

Abstract
Interleukin (IL)-1β is now emerging as a critical cytokine in the pathogenesis of T helper type 17 (Th17)-mediated skin diseases, including psoriasis. Psoriatic keratinocytes are a major source of IL-1β; however, the mechanisms triggering IL-1β processing remain unknown. Recently, an acute-phase protein serum amyloid A (SAA) has been identified as a danger signal that triggers inflammasome activation and IL-1β secretion. In this study, we detected increased SAA mRNA and protein expression in psoriatic epidermis. In cultured keratinocytes, SAA up-regulated the expression of pro-IL-1β and secretion of mature IL-1β. On the transcriptional level, blocking Toll-like receptor-2 (TLR-2), TLR-4 or nuclear factor kappa B (NF-κB) attenuated SAA-induced expression of IL-1β mRNA. SAA up-regulated caspase-1 and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) expression in keratinocytes. Inhibiting caspase-1 activity and silencing NLRP3 decreased IL-1β secretion, confirming NLRP3 as the SAA-responsive inflammasome on the post-transcriptional level. The mechanism of SAA-triggered NLRP3 activation and subsequent IL-1β secretion was found to involve the generation of reactive oxygen species. Finally, the expression of SAA by keratinocytes was up-regulated by IL-17A. Taken together, our results indicate that keratinocyte-derived SAA triggers a key inflammatory mediator, IL-1β, via NLRP3 inflammasome activation, providing new potential targets for the treatment of this chronic skin disease.
Keywords: IL-1β, inflammasome, keratinocytes, psoriasis, serum amyloid A
Introduction
Psoriasis is a chronic inflammatory skin disease of unknown aetiology that affects ∼2% of the general population 1. Interleukin (IL)-1β has been identified as a crucial player in the pathogenesis in psoriasis 1. In particular, IL-23-triggered development of T helper type 17 (Th17) cells, which are pathogenic in psoriasis, depends upon the presence of IL-1β 2,3. In psoriatic skin, keratinocytes are a major source of IL-1β, and IL-1β derived from keratinocytes fosters further Th17 cell-dependent cutaneous inflammation 4,5. The protein level of IL-1β is regulated on both the transcriptional and post-transcriptional levels. Transcription of the IL-1β gene and production of cytosolic pro-IL-1β are dependent upon the activation of nuclear factor-kappa B (NF-κB) via, for example, Toll-like receptors (TLRs) 6. On the post-transcriptional level, generation of mature IL-1β is regulated by inflammasomes, which are immune complexes that are formed upon recognition of certain molecular patterns. The core of the inflammasome is formed by NACHT, LRR and PYD domains-containing protein 3 (NLRP), the most studied member of which is NLRP3 7. Several pathogen-, danger- and disease-associated molecular patterns that trigger the formation of inflammasomes have been discovered 8. In the skin, ultraviolet (UV) exposure and sensitizers activate the inflammasome protein NLRP3 and induce IL-1β processing 9,10. However, in psoriasis, the disease-associated molecular patterns that induce inflammasome formation are not known.
Serum amyloid A (SAA) is one of the most prominent positive acute-phase proteins, which increases greatly in serum due to inflammation, infections, neoplasia and tissue injury 11–13. Recent evidence has indicated that SAA possesses a number of cytokine-like properties. It has been reported that SAA stimulates the release of mature IL-1β from neutrophils, mast cells, macrophages and fibroblasts 14–17. However, whether or not SAA can induce IL-1β production in keratinocytes remains to be investigated.
Although circulating SAA is synthesized by hepatocytes, extra-hepatic synthesis of SAA is up-regulated at local sites of mainly tissue inflammation 18,19. In psoriasis, chronic immune response of the epidermal layer is known to trigger the release of cytokines from epidermal keratinocytes. Based on these findings, we hypothesized that psoriatic epidermal keratinocytes may serve as local producers of SAA, and SAA may function as a danger signal on resident skin cells by inducing mature IL-1β secretion in keratinocytes that further amplify the inflammatory response.
In the present study, we first aimed to examine the local expression of SAA in psoriatic epidermis. We next characterized the putative role of SAA in the production of IL-1β and the activation of NLRP3 inflammasome machinery in keratinocytes. We also demonstrated how IL-17A may, in turn, up-regulate the SAA expression in keratinocytes.
Materials and methods
Reagents
Bay11-7082 was obtained from Cell Signaling Technology (Danvers, MA, USA). Caspase-1 inhibitor (Z-YVAD-fmk) was purchased from Biovision (Milpitas, CA, USA). N-acetyl-l-cysteine (NAC) was obtained from Sigma-Aldrich (St Louis, MO, USA). Anti-human TLR-2 monoclonal antibody (mAb), anti-human TLR-4 mAb and appropriate immunoglobulin (Ig)G1 isotype antibody were purchased from eBioscience (San Diego, CA, USA). Anti-CD36 mAb was obtained from Abcam (Cambridge, MA, USA). For Western blot analysis, we used the following specific antibodies: pro-IL-1β (R&D Systems, Danvers, MA, USA), caspase-1 (Cell Signaling Technology), NLRP3 (Abcam) and β-actin (Cell Signaling Technology). Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgGs (R&D Systems).
Patients and skin samples
Eleven (two females; nine males) unrelated patients with plaque-psoriasis, mean age 42·35 years (range 18–65 years) were recruited from the out-patient and in-patient clinics at the Department of Dermatology, Shanghai Skin Disease Hospital. The mean age of psoriasis onset of the patients was 27·4 years. The Psoriasis Area Severity Index (PASI) score was recorded by the same physician in all cases. The mean PASI score was 25·05 (range 7–68). One patient had PASI lower than 7 (mild disease) and all others had PASI greater than 7 (moderate and severe disease). Ten of the patients had psoriatic nail involvement, two had psoriatic arthropathy and one patient had a positive family history of psoriasis. Punch biopsies (4 mm) were taken from untreated lesional and non-lesional psoriatic skin. The biopsies were taken from non-sun-exposed areas. Prior to taking the biopsies the patients had not received any treatment, including UVB therapy, topical corticosteroids and systemic medication, for at least a month. Eight healthy volunteers free from other dermatoses and from the positive family history of psoriasis were recruited as a control group (six male, two female); mean age 37·2 years (range 20–71 years). For mRNA analyses, biopsies were transferred directly to 1 ml of Trizol and homogenized. All procedures were approved by the Ethic Committee of Shanghai Skin Disease Hospital, and informed written consent was obtained.
Immunohistochemistry
Punch biopsies were obtained from six patients with psoriasis. Two skin biopsies were obtained from healthy control subjects. The tissue samples were formalin-fixed and paraffin-embedded. Staining was performed using the avidin–biotin–horseradish peroxidase method (ABC standard; Vector Laboratories, Burlingame, CA, USA). Antigen retrieval was performed by autoclaving in a 0·01 M sodium citrate buffer (pH 6·0) for 10 min. Monoclonal antibody for human SAA (Abcam) at a dilution of 1 : 100 was used. Colour was developed using diaminobenzidine as a substrate. Slides were examined and images were acquired using a fluorescence microscope (BZ8000; Keyence, Osaka, Japan).
Immunofluorescence
Human keratinocytes were seeded at a concentration of 105 cells per well (six-well plates) onto chamber slides, and cultured for at least for 24 h before stimulation with IL-17A. Cells were fixed in phpsphate-buffered saline (PBS) containing 3·7% paraformaldehyde. The cells were then incubated overnight at 4°C with monoclonal antibody for human SAA. After several washes with PBS, fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG was added and incubated for 1 h at 37°C. The slides were then mounted and observed under fluorescence microscope.
Cell cultures
To help to account for potential donor variability, most experiments were conducted using normal human neonatal keratinocytes isolated from a pool (three to five individuals) of foreskins. The keratinocytes were grown in EpiLife cell culture medium (Cascade Biologics, Portland, OR, USA) containing 0·06 mM Ca2+ and 1 × EpiLife-defined growth supplement (EDGS) at 37°C under standard tissue culture conditions. Stock cultures were maintained for up to five passages in this medium with the addition of gentamicin (10 μg/ml) and amphotericin B (0·25 μg/ml). Cells at 60–80% confluency were stimulated for different time-periods with SAA (1–100 μg/ml; Peprotech, Rocky Hills, NJ, USA) or IL-17A (10–200 ng/ml; R&D Systems).
RNA isolation and real-time reverse transcription–polymerase chain reaction (RT–PCR)
RNA was isolated and purified using the RNeasy Mini kit (Qiagen, Hilden, Germany). For real-time RT–PCR analyses, 1 μg of DNase-treated total RNA was reverse-transcribed. The amplification of the cDNA was accomplished using the ABI Prism 7900HT sequence detection system in the presence of the commercially available SYBR Green PCR Master Mix (Takara, Dalian, China) in a 40-cycle polymerase chain reaction (PCR). The primer sequences were as follows: glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (forward, 5′-GGT CGG AGT CAA CGG ATT TG-3′, reverse, 5′-ATG AGC CCC AGC CTT CTC CAT-3′); SAA (forward, 5′-CGA AGC TTC TTT TCG TTC CTT-3′, reverse, 5′-CAC CAT GGC CAA AGA ATC TC-3′); and IL-1β (forward, 5′-AGC TAC GAA TCT CCG ACC AC-3′, reverse, 5′-CGT TAT CCC ATG TGT CGA AGA A-3′). The denaturing, annealing and extension conditions of each PCR cycle were 95°C for 5 s, 60°C for 20 s and 72°C for 34 s, respectively. The relative expression was calculated using the 2–DDCT method. The mRNA levels of each target gene were normalized to the levels of GAPDH and were represented as fold induction.
Enzyme-linked immunosorbent assay (ELISA)
Keratinocytes were seeded in 12-well plates and grown to 60% confluence. After treatment, supernatants were collected and subsequently centrifuged to pellet cell debris. Supernatants were analysed with ELISA kits for IL-1β (R&D Systems) and SAA (Biosource International, Camarillo, CA, USA).
Western blot analysis
After treatment, keratinocytes were harvested and lysed in 100 μl of lysis buffer [mammalian-protein extraction reagents (M-PER); Thermo Scientific, Waltham, MA, USA] with added protease inhibitors. Protein profiles were separated by electrophoresis in 8–12% sodium dodecyl sulphide (SDS)-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), and specific proteins were detected by the appropriate primary and secondary antibodies. The membranes were developed with an enhanced chemiluminescence system from Amersham and exposed to X-ray film (Fuji Photo Film Co. Ltd, Tokyo, Japan).
Flow cytometry
The keratinocytes were stimulated with SAA for 15 min. The cells then were cooled quickly, fixed and permeabilized with BD Cytofix/Cytoperm™ solution (BD Biosciences), then stained sequentially with phycoerythrin (PE)-labelled phospho-p65 antibody (BD Biosciences). An isotype control mouse IgG was used for gating of phosphoprotein-positive cells. The stained cells were analysed on a LSRII fluorescence activated cell sorter (FACS) analyser (BD Biosciences). For reactive oxygen species (ROS) measurement, treated keratinocytes were incubated with 10 mM CM-H2DCFDA (general oxidative stress indicator) (Invitrogen, Carlsbad, CA, USA) for 30 min at 37°C, harvested and analysed by flow cytometry.
siRNA silencing of NLRP3 expression
To reduce endogenous NLRP3 expression, keratinocytes were transfected with 80 pmol of siRNA oligonucleotides (Santa Cruz Biotechnology, Santa Cruz, CA, USA) following the manufacturer's instructions. The cells were transfected with either a siRNA oligonucleotide against NLRP3 or a non-targeted control siRNA oligonucleotide and incubated for 7 h at 37°C under standard tissue culture conditions. The medium was subsequently replaced, the cells were stimulated with SAA (10 μg/ml) for 24 h, the cell-culture medium was collected for IL-1β ELISA analysis and the cells were harvested for RT–PCR and Western blot analysis.
Statistical analysis
Results were expressed as the mean ± standard error of the mean. One-way analysis of variance was used to compare variances within and among groups. Data were evaluated statistically using post-hoc two-tailed Student's t-tests. Statistical significance was set at P < 0·05.
Results
SAA is increased in psoriatic epidermis
Because of previous suggestions that SAA can be produced in inflammatory tissue 18,19, we hypothesized that SAA may be up-regulated in epidermis of psoriasis patients. We first performed real-time RT–PCR on biopsies from lesional and non-lesional skin of 11 patients with psoriasis vulgaris and from eight normal controls. Compared to non-lesional or normal skin, SAA mRNA was up-regulated significantly in lesional skin of psoriasis patients (Fig. 1a). To determine whether SAA expression was increased at the protein level in psoriatic epidermis, we performed immunohistochemical analysis. Although detectable in the keratinocytes from normal skin, particularly in the basal and suprabasal layers, SAA immunoreactivity was markedly increased in psoriatic keratinocytes (Fig. 1b).
Figure 1.
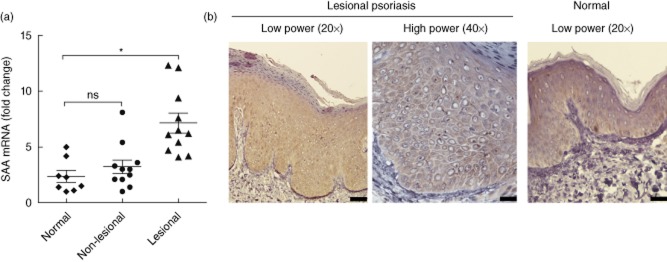
Psoriasis patients have a high abundance of serum amyloid A (SAA) in lesional skin. (a) The expression level of SAA mRNA was quantified by real-time reverse transcription–polymerase chain reaction (RT–PCR) in normal (n = 8), psoriatic non-lesional (n = 11) and lesional (n = 11) epidermal samples, and the data were normalized against the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Gene expression is graphed as mean fold induction over normal skin ± standard error of the mean (*P < 0·05). (b) Immunohistochemical detection of SAA proteins in representative normal and psoriatic lesional epidermis. Scale bars represent 10 μm in figures labelled ×40, and 20 μm in figures labelled ×20.
SAA triggers IL-1β secretion in keratinocytes
We postulated that in-vitro SAA might be an inducer of IL-1β in human keratinocytes. To verify this assumption, neonatal foreskin-isolated normal primary human keratinocytes were stimulated with SAA and the mRNA levels of IL-1β (pro-IL-1β mRNA) were determined by real-time RT–PCR. There was a 14-fold increase in the mRNA expression of IL-1β exposed to 100 μg/ml SAA compared to medium control (Fig. 2a). The induction of pro-IL-1β protein (31 kDa) was determined by Western blot analysis of keratinocyte lysates (Fig. 2b). In addition, the secretion of mature IL-1β was verified by ELISA. The production of mature IL-1β following stimulation with a range of concentrations of SAA was increased significantly (Fig. 2c).
Figure 2.
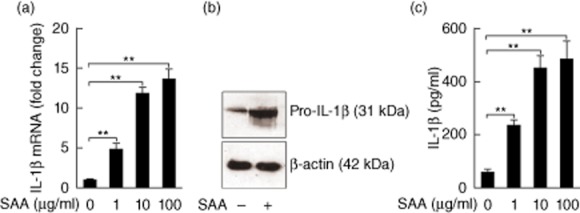
Serum amyloid A (SAA) induces the transcription and secretion of interleukin (IL)-1β in human keratinocytes. Neonatal foreskin-isolated normal primary kerationocytes were stimulated in the presence of various concentrations of SAA (1, 10 and 100 μg/ml) for 24 h. (a) IL-1β gene expression was assessed by real-time transcription–polymerase chain reaction (RT–PCR) and normalized against the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± standard error of the mean (s.e.m.) of three experiments with similar results (**P < 0·01). (b) Normal primary keratinocytes were stimulated with 10 μg/ml SAA for 24 h, and then pro-IL-1β protein (31 kDa) in the cell lysates was determined using Western blot. A single representative experiment is shown from three different experiments. (c) Mature IL-1β secretion into culture supernatant was assessed using enzyme-linked immunosorbent assay (ELISA). The data represent the mean ± s.e.m. of three experiments with similar results (**P < 0·01).
SAA induces expression of IL-1β mRNA via TLR-2, TLR-4 and NF-κB
The biological activity of IL-1β is regulated on both the transcriptional and post-transcriptional levels. First, we elucidated the transcriptional pathways by which SAA induces the up-regulation of IL-1β mRNA. In previous reports, SAA acted as an endogenous ligand for formylpeptide receptor-like 1 (FPRL1), TLR-2, TLR-4 and the scavenger receptor CD36 16. In our study, TLR-2, TLR-4 and CD36 but not FPRL1 were expressed in neonatal foreskin-isolated normal human primary keratinocytes (Supporting information, Fig. S1). Therefore, we tested the involvement of TLR-2, TLR-4 and CD36 in SAA recognition by using neutralizing antibodies. As demonstrated in Fig. 3a, the addition of TLR-2 antibody to the normal primary keratinocyte culture attenuated the SAA-induced IL-1β mRNA response significantly by approximately 53%. Inhibiting TLR-4 signalling by a TLR-4 antibody produced a similar inhibition of IL-1β mRNA by 30%. However, CD36 antibody did not modify the IL-1β mRNA expression (data not shown). Adding TLR-2 and TLR-4 antibodies together inhibited IL-1β mRNA expression by nearly 75%. Activation of NF-κB represents the final step in TLR signalling. Indeed, we found increased phosphorylated forms of NF-κB p65 in SAA-stimulated keratinocytes using flow cytometry (Fig. 3b). Using Bay11-7082, an NF-κB inhibitor, we demonstrated that SAA-induced IL-1β mRNA was suppressed significantly (Fig. 3c). These results suggest that activation of TLR-2/TLR-4–NF-κB signalling is involved in the SAA-mediated induction of IL-1β production.
Figure 3.
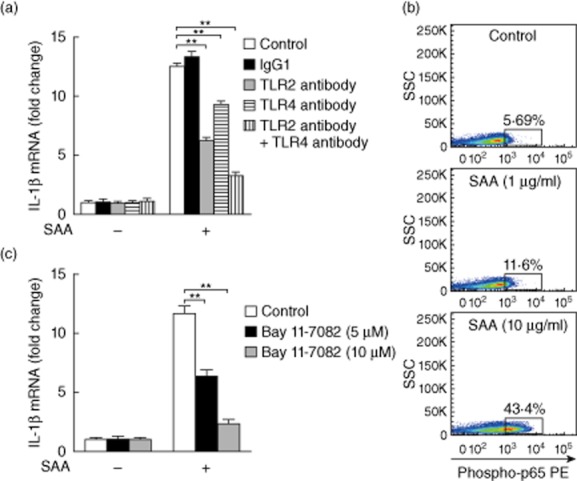
Serum amyloid A (SAA) up-regulates interleukin (IL)-1β transcription in keratinocytes via Toll-like receptor (TLR)-2 and TLR-4. (a) The neonatal foreskin-isolated normal primary keratinocytes were incubated in the presence of SAA (10 μg/ml) in combination with isotype control immunoglobulin (Ig)G1, TLR-2 antibody (10 μg/ml), TLR-4 antibody (20 μg/ml) or TLR-2 antibody + TLR-4 antibody for 24 h. The cells were then analysed for IL-1β mRNA levels by real-time transcription–polymerase chain reaction (RT–PCR). Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± standard error of the mean (s.e.m.) of three experiments with similar results (**P < 0·01). (b) Normal primary keratinocytes were exposed to the indicated concentrations of SAA (1 and 10 μg/ml) for 15 min. Nuclear factor kappa B (NF-κB) p65 phosphorylation was determined by flow cytometry. Values denote phospho-p65+ cells (%) from three independent experiments. (c) Normal primary keratinocytes were treated with SAA (10 μg/ml), in the presence NF-κB inhibitor Bay11-7082 at the indicated concentrations (5 and 10 μM). After 24 h, the cells were analysed for IL-1β mRNA levels by real-time RT–PCR. Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± s.e.m. of three experiments with similar results (**P < 0·01).
SAA activates the NLRP3 inflammasome in keratinocytes
Inflammasome activation results in the processing of pro-IL-1β into mature IL-1β on the post-transcriptional level. Next, we aimed to study the NLRP3 inflammasome complex involved in SAA-triggered IL-1β production. Normal keratinocytes were isolated from neonatal foreskin and cultured in the presence of SAA. Keratinocytes up-regulated the mRNA expression of caspase-1 and NLRP3 in response to SAA (Fig. 4a). SAA induced higher amounts of active caspase-1 subunits (p20) and NLRP3 at protein levels, suggesting NLRP3 inflammasome activation (Fig. 4b). IL-1β secretion following SAA treatment was inhibited in a concentration-dependent manner by Z-YVAD-fmk, the specific inhibitor of caspase-1, confirming the role of caspase-1 in SAA-induced IL-1β secretion (Fig. 4c). To further test the involvement of NLRP3 in SAA-triggered IL-1β release, we adopted RNA interference (siRNA). Transfection with siRNAs efficiently down-regulated NLRP3 mRNA (Fig. 4d) and protein expression (Fig. 4e) in SAA-treated keratinocytes. Knock-down of NLRP3 inhibited SAA-mediated IL-1β release significantly by approximately 65%, indicating a crucial role of the NLRP3 inflammasome in the response of keratinocytes to SAA (Fig. 4f).
Figure 4.
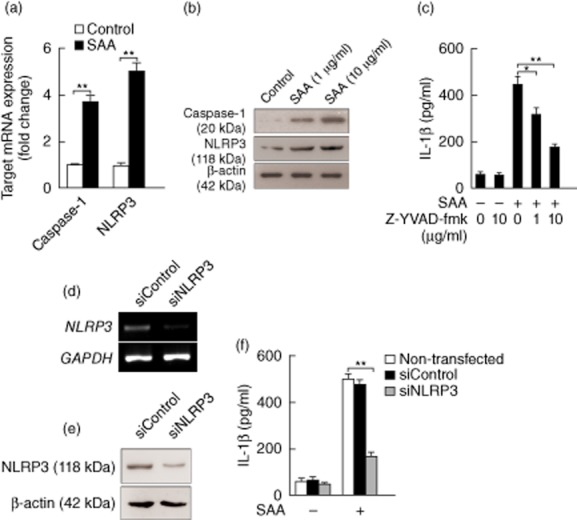
Serum amyloid A (SAA) induction of interleukin (IL)-1β is via NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome in keratinocytes at post-transcriptional level. (a) The neonatal foreskin-isolated normal primary kerationocytes were stimulated with SAA (10 μg/ml) for 24 h, and the mRNA expression of caspase-1 and NLRP3 was determined using real-time transcription–polymerase chain reaction (RT–PCR) and normalized against the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± standard error of the mean (s.e.m.) of three independent experiments (**P < 0·01). (b) Normal primary keratinocytes were treated with indicated concentrations of SAA (1 and 10 μg/ml) for 24 h. The cells were lysed and the whole-cell extracts were assessed by Western blot using antibodies against NLRP3 (118 kDa) and the mature form of caspase-1 (20 kDa). Representative blots of three independent experiments are shown. (c) Normal primary keratinocytes were treated with caspase-1 inhibitor Z-YVAD-fmk at the indicated concentrations (1 and 10 μg/ml) for 30 min and then stimulated with SAA (10 μg/ml). Culture supernatants were collected after 24 h and assayed with IL-1β enzyme-linked immunosorbent assay (ELISA). The data represent the mean ± s.e.m. of three experiments with similar results (*P < 0·05; **P < 0·01). Keratinocytes were transfected with siRNA oligonucleotides specific for NLRP3 (siNLRP3) or a non-specific siRNA oligonucleotide (siControl), and subsequently stimulated with SAA (10 μg/ml) for 24 h, and the levels of NLRP3 mRNA (d) and protein (e) were analysed using RT–PCR and Western blot. One experiment representative of three experiments. (f) siRNA-transfected keratinocytes were then stimulated with SAA (10 μg/ml), and culture supernatants were collected after 24 h and assayed with IL-1β ELISA. The data represent the mean ± s.e.m. of three experiments with similar results (**P < 0·01).
ROS generation is required for SAA-induced IL-1β secretion from keratinocytes
ROS have been proposed to perform an important role in the activation of NLRP3 inflammasome, and SAA has been reported to be an inducer of ROS 20,21. We hypothesized that ROS formation might be involved in SAA-mediated inflammasome activation and IL-1β secretion. To verify this assumption, neonatal foreskin-isolated normal primary human keratinocytes were stimulated with SAA. As determined by 2′,7′-dichlorofluorescein (DCF) labelling and flow cytometry, ROS formation in keratinocytes increased significantly following stimulation with SAA, and preconditioning with the anti-oxidant NAC partially abrogated this effect (Fig. 5a). NAC modulated the expression of caspase-1 and NLRP3 significantly at mRNA and protein levels (Fig. 5b,c). Furthermore, SAA-induced IL-1β secretion was inhibited significantly by NAC. These data indicate that activation of the NLRP3 inflammasome by SAA is dependent upon ROS generation.
Figure 5.
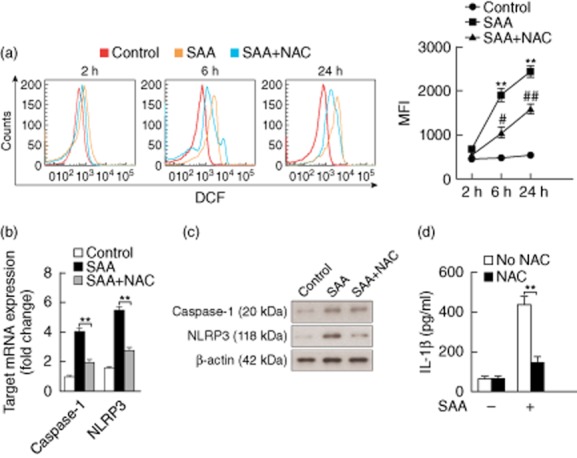
Serum amyloid A (SAA) activation of inflammasome and interleukin (IL)-1β secretion in keratinocytes is regulated by reactive oxygen species (ROS). (a) The neonatal foreskin-isolated normal primary keratinocytes were preincubated for 2, 6 or 24 h with SAA (10 μg/ml) in the presence of 5 mM of N-acetyl-l-cysteine (NAC). The cells were then collected and labelled with 2′,7′-dichlorofluorescein (DCF) and ROS production was measured by flow cytometry. Representative histograms and mean fluorescence intensities (MFI); mean ± standard errors of the mean (s.e.m.) were obtained from five independent experiments (**P < 0·01 versus medium control; #P < 0·05, ##P < 0·01 versus SAA-treated cells). (b) Normal primary keratinocytes were treated with SAA (10 μg/ml) in combination with NAC (5 mM) for 24 h, and the mRNA levels of caspase-1 and NACHT, LRR and PYD (NLRP3) were determined using real-time transcription–polymerase chain reaction (RT–PCR) and normalized against the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± s.e.m. of three experiments with similar results (**P < 0·01). (c) Normal primary keratinocytes were treated with 10 μg/ml SAA for 24 h in the presence of NAC (5 mM). NLRP3 (118 kDa) and the mature form of caspase-1 (20 kDa) were then analysed by Western blot. Representative blots of three independent experiments are shown. (d) Normal primary keratinocytes were treated with SAA (10 μg/ml) in the presence of 5 mM NAC, and then culture supernatants were collected after 24 h and assayed with IL-1β enzyme-linked immunosorbent assay (ELISA). The data represent the mean ± s.e.m. of three experiments with similar results (**P < 0·01).
IL-17A stimulates SAA production in keratinocytes
There is now convincing evidence that IL-1β functions in synergy with IL-23 to promote the production of IL-17A from Th17 cells. Next, we examined whether IL-17A can, in turn, stimulate SAA production in normal keratinocytes. SAA mRNA expression in keratinocytes was increased by IL-17A in a time- and dose-dependent fashion (Fig. 6a), while psoriasin (S100A7) mRNA, an anti-microbial defence protein, was also up-regulated by this treatment (Supporting information, Fig. S2). Increased SAA protein was also observed by immunofluorescence of keratinocytes treated with IL-17A (Fig. 6b). Moreover, the SAA production in keratinocytes following exposure to IL-17A was confirmed by ELISA. This effect was specific to IL-17 receptor A (IL-17RA) signalling as blocking antibodies to IL-17RA, but not isotype-matched antibodies, abrogated the secretion of SAA (Fig. 6c). These observations suggest that SAA-activated inflammasome may promote a positive feed-forward loop, serving as a link between the IL-1β and Th17 pathways.
Figure 6.

Interleukin (IL)-17A induces serum amyloid A (SAA) production from keratinocytes. (a) The neonatal foreskin-isolated normal primary keratinocytes were cultured for 2, 6 and 24 h in the presence of IL-17A at the indicated concentrations (10, 50 and 200 ng/ml), the level of SAA mRNA in the cells was analysed by real-time transcription–polymerase chain reaction (RT–PCR) and the data were normalized for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± standard error of the mean (s.e.m.) of three experiments with similar results (*P < 0·05; **P < 0·01). (b) Normal primary keratinocytes were treated with 200 ng/ml IL-17A for 24 h, then SAA protein was measured by immunofluorescence. Scale bars represent 100 μm. (c) Normal primary keratinocytes were treated with 200 ng/ml IL-17A in the presence of neutralizing anti-IL-17RA antibodies. After 48 h, supernatants were harvested and tested for SAA by enzyme-linked immunosorbent assay (ELISA). The values indicate the mean ± s.e.m. of three experiments (*P < 0·05; **P < 0·01).
Discussion
A hallmark of the acute-phase response is the rapid increase in production of acute-phase proteins, including SAA and C-reactive protein (CRP) 22,23. Acute-phase protein production is a clinical marker for many inflammatory diseases, including atherosclerosis, rheumatoid arthritis and inflammatory bowel diseases 12,24,25. They also participate in the modulation of innate and adaptive immune responses. SAA was shown to promote the development of the Th17 response in a mouse model of asthma 26. SAA was also demonstrated to induce the secretion of IL-23 in peripheral blood monocytes, leading to IL-23-dependent expansion of Th17 cells 27. A recent study showed that SAA can stimulate angiogenesis in psoriatic dermis through Notch-1 signalling, implying a role for SAA in the pathogenesis of psoriasis 28. However, despite these exciting developments in understanding the immunological role of SAA, little is known about the production and function of SAA in psoriasis skin.
In this study, we showed that SAA mRNA and protein was up-regulated in psoriatic epidermis, suggesting that SAA may be not only a circulating acute-phase protein but also a local inflammatory mediator. Although hepatocytes have been reported to be the major source of circulating SAA, the expression of SAA has been detected in various normal tissues such as breast, kidney, skin and intestine, as well as in diseased tissues, including atherosclerotic plaques, brain tissue of patients with Alzheimer's disease and synovial tissue of patients with rheumatoid arthritis 24,29–31. Our observations extend these findings by demonstrating that psoriatic epidermis might be another local source of SAA. It is likely that SAA derived from epidermal keratinocytes contributes to sustaining a local inflammation and hypervascularization in psoriasis. The contribution of keratinocyte-derived SAA to the circulating SAA remains to be determined. To this end, the correlation between circulating SAA and involvement of body surface or PASI should be investigated in further studies. Recently, there have been two reports showing the elevated levels of SAA among psoriatic patients 13,28. However, confirmation of this observation is required in mild, moderate and severe disease. In addition, changes in SAA levels in relation to clinical responses should be assessed in future studies.
Chronic immune responses in the psoriatic epidermis involve the release of various cytokines, including IL-1β. There is strong evidence that the IL-1β pathway contributes to the initiation of Th17 cell-mediated autoinflammatory skin diseases such as psoriasis 4. IL-1β functions in synergy with IL-23 to promote the production of IL-17 and related cytokines from Th17 cells but also from subpopulations of γδ T cells, which may play a pathogenic role in psoriasis 32. Human keratinocytes constitutively synthesize pro-IL-1β and its receptor, IL-1RA, but under normal conditions do not activate and secrete these proinflammatory cytokines. Upon stimulation with UVB irradiation or skin irritant, the active form of IL-1β is released from keratinocytes, particularly involving the activation of NLRP3 inflammasomes. Recently, it was shown that cytosolic DNA is an important disease-associated molecular pattern that can trigger AIM2 inflammasome and IL-1β activation in psoriasis 33.
In this study, we confirmed that induction of pro-IL-1β expression in keratinocytes by SAA was mediated through TLR-2, TLR-4 and NF-κB. These findings were consistent with a previous report, which showed that SAA can signal through TLR-2 and TLR-4 in monocytes 16. Because the acute-phase response is a systemic reaction to infection and tissue injury that protects the host by isolating pathogens and minimizing tissue damage, it is not surprising that SAA can act as a danger signal, which can bind and stimulate the pattern recognition receptors, specifically TLRs.
On the post-transcriptional level, proteolytic activation of IL-1β is regulated by inflammasomes. Although normal activation of the NLRP3 inflammasome contributes to host defence, excessive activation results in the production of IL-1β. In previous reports, the presence of the NLRP3 inflammasome complex and the NLRP3 inflammasome protein components in keratinocytes have been described in both cultured and primary human keratinocytes 9,10,34. In psoriasis, both caspase-1 activity and NLRP3 expression have been reported to be increased in lesional skin 35,36. Consistent with these reports, we showed that caspase-1 and NLRP3 were up-regulated by SAA in keratinocytes, suggesting that SAA could be one of the disease-associated molecular patterns in psoriasis, inducing the activation of IL-1β in keratinocytes by the NLRP3 inflammasome. The identification of such molecular patterns in inflammatory diseases is crucial, and could lead to novel therapies targeting these patterns.
The precise mechanism involved in activation of the NLRP3 inflammasome remains unclear. However, we demonstrated that ROS generation was necessary for NLRP3 inflammasome activation in keratinocytes and subsequent IL-1β release in response to SAA. Skin is a primary target of oxidative stress, and disturbances in the oxidant–anti-oxidant system in skin may play an important role in the pathogenesis of psoriasis 37. SAA and ROS are observed commonly at inflammation sites, and it has been shown recently that SAA stimulates ROS release from neutrophils and fibroblasts. Therefore, we hypothesize that SAA activates keratinocytes in a positive feedback loop producing both inflammatory mediators and ROS.
The role of Th17 responses in the pathogenesis of psoriasis is well established. Cytokines secreted from Th17 cells, particularly IL-17A, induce keratinocytes to express various cytokines, chemokines and anti-microbial peptides, which are proposed to sustain the inflammatory cascade in psoriatic lesions. To our knowledge, this is the first report that IL-17A stimulates SAA expression in keratinocytes, while previous reports have shown that hepatic expression of SAA could be induced notably by tumour necrosis factor (TNF)-α, IL-1β and IL-6 38. Therefore, it is possible that IL-17A may act in synergy with other proinflammatory cytokines in stimulating SAA expression in vivo in epidermis.
In summary, our data indicate that SAA is expressed strongly in psoriatic lesional epidermis, and SAA stimulates keratinocytes to produce IL-1β in an NLRP3 inflammasome-mediated mechanism, thereby providing positive feedback regulation of Th17 responses. As an acute-phase protein with pleiotropic proinflammatory and proangiogenic properties, keratinocyte-derived SAA may be a messenger in the cross-talk between the Th17 and IL-1β pathways.
Acknowledgments
This study was supported by Research Foundation by the Health Bureau of Shanghai City (20124126), Research Foundation by the Shanghai Municipal Science and Technology Commission (12411951702) and Young Talent Project by the Health Bureau of Shanghai City (20114Y064).
Disclosure
The authors declare no conflicts of interest.
Supporting Information
Fig. S1. Keratinocytes express mRNA for Toll-like receptor (TLR)-2, TLR-4 and CD36. Total RNA was prepared from neonatal foreskin-isolated normal primary keratinocytes and was analysed for FPRL1, TLR-2, TLR-4 and CD36 expression by reverse transcription–polymerase chain reaction (RT–PCR). Representative bands of three independent experiments are shown.
Fig. S2. Interleukin (IL)-17A induces S100A7 mRNA expression in keratinocytes. The neonatal foreskin-isolated normal primary keratinocytes were cultured for 2, 6 and 24 h in the presence of IL-17A (50 ng/ml), and then S100A7 mRNA was analysed by real-time reverse transcription–polymerase chain reaction (RT–PCR) and the data were normalized for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± standard error of the mean of three experiments with similar results (*P < 0·05; **P < 0·01).
References
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Zielinski CE, Mele F, Aschenbrenner D. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- Feldmeyer L, Werner S, French LE, Beer HD. Interleukin-1, inflammasomes and the skin. Eur J Cell Biol. 2010;89:638–644. doi: 10.1016/j.ejcb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Guo L, Wei G, Zhu J. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci USA. 2009;106:13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KH, Dunne A. Immune modulation: IL-1, master mediator or initiator of inflammation. Nat Med. 2009;15:1363–1364. doi: 10.1038/nm1209-1363. [DOI] [PubMed] [Google Scholar]
- Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev. 2011;243:152–162. doi: 10.1111/j.1600-065X.2011.01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KH, Dungan LS, Jones SA, Harris J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J Leukoc Biol. 2013;93:489–497. doi: 10.1189/jlb.1012543. [DOI] [PubMed] [Google Scholar]
- Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Gaide O, Pétrilli V. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956–1963. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- Sodin-Semrl S, Zigon P, Cucnik S. Serum amyloid A in autoimmune thrombosis. Autoimmun Rev. 2006;6:21–27. doi: 10.1016/j.autrev.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Migita K, Izumi Y, Jiuchi Y. Effects of Janus kinase inhibitor tofacitinib on circulating serum amyloid A and interleukin-6 during treatment for rheumatoid arthritis. Clin Exp Immunol. 2014;175:208–214. doi: 10.1111/cei.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan S, Atakan N. Is serum amyloid A protein a better indicator of inflammation in severe psoriasis? Br J Dermatol. 2010;163:895–896. doi: 10.1111/j.1365-2133.2010.09907.x. [DOI] [PubMed] [Google Scholar]
- Migita K, Izumi Y, Jiuchi Y. Serum amyloid A induces NLRP-3-mediated IL-1β secretion in neutrophils. PLOS ONE. 2014;9:e96703. doi: 10.1371/journal.pone.0096703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi K, Baumann MH, Kovanen PT, Eklund KK. Serum amyloid A (SAA) activates human mast cells which leads into degradation of SAA and generation of an amyloidogenic SAA fragment. Biochim Biophys Acta. 2006;1762:424–430. doi: 10.1016/j.bbadis.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Niemi K, Teirilä L, Lappalainen J. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186:6119–6128. doi: 10.4049/jimmunol.1002843. [DOI] [PubMed] [Google Scholar]
- Migita K, Koga T, Satomura K. Serum amyloid A triggers the mosodium urate -mediated mature interleukin-1β production from human synovial fibroblasts. Arthritis Res Ther. 2012;14:R119. doi: 10.1186/ar3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly M, Marrelli A, Blades M. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol. 2010;184:6427–6437. doi: 10.4049/jimmunol.0902941. [DOI] [PubMed] [Google Scholar]
- Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci USA. 1994;91:3186–3190. doi: 10.1073/pnas.91.8.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- Hatanaka E, Dermargos A, Armelin HA, Curi R, Campa A. Serum amyloid A induces reactive oxygen species (ROS) production and proliferation of fibroblast. Clin Exp Immunol. 2011;163:362–367. doi: 10.1111/j.1365-2249.2010.04300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- King VL, Thompson J, Tannock LR. Serum amyloid A in atherosclerosis. Curr Opin Lipidol. 2011;22:302–307. doi: 10.1097/MOL.0b013e3283488c39. [DOI] [PubMed] [Google Scholar]
- Chambers RE, Stross P, Barry RE, Whicher JT. Serum amyloid A protein compared with C-reactive protein, alpha 1-antichymotrypsin and alpha 1-acid glycoprotein as a monitor of inflammatory bowel disease. Eur J Clin Invest. 1987;17:460–467. doi: 10.1111/j.1365-2362.1987.tb01143.x. [DOI] [PubMed] [Google Scholar]
- Ather JL, Ckless K, Martin R. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011;187:64–73. doi: 10.4049/jimmunol.1100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Shepard LW, Chen J, Pan ZK, Ye RD. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J Immunol. 2006;177:4072–4079. doi: 10.4049/jimmunol.177.6.4072. [DOI] [PubMed] [Google Scholar]
- Rooney P, Connolly M, Gao W. Notch-1 mediates endothelial cell activation and invasion in psoriasis. Exp Dermatol. 2014;23:113–118. doi: 10.1111/exd.12306. [DOI] [PubMed] [Google Scholar]
- Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J Histochem Cytochem. 1998;46:1377–1384. doi: 10.1177/002215549804601206. [DOI] [PubMed] [Google Scholar]
- Chung TF, Sipe JD, McKee A. Serum amyloid A in Alzheimer's disease brain is predominantly localized to myelin sheaths and axonal membrane. Amyloid. 2000;7:105–110. doi: 10.3109/13506120009146246. [DOI] [PubMed] [Google Scholar]
- O'Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Acute-phase serum amyloid A production by rheumatoid arthritis synovial tissue. Arthritis Res. 2000;2:142–144. doi: 10.1186/ar78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Pantelyushin S. Hiding under the skin: interleukin-17-producing γδ T cells go under the skin? Nat Med. 2012;18:1748–1750. doi: 10.1038/nm.3016. [DOI] [PubMed] [Google Scholar]
- Dombrowski Y, Peric M, Koglin S. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. 2011;3:82ra38. doi: 10.1126/scitranslmed.3002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Sayama K, Tohyama M. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J Allergy Clin Immunol. 2011;127:806–814. doi: 10.1016/j.jaci.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Carlström M, Ekman AK, Petersson S, Söderkvist P, Enerbäck C. Genetic support for the role of the NLRP3 inflammasome in psoriasis susceptibility. Exp Dermatol. 2012;21:932–937. doi: 10.1111/exd.12049. [DOI] [PubMed] [Google Scholar]
- Johansen C, Moeller K, Kragballe K, Iversen L. The activity of caspase-1 is increased in lesional psoriatic epidermis. J Invest Dermatol. 2007;127:2857–2864. doi: 10.1038/sj.jid.5700922. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Mrowietz U, Rostami-Yazdi M. Oxidative stress in the pathogenesis of psoriasis. Free Radic Biol Med. 2009;47:891–905. doi: 10.1016/j.freeradbiomed.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Uhlar CM, Grehan S, Steel DM, Steinkasserer A, Whitehead AS. Use of the acute phase serum amyloid A2 (SAA2) gene promoter in the analysis of pro- and anti-inflammatory mediators: differential kinetics of SAA2 promoter induction by IL-1 beta and TNF-alpha compared to IL-6. J Immunol Methods. 1997;203:123–130. doi: 10.1016/s0022-1759(96)00220-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Keratinocytes express mRNA for Toll-like receptor (TLR)-2, TLR-4 and CD36. Total RNA was prepared from neonatal foreskin-isolated normal primary keratinocytes and was analysed for FPRL1, TLR-2, TLR-4 and CD36 expression by reverse transcription–polymerase chain reaction (RT–PCR). Representative bands of three independent experiments are shown.
Fig. S2. Interleukin (IL)-17A induces S100A7 mRNA expression in keratinocytes. The neonatal foreskin-isolated normal primary keratinocytes were cultured for 2, 6 and 24 h in the presence of IL-17A (50 ng/ml), and then S100A7 mRNA was analysed by real-time reverse transcription–polymerase chain reaction (RT–PCR) and the data were normalized for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Gene expression is graphed as mean fold induction over medium control. The data represent the mean ± standard error of the mean of three experiments with similar results (*P < 0·05; **P < 0·01).