

Abstract
Rheumatoid arthritis (RA) is an autoimmune inflammatory disease that is characterized by increased cardiovascular morbidity and mortality, independent of the traditional risk factors for cardiovascular disease. Although classically known for its role in the regulation of circulatory homeostasis, angiotensin II (Ang II) is recognized to act as a powerful proinflammatory mediator. Some research has showed that Ang II plays important roles in autoimmune diseases, including RA, systemic lupus erythematosus and multiple sclerosis. Ang II blockers prove effective in reducing inflammation and autoimmunity in rheumatic diseases and their relative safety, together with their effects for reducing the cardiovascular disease risk, suggest that Ang II blockers may at least act as effective adjunctive therapy for disease control in patients with RA. The present review focuses systematically on the potential impact of Ang II and its receptors on inflammation and immunomodulation in patients with RA.
Keywords: angiotensin II, immunity, inflammation, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic multi-system inflammatory disease of unknown aetiology, affecting between 0·5 and 1% of the adult population, which is characterized by articular and extra-articular involvement. Patients with RA have a higher risk of mortality when compared with the general population, which is due predominantly to increased cardiovascular disease (CVD) 1,2. Current evidence suggests that both traditional risk factors and disease characteristics contribute to the enhanced CVD risk in RA 3,4. The striking similarities between atherosclerotic vascular disease and states of RA have prompted the hypothesis that enhanced systemic inflammation plays an important role in the pathogenesis of CVD in RA patients 5. It has been shown that CVD risk is increased early in the course of RA, and the absolute CVD risk in RA patients is similar to that in non-RA individuals who are 5–10 years older 6. Consequently, the emphasis of research has shifted from the identification of the increased CVD risk in RA towards the development of effective approaches to reduce this risk. Much more work is needed to develop preventative interventions yielding both inhibition of inflammation and reduction of CVD risk in RA. Therefore, novel therapeutic strategies for RA patients are needed, and drugs targeting the angiotensin pathway, particularly angiotensin II (Ang II) and its receptors, may be considered to be one class of these drugs.
Ang II and its receptors
Ang II, a highly active octapeptide produced by angiotensin-converting enzyme (ACE)-mediated cleavage of angiotensin I, is known classically as a cardiovascular mediator, with a primary role in the regulation of blood pressure and body fluid homeostasis. Ang II has two major receptor subtypes, the Ang II type 1A and 1B receptors (AT1AR and AT1BR) and Ang II type 2 receptor (AT2R), members of the seven transmembranes spanning the G protein-coupled receptor superfamily 7. The AT1R is well known as an activator of the Gq/11 family, but under certain circumstances AT1R may activate Gi/o and G12 8. Ang II also induces its actions through activation of the Ras/Raf/mitogen-activated protein kinase (MAPK) signalling cascade and the Janus cytosolic protein kinase (JAK) signal transducers and activators of the transcription signalling pathway 9. AT2R activates unconventional signalling pathways that generally do not involve coupling to classical regulatory G proteins. AT2R is capable of activating protein tyrosine phosphatase (PTP) such as MAPK phosphatase-1, src homology 2 domain-containing PTP and serine/threonine phosphatase PP2A, depending on the cell types 10.
Human cells express a single AT1R, while two subtypes, AT1AR and AT1BR, with 95% amino acid sequence identity, can be found in rat and mouse. AT1AR, the closest murine homologue to the human AT1R, is expressed in various parts of the body and is associated with their respective functions, such as in the kidney, heart, adrenal cortex, vascular smooth muscle, liver and several other tissues 11. AT1BR is expressed predominantly in the anterior pituitary gland and adrenal zona glomerulosa 12. AT1R is comprised of 359 amino acids, while AT2R comprises 363 amino acids, being ∼30% homologous to AT1R, and are both N-linked glycosylated post-translationally; both receptors have high binding affinities for the Ang II. AT2R is expressed ubiquitously in developing fetal tissue and decreases dramatically after birth, remaining low in various adult tissues, including adrenal medulla, uterus, ovary, vascular endothelium and distinct brain areas 13.
Various components of the renin–angiotensin system (RAS) are expressed on immune cells, and Ang II receptors are present on lymphocytes and macrophages 14. Ang II elicits both its proinflammatory and pro-stress actions mainly through stimulation of AT1R 15. AT1R blockers are known to have direct and indirect anti-inflammatory actions 16. Although data concerning the functions of AT2R are still somewhat controversial, the majority support the concept that AT2R has anti-proliferative, anti-inflammatory and anti-fibrotic effects 13,17. It has been reported that the increased stimulation of AT2R may be responsible for some of the therapeutic effects observed during AT1R blockade 18,19. Moreover, AT1R antagonists are less effective in AT2R-deficient mice, again confirming that AT2R play a pivotal role in the effect of AT1R antagonists 20.
Ang II and inflammatory immune responses
Ang II and inflammation
Although classically known for its role in the regulation of circulatory homeostasis, Ang II is recognized to act as a powerful proinflammatory mediator through stimulation of AT1R 15. In the course of inflammatory processes, excessive local Ang II concentration increases vascular permeability by stimulating the production of prostaglandins and the vascular endothelial cell growth factor (VEGF), which initiates the inflammatory responses 21. Ang II up-regulates adhesion molecules such as P-selectin, intercellular adhesion molecule type 1 and vascular cell adhesion molecule type 1 on vascular endothelial cells and smooth muscle cells, and activates monocytes to adhere to them 22,23. Ang II also up-regulates the expression of monocyte chemoattractant protein type 1 (MCP-1), tumour necrosis factor (TNF)-α, interleukin (IL)-6 and IL-8, which are potent chemoattractants and activators of neutrophils 24. In addition, Ang II increases acute inflammation marker C-reactive protein (CRP) both in mRNA and protein levels in macrophages via AT1R-mediated reactive oxygen species (ROS) production and nuclear factor-kappa B (NF-κB) activation 25. CRP markedly up-regulates AT1R mRNA and increases AT1R numbers expressed on vascular smooth muscle cells 26. Moreover, Ang II can directly stimulate the proliferation and activation of lymphocytes and production of ROS in leucocytes 27. Therefore, Ang II signalling through the AT1R leads to the activation of NF-κB with the subsequent production of proinflammatory cytokines, chemokines and cell adhesion molecules by resident cells, which contribute to the migration of inflammatory cells into sites of tissue injury, thereby amplifying the inflammatory responses.
Ang II and innate immunity
There is increasing evidence that Ang II may activate innate and adaptive immunity 28–30; thus it performs as a potent modulator in the immune system (Fig. 1). The role of RAS as an immunological modulator was first suggested by the presence of Ang II in macrophages 31. The contribution of macrophages in Ang II-induced vascular lesions was evaluated in animals with impairment of innate immunity 32. Monocytes are important target cells of Ang II and express both AT1R and AT2R, which have substantial roles in promoting vascular inflammation and metalloproteinase production 33. In addition, Ang II increases mononuclear cell accumulation through inducing NF-κB activity and MCP-1 expression 34,35. Natural killer (NK) cells are fully equipped with RAS components and are potentially capable of producing and delivering Ang II to sites of inflammation 36. NK cell-derived interferon (IFN)-γ plays an important role in propagating the activation and maturation of monocytes into macrophages and dendritic cells (DCs) 37,38, representing an important amplifying mechanism in the early innate inflammatory response. Ang II can also induce rapid neutrophil infiltration by enhancing CXC chemokines, IL-8 and macrophage inflammatory protein-2 39. Other studies have shown that Ang II via AT1R increases neutrophil migration and stimulates leucocyte–endothelium interactions 40,41.
Figure 1.
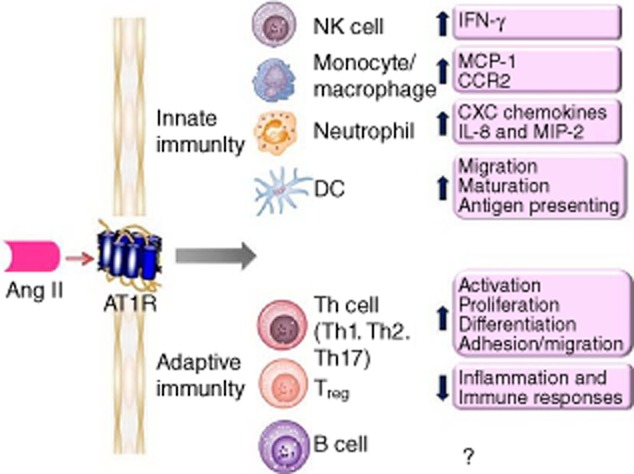
The roles of angiotensin II (Ang II) on innate and adaptive immunity. Ang II via Ang II type 1A receptor (AT1R) signalling in immune cells contributes to the inflammatory responses and activation of the immune system.
The immunoregulatory activity of Ang II is also mediated by activation of DCs, highly specialized antigen-presenting cells responsible for inflammation defence and immune responses. Cultured DCs express both AT1R and AT2R, and Ang II enhances DCs migration, maturation and antigen-presenting ability by interacting with AT1R 29,42. Blockade of AT1R not only inhibits DCs differentiation, but also switches the differentiation of monocytes into macrophage-like cells 43,44. It has been shown that Ang II-induces up-regulation of phenotypical molecules and the increase of inflammatory cytokines in human monocyte-derived DCs via modulating the balance of the negative or positive regulation of the signalling pathways of extracellular signal-regulated kinase (ERK), p38 MAPK and NF-κB 42,45,46. Blockade of AT1R with candesartan, losartan or irbesartan causes poor endocytic and allostimulatory activity in DCs differentiated from human monocytes.
Ang II and adaptive immunity
Evidence suggests that Ang II regulates immune responses by using a calcineurin-dependent pathway through AT1R, indicating a direct effect on T cells. Studies have shown that the ACE inhibitor (ACEI) captopril blocks activation-induced apoptosis in T cells and hence may interfere with clonal deletion and disturb the maintenance of self-tolerance, thus facilitating autoimmunity 47,48. Chronic Ang II infusion augments the expression of CD4+ T cells harbouring the early activation marker CD69 in vivo and in vitro, as well as the tissue-homing proteins CD44 and CCR5 in vitro. Mouse T and B cells isolated from the spleen express AT1AR and Ang II regulates the proliferation of wild-type but not AT1AR-deficient (agt1ar–/–) splenic lymphocytes 49. Activated T cells may themselves generate Ang II locally to influence cell function in an autocrine manner. Ang II, acting through the AT1R on T cells, acts as a co-stimulus for T cell activation, proliferation and differentiation 38,50, and through AT1R Ang II induces an increase in IFN-γ and a reduction in IL-4, suggesting that Ang II may induce differentiation to the T helper type 1 (Th1) phenotype 50. Similarly, Ang II stimulates the production of IFN-γ and TNF-α by peripheral T cells 51 and is capable of stimulating a Th1 response in ApoE-deficient mice 30. Ang II receptor blockers (ARBs) (olmesartan, candesartan and telmisartan) have also been shown to inhibit antigen-specific immune responses for Th1 and Th2 52. Madhur et al. 53 have demonstrated that Ang II also induces Th17 responses. They found that Ang II infusion increases IL-17 production and IL-17 protein in the aortic media. Moreover, vascular dysfunction in response to Ang II is abolished in IL-17–/– mice. In addition, regulatory T cells (Tregs) express AT1R, as do circulating T cell subsets in humans 54,55. In mice receiving chronic Ang II infusion, Treg intravenous administration prevents macrophage and T cell infiltration in aorta. Kvakan et al. 56 demonstrated that transfer of Treg interferes with Ang II-induced inflammatory and immune responses.
Ang II and RA
Animal model studies
The above observations, that Ang II mediates inflammation and modulates T cell-mediated immune responses, suggest a possible role of the peptide in autoimmune diseases. Several studies have shown the beneficial effects of ARB and ACEI use in animal models of induced arthritis. Previously, reports have described the anti-inflammatory activity of the ACEIs in the animal model of acute and chronic arthritis 57,58. It has been demonstrated that the non-thiol ACEI quinapril has significant anti-inflammatory properties, sufficient to suppress the severity of collagen-induced arthritis (CIA), either as prophylaxis or as therapy in established disease 59. Suppression of arthritis by quinapril may be associated with reduced TNF-α production within the joints. Sagawa and colleagues reported that ARB olmesartan suppressed the development of severe arthritis and joint destruction in the CIA model, even when it was administered only after disease onset. They showed that olmesartan significantly suppressed lymphocyte proliferation and IFN-γ production in mice immunized with ovalbumin or type II collagen in Freund's complete adjuvant in vivo 53. In a rat model of adjuvant arthritis (AA), the Ang II infusion induced hypertensive response, endothelial dysfunction and vascular hypertrophy in rats with AA 60. In addition, Ang II-induced expression levels of both AT1R and ACE were significantly enhanced in rats with AA compared with control rats. Both ARBs losartan and irbesartan decreased the levels of superoxide and the expression, activity of NAD(P)H oxidases and improved endothelial dysfunction in antigen-induced arthritis (AIA). However, neither losartan nor irbesartan influenced the clinical severity of arthritis or body weight in rats with AA. More recently, it was reported that losartan reduced local signs of inflammation (pain and oedema) and ameliorated histological joints changes both in murine AIA and rat AA. They observed that losartan decreased neutrophil recruitment, the production of TNF-α, IL-1β and chemokine (CXC motif) ligand 1, and directly inhibited leucocyte–endothelium interactions 61. Mackenzie et al. demonstrated for the first time that AT1R blockade with losartan significantly reversed an impaired endothelium-derived hyperpolarizing factor relaxation in a rat model of AA 62. It seems likely that the mechanism underlying these findings is an AT1R-mediated reduction in the function of connexin components of myoendothelial gap junctions. The results suggest that the angiotensin pathway may be involved in endothelial cell dysfunction, which have important implications in reducing the cardiovascular morbidity and mortality associated with RA 63,64. Refaat et al. showed that losartan and methotrexate combined therapy showed better results than methotrexate and losartan alone. The combined therapy significantly improved paw and liver histopathology, reduced albumin, CRP, nitrite/nitrate concentrations and decreased IL-1β, TNF-α, VEGF, aspartate transaminase and alanine transaminase levels 65. The study showed that losartan increased the efficacy of methotrexate therapy in AA rats with no evidence of toxicity. Moreover, losartan, as a hepatoprotective agent, could decrease the extent of methotrexate-induced hepatotoxicity which is likely to occur after a prolonged period of exposure. Furthermore, our research group reported recently that the losartan-induced therapeutic effects on AA rats in vivo might be correlated with the up-regulation of AT2R and the down-regulation of AT1R 66. Additionally, intra-articular injection of AA rats with AT2R agonist CGP42112 significantly decreased the severity of arthritis. In vitro, CGP42112 effectively inhibited the chemotaxis of IL-1β stimulated-AA monocytes, up-regulated AT2R and down-regulation of AT1R within stimulated AA monocytes. The study suggests strongly that the up-regulation of AT2R might be an additional mechanism by which losartan exerts its therapeutic effects in AA rats.
Human studies
Indeed, AT1R is present and up-regulation in the synovium of patients with RA has been described previously 67,68. It has been postulated that locally generated Ang II acts on synovial angiotensin receptors to modulate synovial hyperplasia. Recently, Kawakami et al. 69 found AT1R and AT2R expression in articular chondrocytes in RA patients and showed that IL-1 was able to regulate the expression of these receptors. Elevated ACE activity has been demonstrated in blood monocytes, rheumatoid nodules and synovial membranes of patients with RA 70–73. Evidence for the increased presence of ACE in tissues from arthritic patients, together with the ability of ACEIs to suppress NF-κB and inflammatory cytokines such as TNF-α and IL-1β, suggest that therapy with tissue-specific ACEIs would be beneficial in arthritis 74.
Martin et al. reported in the early 1980s that the ACEI captopril was clinically beneficial in the treatment of RA in a small open study 75. The drug was assessed for disease-modifying activity in an open trial involving 15 active RA patients who were followed for 48 weeks. Of the 15 patients, 10 patients reported improvement in all clinical symptoms, including reductions in joint symptoms and the number of swollen joints, and reduced levels of CRP at 24 and 48 weeks. However, the clinical benefit of captopril was subsequently attributed to the presence of a thiol group in the compound structure, which was similar to those of the immunosuppressant penicillamine, and not to ACEIs per se. In an open study using the non-thiol-ACEI pentopril, no clinical improvement in patients with RA was observed, although CRP levels decreased significantly at 16 weeks 76. However, the pentopril study may have been powered inadequately to detect significant differences due to the small sample size. In a small, randomized, double-blind study of 11 patients, ACEIs with 10 mg/d ramipril for 8 weeks in addition to current anti-inflammatory treatment markedly improved endothelial function and reduced plasma concentrations of TNF-α and soluble CD40, although other inflammation parameters such as CRP, IL-1, IL-6, myeloperoxidase and erythrocyte sedimentation rate were not influenced 77. In vitro, captopril significantly suppressed TNF-α and IL-1 production in healthy human peripheral blood mononuclear cells stimulated by lipopolysaccharide 61.
There has been little investigation of the potential therapeutic benefits of targeting the pathway with the ARBs. AT1R antagonism losartan use was found to be associated with a significant reduction in CRP and erythrocyte sedimentation rate, key indicators of inflammation, in patients with RA 78. A functional relationship between CRP and Ang II has been demonstrated previously. In addition, in-vitro losartan suppressed TNF-α production from inflamed human synovium in RA patients in a dose-dependent manner 68.
Clinical implication of Ang II interruption in RA
RA has been widely recognized to increase the risk of CVD, with mortality rates from a 1·5- to twofold increase compared with the general population 1,2, but no disease-specific treatment strategies have been agreed upon. Many factors contribute to the elevated CVD risk in RA. Systemic inflammation and its interplay with traditional and nontraditional cardiovascular risk factors appear to have a major role. Recent studies also support the concept of RA as an independent cardiovascular risk factor, analogous to diabetes mellitus, by demonstrating the independent association of RA with both preclinical and overt CVD 79. It has been well established that activation of the RAS plays a major role in the physiology and pathophysiology of the cardiovascular system. Ang II, the main effector molecule of the RAS, contributes to the development of CVD as both a systemic endocrine hormone and a local autocrine/paracrine hormone, producing acute and chronic effects. Ang II regulates not only adhesion molecules expression but also cytokines, chemokines and growth factor secretion within the arterial wall. Although the RAS is involved in inflammation and immune responses of autoimmune disorders, including RA 80, the role of RAS in the pathophysiology of vasculopathies of RA has yet to be established clearly. Ang II is implicated in the up-regulation of proinflammatory cytokines, such as TNF-α, IL-1 and IL-6 42, and conversely 81,82, on the basis of this evidence, may contribute to the pathogenesis of RA. Ang II is not only a chemotactic factor for mononuclear cells, neutrophils, T and B cells but also a growth factor for regulating cellular growth, fibroblast proliferation and angiogenesis 83. Up-regulated proinflammatory cytokines, inflammatory cell infiltration and angiogenesis are the key features of rheumatoid synovitis 84. Furthermore, ACE was localized to fibroblast-like stromal cells and vascular endothelium in the synovial membrane in RA patients 73. Synovial fluid ACE levels were universally increased in RA patients compared to OA patients 72. Locally generated Ang II may act upon synovial AT1R to modulate synovial perfusion and growth present in inflammatory arthritis 67.
Many therapeutic drug options for RA demonstrate conflicting results regarding CVD risk. Early effective anti-rheumatic treatment (e.g. methotrexate and TNF inhibitors) has been shown be associated with a lower CVD 85,86. Some studies have shown that TNF blockade has a transient beneficial effect on CV function 87. Long-term safety analysis of rituximab demonstrated no notable differences in serious CVD events during placebo-controlled periods 88. Tocilizumab, a humanized mAb against the IL-6 receptor, has demonstrated an adverse impact on lipid profiles 89. Similarly, tofacitinib, a new oral JAK inhibitor, recently approved for use in patients with RA, is also associated with significantly increased mean low-density lipoprotein levels compared with placebo 90. Therefore, additional therapeutic strategies are needed to develop defensible interventions yielding both inhibition of inflammation and reduction of CVD risk in RA.
Clinically, RAS blockade exerts potent dual effects, not only through cardiovascular protective effects but also through anti-inflammatory and immunomodulatory properties. Indeed, ARBs and ACEIs have been demonstrated to reduce mortality and morbidity from cardiovascular events among patients with hypertension, ischaemic heart disease and renal disease 91. RA is associated with an increase in CVD risk, whereas hypertension is a major modifiable CVD risk factor with a high prevalence in patients with RA 92. As suggested by the recent recommendations of the European League Against Rheumatism 93, hypertension should be placed at the top of the research agenda for the reduction of CVD risk in RA. Although specific direct evidence in RA is lacking, it seems reasonable to suggest that early detection and aggressive management of hypertension in patients with RA should form part of such a systematic approach. Thus, ARBs and ACEIs may have the therapeutic option of a double effect: anti-hypertensive and anti-inflammatory. Although Ang II interruption will probably never replace anti-rheumatic treatments such as methotrexate and biological agents, ACEIs or ARBs may be the first choice of anti-hypertensive agents in RA patients.
Despite some pharmacological similarities between ACEIs and ARBs, important pharmacological differences may have clinical implications. The ACEIs act by inhibiting ACE that catalyses the conversion of inactive Ang I to active Ang II, resulting in reduced levels of Ang II. A confounding problem with ACEIs is their relative lack of specificity for angiotensin conversion 94. In addition, the development of a persistent dry cough in patients receiving ACEIs therapy is thought to be due primarily to bradykinin or substance P accumulation 95. Furthermore, Ang II also can be produced from chymase, a tissue-specific enzyme originally discovered in mast cells that can make Ang II independent of ACE 96. Thus, ACEI therapy may only partially inhibit Ang II activity.
ARBs avoid this because they selectively inhibit the action of Ang II at the receptor level. ARBs act independently of the Ang II synthetic pathway, allowing a more selective blockade of AT1R-mediated effects of Ang II compared with ACEIs. ARB therapy may also induce anti-inflammatory mechanisms through increased AT2R stimulation. Some researchers believe that stimulation of the AT2R has an opposite effect to that of the AT1R to anti-inflammatory activity 9,19. Under AT1R blockade, the enhanced Ang II levels may result in increased activation of AT2R which, in turn, may confer additional benefit, as persistent AT2R activation is considered to oppose the actions of the AT1R 19. In this way, the ability of Ang II to stimulate AT2R in the presence of AT1R blockade could provide additional complementary therapeutic benefit (Fig. 2). There was a sustained, enhanced effect of combined AT2R stimulation plus AT1R blockade that was greater than AT1R blockade alone 97, together with the absence of functional AT2R desensitization 98. Therefore, AT2R agonists may also be regarded as one of the novel classes of drugs which could be used in the treatment of inflammatory and immune diseases in the future.
Figure 2.
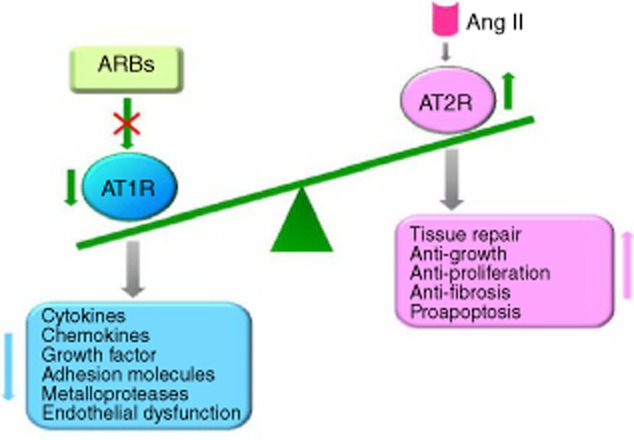
Model of the mechanisms of action of angiotensin II (Ang II) receptor blockers (ARBs). Under Ang II type 1A receptor (AT1R) blockade, the enhancing Ang II levels may result in increased activation of AT2R. In this way, the ability of Ang II to stimulate AT2R in the presence of blockade of AT1R could provide additional complementary therapeutic benefit.
Concluding remarks
Evidence has indicated that Ang II is a potent proinflammatory mediator in autoimmune diseases, which suggests that Ang II blockers may at least act as effectively as adjunctive therapy for disease control in patients with RA. It is noteworthy that statins, similar to ACEIs and ARBs, have positive effects on endothelial progenitor cells and inhibit inflammation, which is desirable in autoimmune rheumatic diseases 99. Because chronic inflammatory diseases are associated with an increased burden of CVD, Ang II inhibitors are of interest from the dual perspectives of disease modification and cardiovascular risk reduction. The evidence reported thus far suggests that ARBs may constitute an appropriate first-line therapy for hypertension in RA. Nevertheless, large, randomized, prospective, placebo-controlled studies are needed to confirm any anti-inflammatory actions of Ang II suppression in patients with RA.
Acknowledgments
The study was supported by the National Natural Science Foundation of China (no. 31200675, 81173075; 81330081), the Natural Science Foundation of Anhui province (numbers 1208085QH158 and 1408085QH173), the Specialized Research Fund for the Doctoral Program of Higher Education (20123420110003) and the Grants for Scientific Research of BSKY from Anhui Medical University (XJ201215).
Disclosure
The authors have declared that no competing interests exist.
References
- Aviña-Zubieta JA, Choi HK, Sadatsafavi M. Risk of cardiovascular mortality in patients with RA: a meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690–1697. doi: 10.1002/art.24092. [DOI] [PubMed] [Google Scholar]
- Meune C, Touzé E, Trinquart L, Allanore Y. High risk of clinical cardiovascular events in RA: levels of associations of myocardial infarction and stroke through a systematic review and meta-analysis. Arch Cardiovasc Dis. 2010;103:253–261. doi: 10.1016/j.acvd.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Dessein PH, Norton GR, Woodiwiss AJ, Joffe BI, Wolfe F. Influence of nonclassical cardiovascular risk factors on the accuracy of predicting subclinical atherosclerosis in RA. J Rheumatol. 2007;34:943–951. [PubMed] [Google Scholar]
- Solomon DH, Kremer J, Curtis JR. Explaining the cardiovascular risk associated with RA traditional risk factors versus markers of RA severity. Ann Rheum Dis. 2010;69:1920–1925. doi: 10.1136/ard.2009.122226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Role of inflammation in atherosclerosis associated with rheumatoid arthritis. Am J Med. 2008;121:S21–31. doi: 10.1016/j.amjmed.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Kremers HM, Crowson CS, Therneau TM, Roger VL, Gabriel SE. High ten-year risk of cardiovascular disease in newly diagnosed rheumatoid arthritis patients: a population based cohort study. Arthritis Rheum. 2008;58:2268–2274. doi: 10.1002/art.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Gines P, Nicolas JM. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118:1149–1156. doi: 10.1016/s0016-5085(00)70368-4. [DOI] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88. doi: 10.1080/08037050310001057. [DOI] [PubMed] [Google Scholar]
- Stoll M, Unger T. Angiotensin and its AT2 receptor: new insights into an old system. Regul Pept. 2001;99:175–182. doi: 10.1016/s0167-0115(01)00246-4. [DOI] [PubMed] [Google Scholar]
- Burson JM, Aguilera G, Gross KW, Sigmund CD. Differential expression of angiotensin receptor 1A and 1B in mouse. Am J Physiol. 1994;267:E260–267. doi: 10.1152/ajpendo.1994.267.2.E260. [DOI] [PubMed] [Google Scholar]
- Oliverio MI, Coffman TM. Angiotensin II receptor physiology using gene targeting. News Physiol Sci. 2000;15:171–175. doi: 10.1152/physiologyonline.2000.15.4.171. [DOI] [PubMed] [Google Scholar]
- Steckelings UM, Kaschina E, Unger T. The AT2 receptor – a matter of love and hate. Peptides. 2005;26:1401–1409. doi: 10.1016/j.peptides.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Nataraj C, Oliverio MI, Mannon RB. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest. 1999;104:1693–1701. doi: 10.1172/JCI7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi C, Paradis P, Schiffrin EL. Role of the renin–angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Dagenais NJ, Jamali F. Protective effects of angiotensin II interruption: evidence for antiinflammatory actions. Pharmacotherapy. 2005;25:1213–1229. doi: 10.1592/phco.2005.25.9.1213. [DOI] [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International Union of Pharmacology: XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- Widdop R, Jones E, Hannan R, Gaspari T. Angiotensin AT2 receptors: cardiovascular hope or hype? Br J Pharmacol. 2003;140:809–824. doi: 10.1038/sj.bjp.0705448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ES, Black MJ, Widdop RE. Angiotensin AT2 receptor contributes to cardiovascular remodelling of aged rats during chronic AT1 receptor blockade. J Mol Cell Cardiol. 2004;37:1023–1030. doi: 10.1016/j.yjmcc.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Mogi M, Li JM, Iwanami J. Angiotensin II type-2 receptor stimulation prevents neural damage by transcriptional activation of methyl methanesulfonate sensitive 2. Hypertension. 2006;48:141–148. doi: 10.1161/01.HYP.0000229648.67883.f9. [DOI] [PubMed] [Google Scholar]
- Williams B, Baker AQ, Gallacher B, Lodwick D. Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension. 1995;25:913–917. doi: 10.1161/01.hyp.25.5.913. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Cerdá-Nicolás M, Naim Abu Nabah Y. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood. 2004;104:402–408. doi: 10.1182/blood-2003-08-2974. [DOI] [PubMed] [Google Scholar]
- Piqueras L, Kubes P, Alvarez A. Angiotensin II induces leukocyte–endothelial cell interactions in vivo via AT(1) and AT(2) receptor-mediated P-selectin upregulation. Circulation. 2000;102:2118–2123. doi: 10.1161/01.cir.102.17.2118. [DOI] [PubMed] [Google Scholar]
- Nobuhiko A, Suganuma E, Babaev VR. Angiotensin II amplifies macrophage-driven atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2143–2148. doi: 10.1161/01.ATV.0000145607.03879.e0. [DOI] [PubMed] [Google Scholar]
- Han C, Liu J, Liu X, Li M. Angiotension II induces C-reactive protein expression through ERK1/2 and JNK signaling in human aortic cells. Atherosclerosis. 2010;212:206–212. doi: 10.1016/j.atherosclerosis.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Wang CH, Li SH, Weisel RD. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation. 2003;107:1783–1790. doi: 10.1161/01.CIR.0000061916.95736.E5. [DOI] [PubMed] [Google Scholar]
- Dandona P, Kumar V, Aljada A. Angiotensin II receptor blocker valsartan suppresses reactive oxygen species generation in leukocytes, nuclear factor-kappa B, in mononuclear cells of normal subjects: evidence of an antiinflammatory action. J Clin Endocrinol Metab. 2003;88:4496–4501. doi: 10.1210/jc.2002-021836. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Gomez-Guerrero C, Shirato I. Susceptibility to T cell-mediated injury in immune complex disease is linked to local activation of rennin–angiotensin system: the role of NF-AT pathway. J Immunol. 2002;169:4136–4146. doi: 10.4049/jimmunol.169.8.4136. [DOI] [PubMed] [Google Scholar]
- Muller DN, Shagdarsuren E, Park JK. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland J, Setton C, Silverstein E. Angiotensin converting enzyme: induction by steroids in rabbit alveolar macrophages in culture. Science. 1977;197:64–65. doi: 10.1126/science.194311. [DOI] [PubMed] [Google Scholar]
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25:2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- Kim MP, Zhou M, Wahl LM. Angiotensin II increases human monocyte matrix metalloproteinase-1 through the AT2 receptor and prostaglandin E2: implications for atherosclerotic plaque rupture. J Leukoc Biol. 2005;78:195–201. doi: 10.1189/jlb.1204715. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Bustos C, Hernández-Presa MA, Lorenzo O, Plaza JJ, Egido J. Angiotensin II participates in mononuclear cell recruitment in the kidney through nuclear factor-kappa B activation and monocyte chemoattractant protein-1 gene expression. J Immunol. 1998;161:430–439. [PubMed] [Google Scholar]
- Ortego M, Bustos C, Hernández-Presa MA. Atorvastatin reduces NF kappa B activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis. 1999;147:253–261. doi: 10.1016/s0021-9150(99)00193-8. [DOI] [PubMed] [Google Scholar]
- Jurewicz M, McDermott DH, Sechler JM. Human T and natural killer cells possess a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol. 2007;18:1093–1102. doi: 10.1681/ASN.2006070707. [DOI] [PubMed] [Google Scholar]
- Kossmann S, Schwenk M, Hausding M. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. 2013;33:1313–1319. doi: 10.1161/ATVBAHA.113.301437. [DOI] [PubMed] [Google Scholar]
- Goldszmid RS, Caspar P, Rivollier A. NK cell-derived interferon-γ orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity. 2012;36:1047–1059. doi: 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte S, Kuttruff S, Waldhauer I, Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80–AICL interaction. Nat Immunol. 2006;7:1334–1342. doi: 10.1038/ni1402. [DOI] [PubMed] [Google Scholar]
- Mateo T, Abu Nabah YN, Abu Taha M. Angiotensin II-induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J Immunol. 2006;176:5577–5586. doi: 10.4049/jimmunol.176.9.5577. [DOI] [PubMed] [Google Scholar]
- Nabah YN, Mateo T, Estellés R. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation. 2004;110:3581–3586. doi: 10.1161/01.CIR.0000148824.93600.F3. [DOI] [PubMed] [Google Scholar]
- Lapteva N, Ide K, Nieda M. Activation and suppression of reninangiotensin system in human dendritic cells. Biochem Biophys Res Commun. 2002;296:194–200. doi: 10.1016/s0006-291x(02)00855-0. [DOI] [PubMed] [Google Scholar]
- Nahmod K, Gentilini C, Vermeulen M. Impaired function of dendritic cells deficient in angiotensin II type 1 receptors. J Pharmacol Exp Ther. 2010;334:854–862. doi: 10.1124/jpet.109.161760. [DOI] [PubMed] [Google Scholar]
- Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247–257. doi: 10.1002/emmm.201000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei-guo Z, Hui Y, Shan L. PPAR-gamma agonist inhibits Ang II-induced activation of dendritic cells via the MAPK and NF-kappa B pathways. Immunol Cell Biol. 2010;88:305–312. doi: 10.1038/icb.2009.100. [DOI] [PubMed] [Google Scholar]
- Nie W, Yan H, Li S. Angiotensin-(1-7) enhances angiotensin II induced phosphorylation of ERK1/2 in mouse bone marrow-derived dendritic cells. Mol Immunol. 2009;46:355–361. doi: 10.1016/j.molimm.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Odaka C, Mizuochi T. Angiotensin-converting enzyme inhibitor captopril prevents activation-induced apoptosis by interfering with T cell activation signals. Clin Exp Immunol. 2000;121:515–522. doi: 10.1046/j.1365-2249.2000.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho dos Santos JS, Menezes CA, Villani FN. Captopril increases the intensity of monocyte infection by Trypanosoma cruzi and induces human T helper type 17 cells. Clin Exp Immunol. 2010;162:528–536. doi: 10.1111/j.1365-2249.2010.04270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- Hoch NE, Guzik TJ, Chen W. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol. 2009;296:R208–216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Nangaku M, Miyata T. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension. 2003;42:31–38. doi: 10.1161/01.HYP.0000075082.06183.4E. [DOI] [PubMed] [Google Scholar]
- Mazzolai L, Duchosal MA, Korber M. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE–/– mice. Hypertension. 2004;44:277–282. doi: 10.1161/01.HYP.0000140269.55873.7b. [DOI] [PubMed] [Google Scholar]
- Sagawa K, Nagatani K, Komagata Y, Yamamoto K. Angiotensin receptor blockers suppress antigen-specific T cell responses and ameliorate collagen-induced arthritis in mice. Arthritis Rheum. 2005;52:1920–1928. doi: 10.1002/art.21040. [DOI] [PubMed] [Google Scholar]
- Akdis CA, Akdis M. Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J Allergy Clin Immunol. 2009;123:735–746. doi: 10.1016/j.jaci.2009.02.030. [DOI] [PubMed] [Google Scholar]
- Zhang JD, Patel MB, Song YS. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110:1604–1617. doi: 10.1161/CIRCRESAHA.111.261768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoumi T, Kasal DA, Li MW. T Regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57:469–476. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- Agha AM, Mansour M. Effects of captopril on interleukin-6, leukotriene B(4), and oxidative stress markers in serum and inflammatory exudate of arthritic rats: evidence of anti-inflammatory activity. Toxicol Appl Pharmacol. 2000;168:123–130. doi: 10.1006/taap.2000.8985. [DOI] [PubMed] [Google Scholar]
- Caspritz G, Alpermann HG, Schleyerbach R. Influence of the new angiotensin converting enzyme inhibitor ramipril on several models of acute inflammation and the adjuvant arthritis in the rat. Arzneimittelforschung. 1986;36:1605–1608. [PubMed] [Google Scholar]
- Dalbeth N, Edwards J, Fairchild S, Callan M, Hall FC. The non-thiol angiotensin-converting enzyme inhibitor quinapril suppresses inflammatory arthritis. Rheumatology (Oxf) 2005;44:24–31. doi: 10.1093/rheumatology/keh398. [DOI] [PubMed] [Google Scholar]
- Sakuta T, Morita Y, Satoh M, Fox DA, Kashihara N. Involvement of the rennin–angiotensin system in the development of vascular damage in a rat model of arthritis: effect of angiotensin receptor blockers. Arthritis Rheum. 2010;62:1319–1328. doi: 10.1002/art.27384. [DOI] [PubMed] [Google Scholar]
- Silveira KD, Coelho FM, Vieira AT. Mechanisms of the anti-inflammatory actions of the angiotensin type 1 receptor antagonist losartan in experimental models of arthritis. Peptides. 2013;46:53–63. doi: 10.1016/j.peptides.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Mackenzie A, Dunning L, Ferrell WR, Lockhart JC. Angiotensin II Type 1 receptor blockade protects endothelium-derived hyperpolarising factor-mediated relaxation in a rat model of monoarthritis. Life Sci. 2013;92:1131–1137. doi: 10.1016/j.lfs.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Dessein PH, Solomon A, Woodiwiss AJ, Norton GR, Tsang L, Gonzalez-Gay MA. Marked independent relationships between circulating interleukin-6 concentrations and endothelial activation in rheumatoid arthritis. Mediators Inflamm. 2013;2013:1–10. doi: 10.1155/2013/510243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Södergren A, Karp K, Boman K. Atherosclerosis in early rheumatoid arthritis: very early endothelial activation and rapid progression of intima media thickness. Arthritis Res Ther. 2010;12:R158. doi: 10.1186/ar3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refaat R, Salama M, Abdel Meguid E, El Sarha A, Gowayed M. Evaluation of the effect of losartan and methotrexate combined therapy in adjuvant-induced arthritis in rats. Eur J Pharmacol. 2013;698:421–428. doi: 10.1016/j.ejphar.2012.10.024. [DOI] [PubMed] [Google Scholar]
- Wang D, Hu S, Zhu J. Angiotensin II type 2 receptor correlates with therapeutic effects of losartan in rats with adjuvant-induced arthritis. J Cell Mol Med. 2013;17:1577–1587. doi: 10.1111/jcmm.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Suzuki T, Knock GA, Blake DR, Polak JM, Wharton J. AT1 receptor characteristics of angiotensin analogue binding in human synovium. Br J Pharmacol. 1994;112:435–442. doi: 10.1111/j.1476-5381.1994.tb13091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, Lockhart JC, Ferrell WR, Gsell W, McLean S, Sturrock RD. Angiotensin II type 1 receptor as a novel therapeutic target in rheumatoid arthritis: in vivo analyses in rodent models of arthritis and ex vivo analyses in human inflammatory synovitis. Arthritis Rheum. 2007;56:441–447. doi: 10.1002/art.22335. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Matsuo K, Murata M. Expression of angiotensin II receptor-1 in human articular chondrocytes. Arthritis. 2012;2012:1–7. doi: 10.1155/2012/648537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M, Fujisawa M, Yamada A. Spontaneous release of angiotensin converting enzyme and interleukin 1 beta from peripheral blood monocytes from patients with rheumatoid arthritis under a serum free condition. Ann Rheum Dis. 1990;49:172–176. doi: 10.1136/ard.49.3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M, Sasano M, Fuzisawa M, Okabe T, Nishizawa K. Constitutive production of angiotensin converting enzyme from rheumatoid nodule cells under serum free conditions. Ann Rheum Dis. 1992;51:741–742. doi: 10.1136/ard.51.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veale D, Yanni G, Bresnihan B, FitzGerald O. Production of angiotensin converting enzyme by rheumatoid synovial membrane. Ann Rheum Dis. 1992;51:476–480. doi: 10.1136/ard.51.4.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DA, Catravas J, Wharton J. Angiotensin converting enzyme in human synovium: increased stromal [(125)I[351A binding in rheumatoid arthritis. Ann Rheum Dis. 2000;59:125–131. doi: 10.1136/ard.59.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzawa M, Satoh J, Sagara M. Angiotensin converting enzyme inhibitors suppress production of tumor necrosis factor-alpha in vitro and in vivo. Immunopharmacology. 1997;36:49–55. doi: 10.1016/s0162-3109(96)00160-9. [DOI] [PubMed] [Google Scholar]
- Martin MF, Surrall KE, McKenna F, Dixon JS, Bird HA, Wright V. Captopril: a new treatment for rheumatoid arthritis? Lancet. 1984;1:1325–1328. doi: 10.1016/s0140-6736(84)91821-x. [DOI] [PubMed] [Google Scholar]
- Bird HA, Le Gallez P, Dixon JS. A clinical and biochemical assessment of a nonthiol ACE inhibitor (pentopril; CGS-13945) in active rheumatoid arthritis. J Rheumatol. 1990;17:603–608. [PubMed] [Google Scholar]
- Flammer AJ, Sudano I, Hermann F. Angiotensin-converting enzyme inhibition improves vascular function in rheumatoid arthritis. Circulation. 2008;117:2262–2269. doi: 10.1161/CIRCULATIONAHA.107.734384. [DOI] [PubMed] [Google Scholar]
- Perry ME, Chee MM, Ferrell WR, Lockhart JC, Sturrock RD. Angiotensin receptor blockers reduce erythrocyte sedimentation rate levels in patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67:1646–1647. doi: 10.1136/ard.2007.082917. [DOI] [PubMed] [Google Scholar]
- van Halm VP, Peters MJ, Voskuyl AE. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: a cross-sectional study, the CARRE Investigation. Ann Rheum Dis. 2009;68:1395–1400. doi: 10.1136/ard.2008.094151. [DOI] [PubMed] [Google Scholar]
- Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin–angiotensin system and cardiovascular risk. Lancet. 2007;369:1208–1219. doi: 10.1016/S0140-6736(07)60242-6. [DOI] [PubMed] [Google Scholar]
- Peng JF, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-α-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–1126. doi: 10.1161/01.res.0000047090.08299.d5. [DOI] [PubMed] [Google Scholar]
- Wassman S, Stumpf M, Stehlow K. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res. 2004;94:534–541. doi: 10.1161/01.RES.0000115557.25127.8D. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–911. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- Micha R, Imamura F, Wyler von Ballmoos M, Solomon DH, Hernán MA. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol. 2011;108:1362–1370. doi: 10.1016/j.amjcard.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, van Sijl AM, Voskuyl AE, Sattar N, Smulders YM. The effects of tumor necrosis factor inhibitors on cardiovascular risk in RA. Curr Pharm Des. 2012;18:1502–1511. doi: 10.2174/138161212799504786. [DOI] [PubMed] [Google Scholar]
- Mäki-Petäjä KM, Hall FC, Booth AD, Wallace SM, Yasmin, Bearcroft PW. Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-alpha therapy. Circulation. 2006;114:1185–1192. doi: 10.1161/CIRCULATIONAHA.105.601641. [DOI] [PubMed] [Google Scholar]
- van Vollenhoven RF, Emery P, Bingham CO., 3rd Long-term safety of rituximab in rheumatoid arthritis: 9 5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann Rheum Dis. 2013;72:1496–1502. doi: 10.1136/annrheumdis-2012-201956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashiri SY, Kawakami A, Yamasaki S. Effects of the antiinterleukin-6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis. Rheumatol Int. 2011;31:451–456. doi: 10.1007/s00296-009-1303-y. [DOI] [PubMed] [Google Scholar]
- Fleischmann R, Kremer J, Cush J. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. doi: 10.1056/NEJMoa1109071. [DOI] [PubMed] [Google Scholar]
- Montecucco F, Mach F. Statins, ACE inhibitors and ARBs in cardiovascular disease. Best Pract Res Clin Endocrinol Metab. 2009;23:389–400. doi: 10.1016/j.beem.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Protogerou AD, Panagiotakos DB, Zampeli E. Arterial hypertension assessed ‘out-of-office’ in a contemporary cohort of rheumatoid arthritis patients free of cardiovascular disease is characterized by high prevalence, low awareness, poor control and increased vascular damage-associated ‘white coat’ phenomenon. Arthritis Res Ther. 2013;15:R142. doi: 10.1186/ar4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, Symmons DP, McCarey D. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325–331. doi: 10.1136/ard.2009.113696. [DOI] [PubMed] [Google Scholar]
- Burnier M, Brunner HR. Angiotensin II receptor antagonists. Lancet. 2000;355:637–645. doi: 10.1016/s0140-6736(99)10365-9. [DOI] [PubMed] [Google Scholar]
- Alderman CP. Adverse effects of the angiotensin-converting enzyme inhibitors. Ann Pharmacother. 1996;30:55–61. doi: 10.1177/106002809603000110. [DOI] [PubMed] [Google Scholar]
- Resende MM, Mill JG. Alternate angiotensin II-forming pathways and their importance in physiological or physiopathological conditions. Arq Bras Cardiol. 2002;78:425–438. doi: 10.1590/s0066-782x2002000400012. [DOI] [PubMed] [Google Scholar]
- Carey RM, Howell NL, Jin XH, Siragy HM. Angiotensin type 2 receptor-mediated hypotension in angiotensin type-1 receptor-blocked rats. Hypertension. 2001;38:1272–1277. doi: 10.1161/hy1201.096576. [DOI] [PubMed] [Google Scholar]
- Widdop RE, Matrougui K, Levy BI, Henrion D. AT2 receptor-mediated relaxation is preserved after long-term AT1 receptor blockade. Hypertension. 2002;40:516–520. doi: 10.1161/01.hyp.0000033224.99806.8a. [DOI] [PubMed] [Google Scholar]
- McCarey DW, McInnes IB, Madhok R. Trial of Atorvastatin in rheumatoid arthritis (TARA): double-blind, randomized placebo-controlled trial. Lancet. 2004;363:2015–2021. doi: 10.1016/S0140-6736(04)16449-0. [DOI] [PubMed] [Google Scholar]