [Google Scholar]
- 3. Gale EA. The rise of childhood type 1 diabetes in the 20th century. Diabetes. 2002;51:3353–3361. [DOI] [PubMed] [Google Scholar]
- 4. Kaprio J, Tuomilehto J, Koskenvuo M, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992;35:1060–1067. [DOI] [PubMed] [Google Scholar]
- 5. Banatvala JE, Bryant J, Schernthaner G, et al. Coxsackie B, mumps, rubella, and cytomegalovirus specific IgM responses in patients with juvenile-onset insulin-dependent diabetes mellitus in Britain, Austria, and Australia. Lancet. 1985;1:1409–1412. [DOI] [PubMed] [Google Scholar]
- 6. Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N. Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of β cells. Clin Immunol. 2004;110:134–144. [DOI] [PubMed] [Google Scholar]
- 7. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781–785. [DOI] [PubMed] [Google Scholar]
- 8. Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179. [DOI] [PubMed] [Google Scholar]
- 9. Yoon JW, Jun HS. Viruses cause type 1 diabetes in animals. Ann NY Acad Sci. 2006;1079:138–146. [DOI] [PubMed] [Google Scholar]
- 10. Rewers M, Zimmet P. The rising tide of childhood type 1 diabetes–what is the elusive environmental trigger? Lancet. 2004;364:1645–1647. [DOI] [PubMed] [Google Scholar]
- 11. von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Microbiology. 2003;1:151–157. [DOI] [PubMed] [Google Scholar]
- 12. Lammi N, Karvonen M, Tuomilehto J. Do microbes have a causal role in type 1 diabetes? Med Sci Monit. 2005;11:RA63–RA69. [PubMed] [Google Scholar]
- 13. Filippi C, von Herrath M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol. 2005;233:125–132. [DOI] [PubMed] [Google Scholar]
- 14. Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2003;19:8–31. [DOI] [PubMed] [Google Scholar]
- 15. Hindersson M, Maria H, Elshebani A, et al. Simultaneous type 1 diabetes onset in mother and son coincident with an enteroviral infection. J Clin Virol. 2005;33:158–167. [DOI] [PubMed] [Google Scholar]
- 16. Mäkelä M, Vaarala O, Hermann R, et al. Enteral virus infections in early childhood and an enhanced type 1 diabetes-associated antibody response to dietary insulin. J Autoimmun. 2006;27:54–61. [DOI] [PubMed] [Google Scholar]
- 17. Jaïdane H, Hober D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008;34:537–548. [DOI] [PubMed] [Google Scholar]
- 18. Helfand RF, Gary HE, Jr, Freeman CY, Anderson LJ, Pallansch MA. Serologic evidence of an association between enteroviruses and the onset of type 1 diabetes mellitus. Pittsburgh Diabetes Research Group. J Infect Dis. 1995;172:1206–1211. [DOI] [PubMed] [Google Scholar]
- 19. Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stene LC, Oikarinen S, Hyöty H, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes. 2010;59:3174–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oikarinen S, Martiskainen M, Tauriainen S, et al. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes. 2011;60:276–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. King ML, Shaikh A, Bidwell D, Voller A, Banatvala JE. Coxsackie-B-virus-specific IgM responses in children with insulin-dependent (juvenile-onset; type I) diabetes mellitus. Lancet. 1983;1:1397–1399. [DOI] [PubMed] [Google Scholar]
- 23. Friman G, Fohlman J, Frisk G, et al. An incidence peak of juvenile diabetes. Relation to Coxsackie B virus immune response. Acta Paediatr Scand Suppl. 1985;320:14–19. [DOI] [PubMed] [Google Scholar]
- 24. Ylipaasto P, Klingel K, Lindberg AM, et al. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet β cells. Diabetologia. 2004;47:225–239. [DOI] [PubMed] [Google Scholar]
- 25. Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes. 2000;49:708–711. [DOI] [PubMed] [Google Scholar]
- 26. Serreze DV, Wasserfall C, Ottendorfer EW, et al. Diabetes acceleration or prevention by a coxsackievirus B4 infection: critical requirements for both interleukin-4 and γ interferon. J Virol. 2005;79:1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horwitz MS, Fine C, Ilic A, Sarvetnick N. Requirements for viral-mediated autoimmune diabetes: β-cell damage and immune infiltration. J Autoimmun. 2001;16:211–217. [DOI] [PubMed] [Google Scholar]
- 28. Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology. 2004;329:381–394. [DOI] [PubMed] [Google Scholar]
- 29. Laitinen OH, Honkanen H, Pakkanen O, et al. Coxsackievirus B1 is associated with induction of β-cell autoimmunity that portends type 1 diabetes. Diabetes. 2014;63:446–455. [DOI] [PubMed] [Google Scholar]
- 30. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature. 2001;413:732–738. [DOI] [PubMed] [Google Scholar]
- 31. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. [DOI] [PubMed] [Google Scholar]
- 32. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. [DOI] [PubMed] [Google Scholar]
- 33. Muzio M, Bosisio D, Polentarutti N, et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol. 2000;164:5998–6004. [DOI] [PubMed] [Google Scholar]
- 34. Heinz S, Haehnel V, Karaghiosoff M, et al. Species-specific regulation of toll-like receptor 3 genes in men and mice. J Biol Chem. 2003;278:21502–21509. [DOI] [PubMed] [Google Scholar]
- 35. Wesch D, Beetz S, Oberg HH, Marget M, Krengel K, Kabelitz D. Direct costimulatory effect of TLR3 ligand poly(I:C) on human γ δ T lymphocytes. J Immunol. 2006;176:1348–1354. [DOI] [PubMed] [Google Scholar]
- 36. Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood. 2005;106:978–987. [DOI] [PubMed] [Google Scholar]
- 37. McCall KD, Harii N, Lewis CJ, et al. High basal levels of functional toll-like receptor 3 (TLR3) and noncannonical Wnt5a are expressed in papillary thyroid cancer (PTC) and are coordinately decreased by phenylmethimazole together with cell proliferation and migration. Endocrinology. 2007;148:4226–4237. [DOI] [PubMed] [Google Scholar]
- 38. Harii N, Lewis CJ, Vasko V, et al. Thyrocytes express a functional toll-like receptor 3(TLR3): overexpression can be induced by viral infection, reversed by phenylmethimazole and is associated with Hashimoto's autoimmune thyroiditis. Mol Endocrinol. 2005;19:1231–1250. [DOI] [PubMed] [Google Scholar]
- 39. Yang X, Murthy V, Schultz K, Tatro JB, Fitzgerald KA, Beasley D. Toll-like receptor 3 signaling evokes a proinflammatory and proliferative phenotype in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;291:H2334–H2343. [DOI] [PubMed] [Google Scholar]
- 40. Ciencewicki J, Brighton L, Wu WD, Madden M, Jaspers I. Diesel exhaust enhances virus- and poly(I:C)-induced toll-like receptor 3 expression and signaling in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1154–L1163. [DOI] [PubMed] [Google Scholar]
- 41. Lafon M, Megret F, Lafage M, Prehaud C. The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J Mol Neurosci. 2006;29:185–194. [DOI] [PubMed] [Google Scholar]
- 42. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. [DOI] [PubMed] [Google Scholar]
- 43. Rasschaert J, Ladrière L, Urbain M, et al. Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-γ-induced apoptosis in primary pancreatic β-cells. J Biol Chem. 2005;280:33984–33991. [DOI] [PubMed] [Google Scholar]
- 44. Giarratana N, Penna G, Amuchastegui S, Mariani R, Daniel KC, Adorini L. A vitamin D analog down-regulates proinflammatory chemokine production by pancreatic islets inhibiting T cell recruitment and type 1 diabetes development. J Immunol. 2004;173:2280–2287. [DOI] [PubMed] [Google Scholar]
- 45. Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of toll-like receptors on pancreatic islets. J Immunol. 2004;172:3173–3180. [DOI] [PubMed] [Google Scholar]
- 46. Devendra D, Eisenbarth GS. Interferon α–a potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clin Immunol. 2004;111:225–233. [DOI] [PubMed] [Google Scholar]
- 47. Lang KS, Recher M, Junt T, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–145. [DOI] [PubMed] [Google Scholar]
- 48. Shibasaki S, Imagawa A, Tauriainen S, et al. Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr J. 2010;57:211–219. [DOI] [PubMed] [Google Scholar]
- 49. Miranda-Hernandez S, Baxter AG. Role of toll-like receptors in multiple sclerosis. Am J Clin Exp Immunol. 2013;2:75–93. [PMC free article] [PubMed] [Google Scholar]
- 50. Laska MJ, Troldborg A, Hansen B, et al. Polymorphisms within toll-like receptors are associated with systemic lupus erythematosus in a cohort of Danish females. Rheumatology. 2014;53:48–55. [DOI] [PubMed] [Google Scholar]
- 51. Midgley A, Thorbinson C, Beresford MW. Expression of toll-like receptors and their detection of nuclear self-antigen leading to immune activation in JSLE. Rheumatology. 2012;51:824–832. [DOI] [PubMed] [Google Scholar]
- 52. Webb SR, Loria RM, Madge GE, Kibrick S. Susceptibility of mice to group B coxsackie virus is influenced by the diabetic gene. J Exp Med. 1976;143:1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wong FS, Hu C, Zhang L, et al. The role of toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann NY Acad Sci. 2008;1150:146–148. [DOI] [PubMed] [Google Scholar]
- 54. Kanno T, Kim K, Kono K, Drescher KM, Chapman NM, Tracy S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J Virol. 2006;80:5637–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oshiumi H, Okamoto M, Fujii K, et al. The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J Immunol. 2011;187:5320–5327. [DOI] [PubMed] [Google Scholar]
- 56. Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest. 2009;119:1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thivolet C, Bendelac A, Bedossa P, Bach JF, Carnaud C. CD8+ T cell homing to the pancreas in the nonobese diabetic mouse is CD4+ T cell-dependent. J Immunol. 1991;146:85–88. [PubMed] [Google Scholar]
- 58. Yoon JW, Jun HS. Autoimmune destruction of pancreatic β cells. Am J Ther. 2005;12:580–591. [DOI] [PubMed] [Google Scholar]
- 59. Yagi H, Matsumoto M, Kunimoto K, Kawaguchi J, Makino S, Harada M. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur J Immunol. 1992;22:2387–2393. [DOI] [PubMed] [Google Scholar]
- 60. Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. [DOI] [PubMed] [Google Scholar]
- 61. Nagata M, Yoon JW. Studies on autoimmunity for T-cell-mediated β-cell destruction. Distinct difference in β-cell destruction between CD4+ and CD8+ T-cell clones derived from lymphocytes infiltrating the islets of NOD mice. Diabetes. 1992;41:998–1008. [DOI] [PubMed] [Google Scholar]
- 62. Chen W, Wahl SM. TGF-β: the missing link in CD4+CD25+ regulatory T cell-mediated immunosuppression. Cytokine Growth Factor Rev. 2003;14:85–89. [DOI] [PubMed] [Google Scholar]
- 63. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor β. J Exp Med. 2001;194:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. You S, Belghith M, Cobbold S, et al. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes. 2005;54:1415–1422. [DOI] [PubMed] [Google Scholar]
- 65. McCall KD, Schmerr MJ, Thuma JR, et al. Phenylmethimazole suppresses dsRNA-induced cytotoxicity and inflammatory cytokines in murine pancreatic β cells and blocks viral acceleration of type 1 diabetes in NOD mice. Molecules. 2013;18:3841–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]