

Abstract
Hydrogels are proving to be an excellent class of materials for biomedical applications. The molecular self-assembly of designed MAX1 β-hairpin peptides into fibrillar networks has emerged as a novel route to form responsive hydrogels. Herein, computational modeling techniques are used to investigate the relative arrangements of individual hairpins within the fibrils that constitute the gel. The modeling provides insight into the morphology of the fibril network, which defines the gel’s mechanical properties. Our study suggests polymorphic arrangements of the hairpins within the fibrils; however, the relative populations and the relative conformational energies of the polymorphic arrangements show a preference toward an arrangement of hairpins where their turn regions are not capable of forming intermolecular interaction. Repulsive intramolecular electrostatic interactions appear to dictate the formation of fibrils with shorter, rather than longer, persistent lengths. These repulsive intramolecular interactions also disfavor the formation of fibril entanglements. Taken together, the modeling predicts that MAX1 forms a network containing a large number of branch points, a network morphology supported by the formation of short fibril segments. We posit that, under static conditions, the preferred branched structures of the MAX1 peptide assembly result in a cross-linked hydrogel organization. At the same time, the shear stress leads to short fibrillar structures, thus fluidic hydrogel states.
Introduction
Hydrogel materials are used in tissue engineering,1−3 microfluidics,4,5 and drug delivery.6−8 A promising approach for the design of hydrogels is the use of self-assembling peptides in which noncovalent interactions between molecules drive the assembly and formation of supramolecular fibril networks that define the material.9,10 Using peptides as building blocks, one can form self-assembled structures that result in a percolated hydrogel network. Schneider and co-workers11−18 have designed a class of peptides that undergo triggered folding into facially amphiphilic β-hairpins that subsequently self-assemble to form self-supporting, rigid hydrogels. Recently, the kinetic and mechanical rigidity of amphiphilic β-hairpins were investigated by Chen et al.19 MAX1 is a 20-residue peptide with the sequence VKVKVKVKVDPPTKVKVKVKV–NH2. It forms two β-strands consisting of alternating hydrophobic valine and hydrophilic lysine residues connected by a tetrapeptide sequence (−VDPPT−) designed to adopt a type II′ turn structure. In its folded state, hairpin amphiphilicity is manifested in the composition of its faces: one hydrophobic valines, the other hydrophilic lysine residues (Figure 1A). Importantly, this amphiphilic folded state is highly prone to self-assembly, forming a network of β-sheet-rich fibrils. It is known that temperature, pH, and ionic strength trigger the self-assembly of MAX1 peptides. Previously, it was shown that MAX1 self-assembles under physiological conditions (pH 7.4 and a temperature of 37°).20 Small angle neutron scattering (SANS) and transmission electron microscopy (TEM) studies demonstrated that the hairpin self-assembles laterally, forming a network of intermolecular hydrogen bonds that define the long axis of a given fibril. All the β-strands of the assembled hairpins are in register, affording fibrils with distinct diameters of ∼3 nm.11,20 MAX1 facial self-assembly can also take place by burial of the valine-rich face to form a bilayer that defines the thickness of a given fibril. Along the long axis of a given fibril, bilayer formation occurs regularly with one hairpin docked and in register with its partner, an arrangement that shields the maximum surface area of the valine side chains from water (Figure 1B). It was suggested that imperfections in this mechanism can occur where the face of one hairpin is rotated relative to that of its bilayer partner, leading to nascent fibril growth in three dimensions, forming an interfibril cross-link (branch point). These branch points are noncovalent physical cross-links that define the mechanical rigidity of the gel.13 Although light scattering and microscopy studies clarified the network and the local fibril morphology, a detailed molecular-level model of how the peptides arrange themselves within the fibrils remained elusive. Herein, we constructed variant models of MAX1 peptide arrangements and investigated the stability of each variant model. Three sets of models were examined. The first models the β-hairpin arrangement along the long axis of fibrils constituting the network. The second models the β-hairpin arrangement within the branch point regions of the network. The third arrangement is an amyloid-like cross-β structure, i.e., a β-arch structure (Figure 1C and D).
Figure 1.

Building blocks of the MAX1 hydrogel are composed of β-hairpin structures, not by an amyloid-like cross-β arch. (A) A folded β-hairpin structure monomer of MAX1 peptide which presents intramolecular hydrogen bonds. (B) Two folded β-hairpin structures of MAX1 peptide forming bilayer with a hydrophobic surface of valine residues. (C) A proposed model for a folded amyloid-like β-arch monomer of MAX1 peptide which does not present intramolecular hydrogen bonds. (D) Two folded β-arch structures of MAX1 peptide are assembled into an amyloid-like cross-β structure with intermolecular hydrogen bonds. (E) A simulated self-assembled amyloid-like β-arch structure presents an unstable cross-β structure. (F and G) A simulated self-assembled β-hairpin structure illustrates a structurally stable fibrillar state. (F) A view along the fibril axis and (G) a view from the side of the fibril. In parts A–C, valine and lysine residues are colored as blue and red, respectively, and serine residues are colored green.
Through extensive simulations, we found that the β-hairpins exhibit polymorphic packing along the fibril long axis and in the branch points. Previously, it was hypothesized that the cross-linking phenomenon in MAX1 hydrogel is due to fibril imperfections. Our simulations show that β-hairpin arrangements of branching structures are preferred over β-hairpin arrangements of long fibril structures. Therefore, the hairpins are more favored to form branch points than to pack along the long fibril axis. We thus conclude that short persistence lengths of the self-assembled hairpins exist within the gel. In addition, all possible arrangements illustrate that the type II′ turns that directly define the secondary structure of the β-hairpins do not form intermolecular interactions between the β-hairpins in the bilayer fibrils and thus do not contribute to the network morphology.
Materials and Methods
MD simulations of the solvated variant models M1–M8 and other tested models were performed in the NPT ensemble using the NAMD program21 with the CHARMM27 force field22,23 for 60 ns. The models were explicitly solvated with TIP3P water molecules.24,25 The Langevin piston method21,26,27 with a decay period of 100 fs and a damping time of 50 fs was used to maintain a constant pressure of 1 atm. The temperature (330 K) was controlled by a Langevin thermostat with a damping coefficient of 10 ps–1.21 The short-range van der Waals (VDW) interactions were calculated using the switching function, with a twin range cutoff of 10.0 and 12.0 Å. Long-range electrostatic interactions were calculated using the particle mesh Ewald method with a cutoff of 12.0 Å for all simulations.28,29 The equations of motion were integrated using the leapfrog integrator with a step of 2 fs. All initial variant models were energy minimized and then solvated in a TIP3P water box with a minimum distance of 15 Å from any edge of the box to any β-hairpin atom. Any water molecule within 2.5 Å of the β-hairpin was removed. Counterions were added at random locations to neutralize the β-hairpins’ charge.
The solvated systems were energy minimized for 2000 conjugated gradient steps, where the distance between the β-sheets in the β-hairpins is fixed in the range 2.2–2.5 Å. The counterions and water molecules were allowed to move. The hydrogen atoms were constrained to the equilibrium bond using the SHAKE algorithm.30 The minimized solvated systems were heated to 200 K, where all atoms were allowed to move. Then, the systems were heated from 200 to 250 K for 300 ps and equilibrated at 330 K for 300 ps. All simulations ran for 60 ns, and structures were saved every 10 ps for analysis. These conditions (330 K and 60 ns of time scales) are applied to test the stabilities of all variant models. All simulations were performed at physiological pH. We applied a higher temperature than the physiological temperature (310 K), aiming to investigate the stability of the variant models. The self-assembly of MAX1 peptides is triggered by the higher temperature; therefore, the choice of 330 K for MD simulations is a reasonable choice.
To obtain the relative structural stability of the variant models, the trajectories during the last 5 ns were first extracted from the explicit MD simulation excluding water molecules. The solvation energies of all systems were calculated using the generalized Born method with molecular volume (GBMV).31,32 In the GBMV calculations, the dielectric constant of water was set to 80.0. The solvent-accessible surface area (SASA) term factor was set to 0.00592 kcal/mol·Å2. Each variant is minimized 1000 cycles, and the conformation energy is evaluated by grid-based GBMV. The minimization does not change the conformations of each variant but only relaxes the local geometries due to thermal fluctuation which occur during the MD simulations. A total of 4000 conformations (500 conformations for each of the 8 examined conformers) were used to construct the free energy landscape of the conformers and to evaluate the conformer probabilities by Monte Carlo (MC) simulations.
Results and Discussion
Polymorphic Packing of β-Hairpin Bilayer Fibril and Cross-Linking Structures
We based our constructed models on the overall structure that had been proposed previously by Schneider and co-workers: the MAX1 peptide is composed of two β-strands of alternating lysine and valine residues, connected by a four-residue type II′ β-turn.11,12,20 The model of MAX1 peptide based on the β-arch structure of Aβ33 was examined (Figure 1C and D) by applying MD simulations to the self-assembled peptides. As one can see from Figure 1E, after simulations of 26 ns, the self-assembled β-arch structure MAX1 is unstable due to the loss of hydrogen bond interactions along the fibril axis (Figure 1E). We therefore conclude that the folded state of MAX1 peptide is not a β-arch structure.
Previously, it was suggested that the MAX1 peptide self-assembles to form fibrils that cross-link through branch points.34 Each fibril is composed of a bilayer of hairpins that hydrogen-bond along the long axis of a given fibril (Figure 1B). We investigated four possible arrangements of the β-hairpins constituting the fibril: variants M1–M4 (Figure 2A). They differ in the arrangement and orientation of the turn regions. In models M1 and M2, the turn regions in each layer are organized in the same direction. In the bilayer of M1, the turn regions are organized in the same direction, while in M2 they are organized in opposite directions along the fibril axis. In models M3 and M4, the turn regions are organized in opposite directions in each layer. In the bilayer of M3, the turn regions are organized in the same direction, and in M4, the turn regions are organized in opposite directions along the fibril axis. We further examined two possible arrangements in the branching point region: variants M5 and M7 and variants M6 and M8 (Figure 2B). In models M5 and M7, the turn regions are organized in the same direction in each layer. In models M6 and M8, the turn regions are organized in opposite directions in each layer. While in M5 and M6 each of the opposing monolayers illustrate that the hydrophobic residues are exposed to the solvent, in M7 and M8, the opposing monolayers demonstrate that the hydrophobic residues are not exposed to the solvent. Each model of the fibril long axis comprises 16 β-hairpins. The number of possible organizations at the branching points is large, because the range of angles (0–90°) at which two layers in the self-assembled peptide can be oriented toward each other is large. We considered a single angle, 90°, between the two layers.
Figure 2.

Illustration of the variant models of the arrangements of the MAX1 hairpins in the gel network morphology. The secondary structure of the hairpins is based on the experimentally designed peptide.11,12,20 The variant models M1–M4 illustrate arrangement along the peptide fibril growth and in variant models M5–M8 show arrangement in the cross-link branch point regions.
After 60 ns simulations, all eight self-assembled hairpin models illustrated structurally stable fibrillar cross-β structures. Interestingly, all M1–M4 fibrillar structural models presented small RMSDs (Figure 3). While the fibrillar structural models M1, M3, and M4 are relatively rigid, the fibrillar model M2 is slightly flexible. The branching models M5–M8 are relatively less rigid than the fibrillate models M1–M4. Interestingly, models M5 and M6, that illustrate that the hydrophobic residues are exposed to the solvent, showed relatively large RMSD values (∼2 Å) compared with the RMSD values of models M7 and M8 (∼1.2 Å) where the hydrophobic residues are not exposed to the solvent. The RMSD values of 2 Å are in the range observed in protein structures. In summary, we propose that the arrangements in branching point regions in which the hydrophobic residues are not exposed to the solvent are relatively flexible compared to the fibrillar models and present higher rigidity than the arrangements in the branching point region in which the hydrophobic residues are exposed to the solvent.
Figure 3.
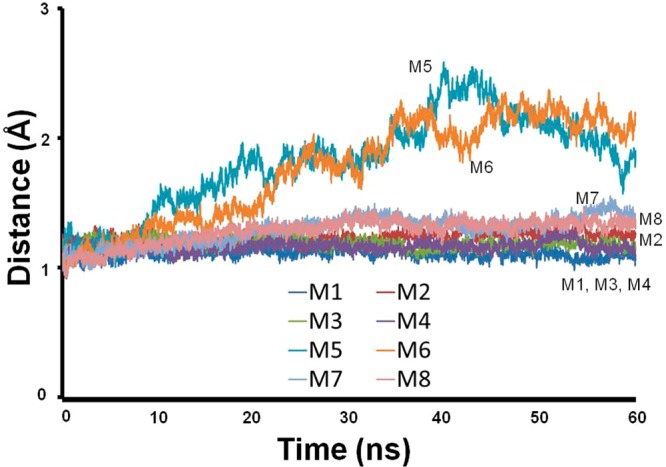
RMSD values along the MD simulations indicating that the fibrillar models are relatively more rigid than the branching models.
The high structural stabilities of the fibrillar models M1–M4 are due to the well protected backbone hydrogen bond networks that are shielded from the solvent. The lysine-rich surface in the fibrillar models is highly charged, while the hydrocarbon side chain of the lysine residues provides a hydrophobic environment for the backbone hydrogen bond networks. To examine this claim, we computed the solvation backbone values for all 20 residues within the β-hairpin for each of the fibrillar models M1–M4 and the branching models M5–M8 (Figure 4). The backbone solvation values for each residue are similar for all eight models, with slight deviations for models M6 and M7, which illustrate hydrophobic residues that are exposed to the solvent. As one can see from Figure 4, as expected among all 20 residues in the β-hairpin, the residues in the C- and N-termini Val1 and Val20 and the residues in the turn regions Pro11 illustrate relatively high solvation values. Additionally, one can see an alternative pattern of backbone solvation along the two β-strands of the hairpin in line with the alternating sequence of valine and lysine residues. Valine residues illustrate relatively small backbone solvation values compared to the lysine residues. At the same time, the backbone solvation values for both valines and lysines are relatively small, indicating the well protected backbone hydrogen bond networks that are shielded from the solvation for all models.
Figure 4.

Average water molecules around each side chain Cβ carbon and backbone atoms (within 4 Å) for models M1–M8.
Table 1 summarizes the conformational energies of the eight variants of the self-assembled β-hairpins computed by the generalized Born method with the molecular volume (GBMV) method and the populations using Monte Carlo (MC) simulations. Among the four possible arrangements of the β-hairpins constituting the fibril, variants M1–M4, M2 and M4 have the highest and similar energies and the highest population and M2 is slightly more stable energetically than M4 and dramatically more stable than the other models M1 and M3. Among the possible M5–M8 arrangements in the branching point region, variants M6 and M8 are dramatically more favored energetically than M5 and M7.
Table 1. Conformational Energies and the Populations of the Simulated Variant Models of the Hairpin MAX1a.
| variant | energy (kcal/mol) | energy differenceb (kcal/mol) | populations (%) |
|---|---|---|---|
| M1 | –5799.3 (±221.2) | 139.3 (±13.1) | 10.2 |
| M2 | –5876.2 (±239.6) | 62.4 (±13.8) | 13.6 |
| M3 | –5793.1 (±237.4) | 145.3 (±13.7) | 10.1 |
| M4 | –5829.0 (±216.8) | 109.6 (±13.0) | 11.2 |
| M5 | –5845.1 (±237.9) | 93.5 (±13.7) | 12.0 |
| M6 | –5938.6 (±193.7) | 0.0 | 15.7 |
| M7 | –5836.0 (±252.0) | 102.6 (±14.2) | 12.1 |
| M8 | –5911.4 (±227.6) | 27.2 (±13.4) | 15.1 |
Conformational energies were computed using the GBMV calculations (refs (31 and 32)). Standard deviation values are presented in parentheses.
The standard deviation of the energy difference is calculated by σx1–x2 = sqrt[σ12/n1 + σ22/n2], where x1 and x2 are the means of two samplings and n1 and n2 are the sizes of the sampling (500).
We therefore suggest that these constructed variant models may provide insight into the mechanisms of the self-assembly of the β-hairpins within the hydrogel network’s morphology. β-hairpins self-assemble to form polymorphic arrangements with a preference toward arrangement along the fibril axis and a preference toward arrangement within the branching point regions. While along the fibril axis the preferred self-assembling occurs when the turn regions oriented in the same directions in each layer and in opposite directions in the bilayer (e.g., model M2), within the branching point regions, preferred self-assembling occurs when the turn regions are organized in opposite directions in each layer (e.g., models M6 and M8).
Short Persistence Lengths of Hairpins Are Favored along the Bilayer Fibril Axis
The persistence lengths of the self-assembled fibril of MAX1 hydrogels were previously estimated from rheological experiments to be 55 nm.13 The interactions that control the self-assembly along the fibril bilayer include electrostatic and hydrophobic interactions, and hydrogen bonding between individual β-hairpins. Electrostatic interactions have also been found to influence the persistence length.
Electrostatic frustration may affect the organization and persistence length of the assembly of the peptide. To test the effect of electrostatic frustration on the persistence length along the fibril growth of the 16 β-hairpins, pairs of β-hairpins were removed from the most stable model M2, that is, 14 β-hairpins and a dimer of β-hairpins. The total energies of M2 and its small β-hairpin pair “fragments” were computed with the GBMV method. Figure 5 demonstrates the total energies of M2 and its “fragment” pairs. Energetically, short pairs of the self-assembled hairpins are more favored than the longest self-assembled hairpin, indicating that electrostatic repulsion might overcome hydrogen bonding and hydrophobic interactions permitting further fibril elongation. The energy gap between the 16 β-hairpins M2 and its pairs of “fragments” reveals that the preferred size is between 6 and 10 hairpins (Figure 5C). Figure 5B illustrates a similar scenario; the sum of the conformational energies of three pairs of M2 “fragments” is less than the conformational energy of the 16. Therefore, the electrostatic frustration disfavors long-axis fibril growth and it is more likely that short persistence lengths are formed, suggesting that the large numbers of cross-links in the network morphology is due to the preference of short persistence lengths. Therefore, the shorter persistence lengths may contribute to a stiffer gel material, since the fibrillar structural models are relatively more rigid (Figure 3).
Figure 5.

Sum of energies of self-assembled short persistence lengths and the energy of model M2 that consists of 16 monomers. Parts A and B demonstrate that short persistence lengths along the fibril’s growth are energetically more favored than the longer fibril (consisting of 16 monomers). As seen in part B, the most favored short persistence lengths for model M2 are octamers and two tetramers. (C) The energy gap between the self-assembled model M2 and the self-assembled short persistence length. The energy gap increases with the formation of shorter fragments.
Electrostatic Repulsion of Bilayer Fibrils in the Network Morphology Disfavors the Formation of Entanglements
In addition to the bilayer and the interfibril cross-linking, i.e., the branch points within the network, it has also been suggested that, at a sufficiently high hairpin concentration, entanglements of the fibrils may also contribute to the mechanical rigidity of the gel.11,13,20,35,36 Our simulations demonstrate that the short persistence length of the fibrils and interfibril cross-linking strongly contribute to the mechanical rigidity of the fibril. To test the hypothesis that the entanglements of the fibrils also contribute to the mechanical rigidity of the gel, we simulated two self-assembled fibrils that are in close and far proximity from each other (Figure 6A). To this end, we applied the most stable variant, M2. The simulations illustrate that electrostatic repulsions of the lysines between the two fibrils prevent the formation of fibril entanglements. The strong electrostatic repulsions lead to the “breaking” of one fibril, reflecting that fibril entanglements cannot be achieved when two lysine-rich surfaces are in contact (Figure 6B). Since it has been proposed that the formation of the entanglements of the fibrils may occur at high concentrations of salt ions, we increased the concentrations of the counterions in the simulations while keeping the system neutralized. Interestingly, the simulations at high counterion concentrations illustrated a similar scenario of electrostatic repulsions of the lysines that prevent the formation of the fibril entanglements. We conclude that the electrostatic repulsions of the lysines within the fibrils disfavor the formation of entanglements and therefore help to define the percolated nature of the network. In the network morphology, while the interfibril cross-linking contributes to the material’s mechanical rigidity, it is unlikely that the fibrils form entanglements. This conclusion and our results that (1) MAX1 prefers to have short fibrils and (2) branched models are more stable than a long MAX1 fibril point to a cross-linking mechanism for MAX1 hydrogel (Figure 6C). We suggest that the MAX1 hydrogel cross-linking is not caused by “imperfection” or entanglements of MAX1 fibril; rather, the cross-linking state is one of the low energy and stable polymorphic states of MAX1 peptide ensembles. Our mechanism is consistent with several experimental observations.17,19 MAX1 hydrogel can become fluidic under shear stress and immediately resume its gel state when the shear stress is relieved. In our mechanism, shear stress provides the energy to reach the available short fibrillar structures and thus yield a fluidic hydrogel. When the shear energy disappears, the cross-linking state again becomes more populated. The entanglement mechanism can explain the fast restoration of the gel state immediately after the shear stress stops. Our mechanism does not require converting all branching states to short fibrillar structures; it suffices to provide enough energy to break a large gel domain into a smaller one. Consistently, experimental works have shown that random orientation of fibrillar structures does not change under shear stress.17 Our mechanism also implies that stronger hydrophobic interactions can increase the hydrogel mechanical stiffness, since the cross-linking is still provided by hydrophobic surfaces rather than entanglement through lysine-rich surface contact. In support of this, recent experiments have shown that, after the valine residues of MAX1 were replaced by the more hydrophobic isoleucines, the resulting gels displayed higher mechanical stiffness.19
Figure 6.

A proposed cross-link mechanism of MAX1 peptide hydrogel. (A) Initial structure of two MAX1 fibrils that are contacted with 90° between them. (B) Simulated structure of the two contacted MAX1 fibrils demonstrating that the strong electrostatic repulsion between the lysine-rich surfaces does not allow two stable MAX1 fibrils to form an entangled complex. (C) Shear forces provide energy to flow and therefore yield for structural transitions. Under static conditions, the branching structures of the MAX1 peptide assembly are more favored and thus provide cross-linking of the hydrogel. However, the shear stress provides energy flow, leading to short fibrillar structures and therefore yields to a fluidic hydrogel.
Conclusions
MAX1 undergoes triggered self-assembly at the nanoscale to form a physically cross-linked network of fibrils with a defined cross section.11−13,20 The folded state of MAX1 is characterized by a type II′ β-turn (−VDPPT−), which connects two amphiphilic β-strands of alternating lysine and valine residues, resulting in a hairpin with one hydrophilic, lysine-rich face and one hydrophobic, valine-rich face.11 This conformation rapidly self-assembles following an external stimulus, yielding a mechanically rigid hydrogel. How the hairpins self-assemble and how the branch points are formed are elusive. To understand the structural features that contribute to the network’s morphology and the material’s mechanical rigidity of the gel, it is necessary to investigate the structure of the network morphology at the molecular level. Recently, the morphologies of the self-assembly of amphiphilic β-sheet peptides into various organizations have been investigated at high resolution.37
Herein, using all-atom molecular dynamics (MD) simulations in explicit solvent, we investigated the network morphology of the hairpins in the fibril’s growth domains and at the branch points. The stable variant models of the self-assembled β-hairpins were based on the secondary structure which has been designed by Schneider and co-workers.11−15 Overall, our study provides insight into the molecular network morphology of the hairpins in the gel state. The preferred conformation of the MAX1 peptide is the β-hairpin structure, rather than the amyloid-like β-arch structure. It is interesting to note that there is similar temperature sensitivity between MAX1 peptide gel formation and some amyloid formations. Because hydrophobic interactions have higher contributions at higher temperatures,12 they can trigger the formation of the MAX1 hydrogel. Similarly, tau protein amyloids also form at high temperature which can be reversed to small oligomers at low temperature.38 However, there is an important difference between the temperature sensitivities of the monomer conformation of MAX1 peptide and tau protein. With increasing temperature, the MAX1 peptide monomer changes from a random conformation into a β-hairpin. On the other hand, tau peptides K19 and K18 present metastable secondary structure states39 (both β-strand and α-helix) at room temperature, and the β-strand percentage decreases with increasing temperature. For amyloids, the rate limiting step of self-assembly depends on the nucleus size.40 Thus, the preferred hydrophobic interactions at higher temperatures accelerate the nucleus formation of tau oligomer. While it is possible that formation of the MAX1 hydrogel is also limited by the nucleus size, the monomeric folding into the β-hairpin conformation could be a prerequisite for the formation of the MAX1 hydrogel nucleus.
Several important observations emerged from our study. First, simulations of the hairpins exhibit polymorphic packing of hairpins along the long fibrillar axis and within the branch point regions of the network. Our computations suggest a slight preference for turn regions of neighboring hairpins in the bilayer to be organized in opposing directions, as demonstrated by the variant model M2. Second, short persistence lengths along the fibril growth indicate that packing of hairpins within the branch points is preferred over packing along the fibril long axis. Electrostatic frustration disfavors long-axis fibril growth; in the network morphology, it is thus more likely that short persistence lengths are formed. These observations indicate that both the short persistence and the large number of cross-linked branch points may be largely responsible for contributing to the material’s mechanical rigidity.13,41 A network morphology that is comprised of a large number of branch points and fibrils of shorter persistence lengths leads to a stiffer material. Our results indicate that small sizes of self-assembled peptides, i.e., short persistence lengths along the fibril growth, are energetically preferred over longer fibril sizes. This indicates that the rate limiting step for formation of the hydrogel depends on the nucleus size. Finally, electrostatic repulsions between fibrils due to the hydrophilic lysine-rich face disfavor the formation of fibril entanglements and consequently assist in defining the percolated nature of the network morphology.
In summary, we confirmed that the building block of MAX1 hydrogel is a β-hairpin structure, not an amyloid-like β-arch structure. The self-assembled β-hairpin structure of MAX1 illustrates a well-packed cross-β structure. Recent MD simulations have demonstrated cross-β structures in a small collection of peptide molecules.42,43 Our MD simulations and energy estimation presented a polymorphic energy landscape of the MAX1 peptide assembly, clarifying the mechanism of structural transition in the hydrogel (Figure 6C). It was hypothesized that the MAX1 hydrogel cross-linking is due to imperfections of fibrils. Herein, we examined this hypothesis and found that branching structures are more stable than long fibrillar structures. Finally, under static conditions, the preferred branching structures of the MAX1 peptide assembly provide the cross-linking of the hydrogel. However, shear stress provides the energy to convert some branching states into short fibrillar structures, thereby breaking large gel domains into smaller ones and yielding a fluidic hydrogel.
Acknowledgments
We thank Yoav Atsmon-Raz for the RMSD analysis for all models. All simulations have been performed using the high-performance computational facilities of the Miller lab in the BGU HPC computational center and the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). The support of the BGU HPC computational center staff is greatly acknowledged. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E and by the Israel Binational Science Foundation Grant No. 2011128. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Supporting Information Available
The atomic coordinates of models M1–M8 (files jp511485n_si_002.pdb through jp511485n_si_009.pdb). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rajagopal K.; Schneider J. P. Self-Assembling Peptides and Proteins for Nanotechnological Applications. Curr. Opin. Struct. Biol. 2004, 14, 480–486. [DOI] [PubMed] [Google Scholar]
- Lee K. Y.; Mooney D. J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 1869–1879. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Hubbell J. A. Synthetic Biomaterials as Instructive Extracellular Microenvironments for Morphogenesis in Tissue Engineering. Nat. Biotechnol. 2005, 23, 47–55. [DOI] [PubMed] [Google Scholar]
- Beebe D. J.; Moore J. S.; Bauer J. M.; Yu Q.; Liu R. H.; Devadoss C.; Jo B. H. Functional Hydrogel Structures for Autonomous Flow Control Inside Microfluidic Channels. Nature 2000, 404, 588–590. [DOI] [PubMed] [Google Scholar]
- Eddington D. T.; Beebe D. J. Flow Control with Hydrogels. Adv. Drug Delivery Rev. 2004, 56, 199–210. [DOI] [PubMed] [Google Scholar]
- Lin C. C.; Metters A. T. Hydrogels in Controlled Release Formulations: Network Design and Mathematical Modeling. Adv. Drug Delivery Rev. 2006, 58, 1379–1408. [DOI] [PubMed] [Google Scholar]
- de Las Heras Alarcon C.; Pennadam S.; Alexander C. Stimuli Responsive Polymers for Biomedical Applications. Chem. Soc. Rev. 2005, 34, 276–285. [DOI] [PubMed] [Google Scholar]
- Alarcon C. D. H.; Pennadam S.; Alexander C. Stimuli Responsive Polymers for Biomedical Applications. Chem. Soc. Rev. 2005, 34, 276–285. [DOI] [PubMed] [Google Scholar]
- Xu C. Y.; Kopecek J. Self-Assembling Hydrogels. Polym. Bull. 2007, 58, 53–63. [Google Scholar]
- Yang Z.; Xu B. Supramolecular Hydrogels Based on Biofunctional Nanofibers of Self-Assembled Small Molecules. J. Mater. Chem. 2007, 17, 2385–2393. [Google Scholar]
- Schneider J. P.; Pochan D. J.; Ozbas B.; Rajagopal K.; Pakstis L.; Kretsinger J. Responsive Hydrogels from the Intramolecular Folding and Self-Assembly of a Designed Peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [DOI] [PubMed] [Google Scholar]
- Pochan D. J.; Schneider J. P.; Kretsinger J.; Ozbas B.; Rajagopal K.; Haines L. Thermally Reversible Hydrogels Via Intramolecular Folding and Consequent Self-Assembly of a De Novo Designed Peptide. J. Am. Chem. Soc. 2003, 125, 11802–11803. [DOI] [PubMed] [Google Scholar]
- Ozbas B.; Rajagopal K.; Schneider J. P.; Pochan D. J. Semiflexible Chain Networks Formed Via Self-Assembly of Beta-Hairpin Molecules. Phys. Rev. Lett. 2004, 93, 268106. [DOI] [PubMed] [Google Scholar]
- Haines L. A.; Rajagopal K.; Ozbas B.; Salick D. A.; Pochan D. J.; Schneider J. P. Light-Activated Hydrogel Formation Via the Triggered Folding and Self-Assembly of a Designed Peptide. J. Am. Chem. Soc. 2005, 127, 17025–17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretsinger J. K.; Haines L. A.; Ozbas B.; Pochan D. J.; Schneider J. P. Cytocompatibility of Self-Assembled Beta-Hairpin Peptide Hydrogel Surfaces. Biomaterials 2005, 26, 5177–5186. [DOI] [PubMed] [Google Scholar]
- Sathaye S.; Zhang H.; Sonmez C.; Schneider J. P.; MacDermaid C. M.; Von Bargen C. D.; Saven J. G.; Pochan D. J. Engineering Complementary Hydrophobic Interactions to Control beta-Hairpin Peptide Self-Assembly, Network Branching, and Hydrogel Properties. Biomacromolecules 2014, 15, 3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.; Altunbas A.; Yucel T.; Nagarkar R. P.; Schneider J. P.; Pochan D. J. Injectable Solid Hydrogel: Mechanism of Shear-Thinning and Immediate Recovery of Injectable Beta-Hairpin Peptide Hydrogels. Soft Matter 2010, 6, 5143–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Jang H.; Harlen K.; Puri A.; Nussinov R.; Schneider J. P.; Blumenthal R. Mechanism of Membrane Permeation Induced by Synthetic Beta-Hairpin Peptides. Biophys. J. 2013, 105, 2093–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Gu Y.; Deng L.; Han S.; Sun X.; Chen Y.; Lu J. R.; Xu H. Tuning Gelation Kinetics and Mechanical Rigidity of Beta-Hairpin Peptide Hydrogels Via Hydrophobic Amino Acid Substitutions. ACS Appl. Mater. Interfaces 2014, 6, 14360–14368. [DOI] [PubMed] [Google Scholar]
- Ozbas B.; Kretsinger J.; Rajagopal K.; Schneider J. P.; Pochan D. J. Salt-Triggered Peptide Folding and Consequent Self-Assembly into Hydrogels with Tunable Modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar]
- Kale L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. Charmm - A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar]
- Mahoney M. W.; Jorgensen W. L. A Five-Site Model for Liquid Water and the Reproduction of the Density Anomaly by Rigid, Nonpolarizable Potential Functions. J. Chem. Phys. 2000, 112, 8910–8922. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar]
- Feller S. E.; Zhang Y. H.; Pastor R. W.; Brooks B. R. Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Lee M. S.; Salsbury F. R.; Brooks C. L. Novel Generalized Born Methods. J. Chem. Phys. 2002, 116, 10606–10614. [Google Scholar]
- Lee M. S.; Feig M.; Salsbury F. R.; Brooks C. L. New Analytic Approximation to the Standard Molecular Volume Definition and Its Application to Generalized Born Calculations. J. Comput. Chem. 2003, 24, 1348–1356. [DOI] [PubMed] [Google Scholar]
- Ma B.; Nussinov R. Stabilities and Conformations of Alzheimer’s Beta -Amyloid Peptide Oligomers (Aβ16–22, Aβ16–35, and Aβ10–35): Sequence Effects. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlusty T.; Safran S. A. Defect-Induced Phase Separation in Dipolar Fluids. Science 2000, 290, 1328–1331. [DOI] [PubMed] [Google Scholar]
- Branco M. C.; Nettesheim F.; Pochan D. J.; Schneider J. P.; Wagner N. J. Fast Dynamics of Semiflexible Chain Networks of Self-Assembled Peptides. Biomacromolecules 2009, 10, 1374–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. C.; Pochan D. J.; Wagner N. J.; Schneider J. P. Macromolecular Diffusion and Release from Self-Assembled Beta-Hairpin Peptide Hydrogels. Biomaterials 2009, 30, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz Y.; Rubinov B.; Matmor M.; Rapaport H.; Ashkenasy G.; Miller Y. Effects of Mutations In De Novo Designed Synthetic Amphiphilic Beta-Sheet Peptides on Self-Assembly of Fibrils. Chem. Commun. 2013, 49, 6561–6563. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Dinkel P.; Yu X.; Margittai M.; Zheng J.; Nussinov R.; Wei G.; Ma B. Molecular Insights Into the Reversible Formation of Tau Protein Fibrils. Chem. Commun. (Cambridge, U. K.) 2013, 49, 3582–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Ma B.; Nussinov R.; Wei G. Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. J. Phys. Chem. Lett. 2014, 5, 3026–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P.; Derreumaux P. Understanding Amyloid Fibril Nucleation and Abeta Oligomer/Drug Interactions from Computer Simulations. Acc. Chem. Res. 2014, 47, 603–611. [DOI] [PubMed] [Google Scholar]
- MacKintosh F. C.; Kas J.; Janmey P. A. Elasticity of Semiflexible Biopolymer Networks. Phys. Rev. Lett. 1995, 75, 4425–4428. [DOI] [PubMed] [Google Scholar]
- Morriss-Andrews A.; Bellesia G.; Shea J. E. Beta-Sheet Propensity Controls the Kinetic Pathways and Morphologies of Seeded Peptide Aggregation. J. Chem. Phys. 2012, 137, 145104. [DOI] [PubMed] [Google Scholar]
- Larini L.; Shea J. E. Role of Beta-Hairpin Formation in Aggregation: the Self-Assembly of the Amyloid-Beta(25–35) Peptide. Biophys. J. 2012, 103, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.