

Abstract
The most potent killing machinery in our immune system is the cytotoxic T lymphocyte (CTL). Since the possibility for self-destruction by these cells is high, many regulatory activities exist to prevent autoimmune destruction by these cells. A tumour (cancer) grows from the cells of the body and is tolerated by the body's immune system. Yet, it has been possible to generate tumour-associated antigen (TAA) -specific CTL that are also self-antigen specific in vivo, to achieve a degree of therapeutic efficacy. Tumour-associated antigen-specific T-cell tolerance through pathways of self-tolerance generation represents a significant challenge to successful immunotherapy. CD4+ CD25+ FoxP3+ T cells, referred to as T regulatory (Treg) cells, are selected in the thymus as controllers of the anti-self repertoire. These cells are referred to as natural T regulatory (nTreg) cells. According to the new consensus (Nature Immunology 2013; 14:307–308) these cells are to be termed as (tTreg). There is another class of CD4+ Treg cells also involved in regulatory function in the periphery, also phenotypically CD4+ CD25±, classified as induced Treg (iTreg) cells. These cells are to be termed as peripherally induced Treg (pTreg) cells. In vitro-induced Treg cells with suppressor function should be termed as iTreg. These different Treg cells differ in their requirements for activation and in their mode of action. The current challenges are to determine the degree of specificity of these Treg cells in recognizing the same TAA as the CTL population and to circumvent their regulatory constraints so as to achieve robust CTL responses against cancer.
Keywords: regulatory T cells, tumour immunology, vaccination
Introduction
Regulatory T (Treg) cells are thought to exert regulatory function in the entire immune system, ranging from regulation of immune responses against self-antigen, infectious agents, tumour antigens and transplantation antigens. Although the literature on Treg cells in the entire immune response system is substantial, many important issues on Treg cells (their ontogeny, mechanism of activation, mode of action, specificity, etc.) remain to be fully elucidated. The suppression of active specific immune response represents a significant challenge to cancer immunotherapy.1 Nonetheless, the following points were accepted by the immunologists
Regulatory T cells are primarily classified as CD4+ CD25+ FoxP3+ T cells. Although several types of CD4+ T cells are capable of exerting regulatory function, a particular class of Treg cells, bearing the CD4 and CD25 and FoxP3 markers and seemingly selected by the thymus, serves as natural T regulatory cells (nTreg, now to be termed tTreg). These tTreg cells differ from inducible Treg cells (iTreg, now to be termed pTreg) that are generated in the periphery from CD4+ CD25− precursors.2–7
In addition to expressing CD25, tTreg cells express cytolytic T lymphocyte antigen 4 (CTLA-4), glucocorticoid-inducible tumour necrosis factor receptor family-related (GITR), and FoxP3 (a fork head family transcriptional regulator).8–10 Although none of the aforementioned markers is a distinct marker of the lineage, FoxP3 has become useful as a marker as well as a critical factor for the ‘differentiation’ of tTreg cells.11,12
The tTreg cells need to be activated via their T-cell receptors (TCR) but they do not need simultaneous TCR signalling and co-stimulation. These cells function in a contact-dependent manner and suppress non-specifically in a bystander fashion.13,14
The role of tTreg cells has mostly been ascribed to maintaining ‘self-tolerance’ in the periphery. Although other types of Treg cells can be induced (pTreg) from CD4+ CD25− precursors to suppress and/or dampen immune responses to antigens – ‘self or non-self’ – the regulatory role of tTreg/pTreg cells has been attributed to the control of immune responses against infectious agents, tumours and transplantations.15
In addition to the experimental observations regarding tTreg cells, other observations regarding other immune cells with similar phenotype have been elucidated. For example, ROR+ T helper type 17 (Th17) cells have been found to be capable of autoimmune destruction. Another class of cells known as T follicular helper cells also bears the same CD4 markers as tTreg cells. These cells play an important role in the CD4+ population by helping the activation and differentiation of B cells into immunoglobulin-secreting cells. Unlike pathogen-derived antigens, cancer-associated antigens are released in the body in a slow and continuous manner. The response of B cells with the help from T follicular helper cells might provide a slow and steady antibody reaction against the cancer antigens that could provide additional help for vaccine-induced CTL activity. Recent research has suggested that tumours with T follicular helper cell infiltration were associated with increased survival rates, which can be used in propagating vaccine-induced CTL activity for better prognoses.16 Although these cells are helpers (not suppressors), appropriate utilization of these cells could provide extra help for mitigating activation and expansion of Treg cells.
The above issues concerning Treg cells have been covered in many important review articles.17–21 We will discuss here the role of Treg cells, especially nTreg (tTreg), iTreg (pTreg) and in vitro-induced iTreg in tumour immunity, from the viewpoint that ‘tumour immunity’ and ‘autoimmunity’ can be viewed as essentially the same process. We will proceed with this viewpoint because the idea of generating an immune response against tumour antigens is similar to breaking ‘tolerance’ for self-antigens, as most tumour-associated antigens (TAA) are also self-antigens. While the classifications of Treg cells discussed above have been used in the literature, the newest recommendations for the nomenclature of such cells needs be used to avoid any ambiguity.22 These recommendations include the use of thymus-derived Treg (tTreg previously termed as nTreg) and peripherally derived Treg (pTreg) cells in place of natural Treg cell and induced Treg cell, respectively. In addition, the term ‘in vitro-induced Treg cell’ should be used to distinguish between the populations generated in vivo and those generated in vitro.22 Treg cells as such will be considered as T cells with suppressor activity. More detail about this consensus and the nomenclature is described in a review article.23
In this article, we also intend to discuss the promising advances in US Food and Drug Administration-approved agents in use for clinical trials to combat cancer by interfering with Treg cell activities. Several clinical trials showed that cyclophosphamide, fludarabine, CTLA-4 antibodies, and programmed cell death 1 (PD1) blockers have had considerable success in circumventing Treg cells through different mechanisms of action.24–27 We will discuss the use of existing chemotherapeutic agents for the removal of CD4+ T cells that initiate suppressor activity in CD8+ T cells by cyclophosphamide and also the inhibition of pTreg cell expansion with simultaneous fludarabine-maintained CTL activity.24,25 We will also include current clinical studies trying to block the signalling molecules CTLA-4 and PD1 that down-regulate the effector response, and show great promise in the enhancement of the anti-tumour immune response.26,27 Some of these chemotherapeutic agents in low doses were found to block the expansion of the Treg cells and in turn prolong the cytotoxic activity of the killer cells. However, the exact mechanism of how the drugs work is not yet known.
Immune response against tumours
In the early twentieth century, Paul Ehrlich first introduced the idea that there could be an immune response against tumours, which was later proven by several scientists in animal and human tumour systems.28–31 As a result of Ehrlich's influential hypothesis and subsequent important studies, it is now clear that many cancer patients can mount serological as well as cellular immune responses against their own cancer cells. Moreover, it has been determined that any cancer-bearing host can respond to a large number of cancer-associated antigens or TAA as well as antigenic epitopes, whether they are classified as self or mutated.31–34 It is also clear that vaccination with some of these TAA epitopes, administered with or without an adjuvant or presented by ex vivo cultured antigen-presenting cells (APC) or dendritic cells (DC), could induce serological and CTL responses.31,33–35 This raises the question of how the immune activation/expansion is controlled in the periphery. Later, this question was clarified when it became clear that tumour cells are quite capable of using multiple methods of escaping a host immune response.36–41 Here, we will concentrate on the role of natural (tTreg + pTreg) or induced (pTreg or in vitro-induced iTreg) cells in controlling host immune responses against tumours.
T regulatory cells and the anti-tumour immune response
North's group first demonstrated CD4+ T cells functioning as suppressor cells in an anti-tumour response in a murine model in vivo.42,43 Regulation of an immune response by CD4+ T cells was also demonstrated in a human tumour model in vitro.40,44,45 In a human melanoma model, CD4+ regulatory T cells were isolated from lymph nodes, tumour tissues and blood. These cells suppressed the CD8+ CTL response in an in vitro CTL generation assay.37,45,46 The CD4+ Treg cells generated from in vitro cultures also expressed CD25, up-regulated CD25 upon subsequent stimulation, and functioned in MHC class II restricted fashion mostly by elaborating interleukin-10 (IL-10).44,45 These in vitro observations on suppression of anti-tumour CTL by CD4+ T cells in humans, however, could not establish the biological significance because they were exclusively in vitro studies and the specificity of these CD4+ Treg cells could not be clarified. We would like to emphasize our work,45 where we showed that immunization of melanoma patients with synthetic peptide or tumour-lysate-loaded APC-based vaccines could lead to the expansion of epitope-specific CD8+ CTL cells, in vivo. However, repetitive vaccinations also induced IL-10-producing CD4+ regulatory cells, in vivo.45 Subsequent research by Wang et al.47 showed that this type of CD4+ Treg cells exhibit specificity for an epitope derived from the tumour-associated but self-antigen LAGE-1. At the same time as Wang's research, Sakaguchi's group demonstrated that the removal of CD4+ CD25+ T cells as well as injecting monoclonal antibody against CD25 to the mice could induce anti-tumour response and enunciated a ‘common basis’ between tumour immunity and autoimmunity. These basic observations were further supported by other laboratories45,46,48,49 and generated additional information that showed that these CD4+ CD25+ Treg cells interfere with the generation of long-lasting tumour immunity. Furthermore, removal of the Treg cells results in better outcome from tumour immunotherapy, prompting investigations into Treg cell circumvention.50,51
A number of studies in human and mouse tumour models, described the increased frequencies of CD4+ CD25+ Treg cells in blood, malignant effusions, draining lymph nodes and tumour tissues, so implicating impaired immune responses to cancer due to a higher frequency and/or hyperactivity of Treg cells.50–55 Freshly isolated CD4+ CD25+ T cells from cancer patients, or from patients receiving immunotherapy, were found to suppress the proliferation of CD4+ CD25− T cells in vitro, an assay that has been extensively used to assess Treg cell function in vitro.56–58 The results from these studies suggested that a higher frequency of and/or hyperactivity in the CD4+ CD25+ T-cell population might have a negative effect on anti-tumour response. Furthermore, a stronger correlation between Treg cell activities and impaired tumour immunity in human tumour models has emerged from two groups of investigators.59,60 The results described by Curiel et al.61 reported that CD4+ CD25+ GITR+ FoxP3+ T cells preferentially accumulate in ovarian tumours and in malignant ascites seemingly attracted by CCL22 elaborated by tumour cells and macrophages in tumour beds. Curiously, these cells tend not to accumulate in lymph nodes. In addition, Curiel's group has also shown that such accumulation of CD4+ CD25+ GITR+ FoxP3+ Treg cells in tumour sites correlates with poor outcome.61 In contrast, Viguier et al.62 has found higher accumulation of Treg cells in draining lymph nodes infiltrated by melanoma cells and both IL-10-producing Tr1 type Treg cells and IL-10-negative Treg cells in tumour-infiltrated nodes. As such, they have suggested that both tTreg and pTreg cells contribute to the local immunosuppressive milieu.
Mechanism of Treg-based suppression of anti-tumour immunity
The literature on Treg cell activities in anti-tumour immunity is substantial. Yet, the mechanism behind Treg cell-based regulation of anti-tumour immunity is poorly understood. This is because both tTreg and pTreg cells can be involved in the suppression of anti-tumour immunity. However, the key differentiation factor between these two types of Treg cells (specifically in their requirements for activation as well as in their mode of action) is unknown. Hence, a critical review of the mechanism of activation and relative efficacy under which Treg cells operate to control anti-tumour immunity will be useful. To control the negative roles of Treg cells, it is imperative to delineate how the Treg cell-based regulatory arm operates in anti-tumour immunity. It is also important to determine which of the two Treg cells (tTreg versus pTreg) pose more of a constraint in anti-tumour immunity.63–66 Hence the most important, question is ‘how can we circumvent these regulatory constraints’?
While the topic of mechanism of activation has been extensively addressed with Treg cells involved in controlling autoimmunity, these cells have not been systematically studied in the tumour immunity model. There is the existence of the common basis in autoimmunity and tumour immunity. The information generated in the autoimmunity model might be extrapolated in the tumour immunity model as well. It is generally understood that with a CD4+ T-cell classification, tTreg cells as well as pTreg cells need to be activated by MHC class II bound epitopes on APC/DC. The caveat in this fundamental construct is that while pTreg cells need both TCR ligand and co-stimulation, tTreg cells need only a TCR-driven signal for functional activation. Nonetheless, as only a limited class of cells express MHC class II molecules, tTreg cells need APC for their activation.14 Hence, from an operational viewpoint, APC are indispensable in their dual functions of activating naive effector T cells and Treg cells. Admittedly, the nature of the microenvironment that induces tolerance is yet to be fully understood. Whatever the underlying mechanism of antigen presentation that leads to the activation and polarization of Tr1 or Th3 type T cells might be, the fact remains that the induction of a pTreg response from CD4+ CD25− precursors is a process that requires antigen presentation. In this regard, the important work by Mempel et al.67 in Immunity is worth mentioning. In that article, they have demonstrated how CTL interact with antigen-presenting target cells in the presence or absence of activated Treg cells by using multiphoton intravital microscopy in lymph nodes of anaesthetized mice. They have shown that non-regulated CTL killed their targets at a 6·6-fold faster rate than regulated. Other than this compromised killing activity, regulated CTL exhibited no defect in proliferation, induction of cytotoxic effector molecules and secretory granules, in situ motility, or ability to form antigen-dependent conjugates with target cells etc. Furthermore, after the regulated CTL are detached from the Treg cells, the regulated CTL regain their killing efficiency.67
Until now extensive studies could not define the requirements for the activation of tTreg cells. In fact, the literature on this subject is confusing and, at times, contradictory. It is believed that tTreg cells are selectively ‘anergic’, but they are anergic only to ‘weak’ TCR signals (e.g. to soluble anti-CD3 antibody or to phytohaemagglutinin) and not to ‘strong’ stimuli (to plate-bound anti-CD3 antibody or to phytohaemagglutinin plus PMA).68 It has been shown that tTreg cells can be expanded in cultures. The in vitro-expanded tTreg cells function as more potent suppressors and are believed to be driven by self-peptides and require only TCR stimulation for functional activation. At the same time, these cells seem to require TCR signals and co-stimulation for their maintenance. It has been proposed that IL-2 plays a ‘critical role’ for their functional activation, although these cells are unable to synthesize IL-2 and have been shown to prevent IL-2 synthesis by the effector cells. Moreover, it has been shown that the strength of activation signals for the effector cells is an important determinant as to whether tTreg cells could possibly block the effector cell activation or not. It is not, however, clear if a ‘strong’ signal makes effector cells refractory to the tTreg cells or if the robustness of the effector cell response turns them off by some unknown mechanism.68–71,57 Presently, it is apparent that while the activation requirements for tTreg cells are not the same as that of naive T cells, the rule that governs their activation in the regulation of tumour immunity is yet to be established. The activation of such cells might vary depending upon the type of tumour and affected organ.
At present, our ability to appropriately describe the development and function of the unique Treg cells, how FoxP3 is controlled, and what type of microenvironment or physiological inducers is involved, is poor. The recent finding by Strainic et al.72 provides evidence that suppressive FoxP3+ iTreg (in vitro studies) cells can be generated when human naive CD4+ T cells are activated in co-cultures with DC by combined treatment with anti-CD3 plus IL-2 when C3aR, C5aR, or their cognate ligand are targeted pharmacologically. In contrast to transforming growth factor-β (TGF-β) -generated human iTreg cells, these iTreg cells do not produce IL-2 or proliferate after stimulation, similar to the results obtained with mice. This observation is important, because this may suggest a direction for a new option to block pTreg cells to prolong the therapeutic vaccine-induced CTL activity. The primary mode of action of pTreg cells is through IL-10 and TGF-β – two powerful immune-suppressive cytokines with broad inhibitory properties on T cells, APC and other immune cells. Hence, these cells function in a contact-independent manner. As their action is primarily cytokine mediated, they can suppress priming of the effector cells as well as their effector function. A large body of information also exists on the molecular mechanism underlying the suppressive effects of these cytokines. Several groups have shown that CD4+ CD25+ Treg cells act through cytokine-independent as well as cytokine-dependent manner, in vivo. Various investigators have also reported human CD4+ CD25+ Treg cells to be functioning via IL-10 or TGF-β in a contact-independent manner and also in contact-dependent manner.73–75
Although the role of CTLA-4 has also been controversial76,13 the use of the antibody against CTLA-4 in a number of clinical trials showed promising results. Whether the effect is directly via CTLA-4 or not is yet to be clearly explored. CTLA-4 pathway and Treg cells are essential for immune homeostasis76,77. The use of anti-CTLA-4 antibody in tumour therapy and transfer of Treg cell for use in autoimmunity and transplantation settings, are well known now. Although Foxp3 and CTLA-4 direct independent programmes of immune regulation, there are significant overlaps. Walker, in his article,78 has discussed this in detail to possibly establish the fact that autoimmunity and cancer are two sides of the same coin.
It has also been shown that tTreg cells could down-regulate the expression of co-stimulatory molecules on APC, hence blocking the expansion of effector T cells.56,79–81 The major effect of tTreg cells is thought to be mediated through a non-cognate T–T interaction. The nTreg cells could also inhibit APC function and interfere with the generation of immune response by blocking the activation of APC cells.80–82 Modulation of APC or DC functions with various agents is now feasible but in this article, we will not be discussing those points.
Which Treg cell is more of a constraint in anti-tumour immunotherapy: tTreg or pTreg?
Currently, there are no direct comparisons of tTreg and pTreg cells as a constraint in the immunotherapy of tumours. A relatively high tTregs : effector cell ratio and a relatively weak effector T-cell activation signal are needed to elicit regulation by tTreg cells. The tTreg cells are essentially ineffective when the effector cells are stimulated with ‘strong signals’ (such as plate-bound anti-CD3 antibody, phytohaemagglutinin plus PMA, or when IL-2 is added to the culture) and tTreg cells do not appear to pose a major constraint in anti-tumour immunity in the face of an optimal activation signal. However, the role of CD4+ CD25+ Treg cells on autoimmunity in the animal model is indisputable. With this in mind, we propose that tTreg cells may also be able to regulate an anti-tumour response particularly at steady state. Tumour-associated but self-antigen presentation in steady state is unlikely to be ‘optimum’ and rarely the TAA would be viewed as ‘dangerous.’ Tissue invasion by tumour cells and tumour cell growth seldom present with the same ‘danger’ as do invading pathogens. However, suboptimal stimulation may lead to a low-level effector T-cell activation that is likely to contract on its own or be brought down by tTreg cells, when needed. The tTreg cells, however, may not be able to abort a full-blown effector T-cell activation orchestrated by optimal stimulation with all the right ingredients (antigen presentation by fully activated APC, provision of co-stimulation, inflammatory backdrop, etc.). Indeed, it has been shown that tTreg cells do not regulate effector T-cell activation to a significant level, when the TCR signal is robust.44,70
The most recent research detailing the essential role of the Nr4a family of transcription factors controlling FoxP3 expression opens a new view into developing therapeutic agents targeting FoxP3 expression.83 The dual role of Nr4a transcription factors promoting FoxP3 expression, especially in tTreg cells, is regulated by the strength of TCR signalling, which increases Nr4a expression in thymocytes, resulting in either the trans-activation of FoxP3 expression or initiation of apoptosis during negative selection. The differentiation of naive CD4 T cells into Th2 cells requires IL-4 in the local microenvironment. It is evident that in the microenvironment, partially activated CD4+ T cells produce IL-4 that drives those cells towards Th2 type.84 Since a strong Th1-type response helps the expansion of specific CTL, blocking the Th2 differentiation pathway could generate strong Th1-type CD4 cells. Although the ability of the Nr4a family to suppress the production of IL-4 is known, further research must be conducted to understand the mechanisms of this action. The selective and specific manipulation of the molecular patterns constituting the Nr4a family could damage the ability to modulate FoxP3 expression. However, because of the molecular structure of the Nr4a proteins with an occupied ligand-binding pocket and charged interaction surface, further identification of the molecular patterns constituting the Nr4a family composition must be concluded before attempts at unique targeted therapy design.
It may be argued that pTreg cells are likely to be more efficient by virtue of their mode of action and the cytokines they produce, IL-10 and TGF-β, which are two formidable immunosuppressive cytokines. We have compared the regulatory properties of freshly isolated CD4+ CD25+ T cells and CD4+ CD4− T cells in an in vitro CTL generation protocol44 against a tumour-associated but essentially self-epitope such as the Mart-127–35 peptide presented by fully activated DC. In these experiments, freshly isolated CD4+ CD25+ T cells were found to be quite inefficient—on a per cell basis—compared with induced Treg cells in culture from CD4+ CD25− cohorts, in preventing activation and amplification of the epitope-specific CTL. In the article, Chattopadhyay et al.44 further showed that freshly isolated CD4+ CD25+ cells are potent and effective in suppressing the proliferation of CD4+ CD25− effector T cells, but they do not affect the activation and proliferation of self but melanoma-associated Mart-127–35-specific CD8+ T cells stimulated by the respective peptide-loaded mature DC in vitro. In contrast, in vitro-induced iTreg cells are very effective in inhibiting the induction of Mart-127–35 epitope-specific CTL. This in vitro study points towards a new focus on the inhibition of iTreg cells (or comparing this with peripherally induced pTreg cells) because of their vital role as a constraint in the immune system during optimal stimulation of CTL precursors (tumour vaccine induced in vivo or in vitro stimulation in culture). Hence, future research studies need to be designed to combat both types of Treg cells in two types of microenvironments: by targeting tTreg cells in suboptimal T-cell stimulation responses and attacking iTreg cells (in vitro) or pTreg cells (in vivo) in optimal T-cell stimulation responses as depicted in Fig.1.
Figure 1.
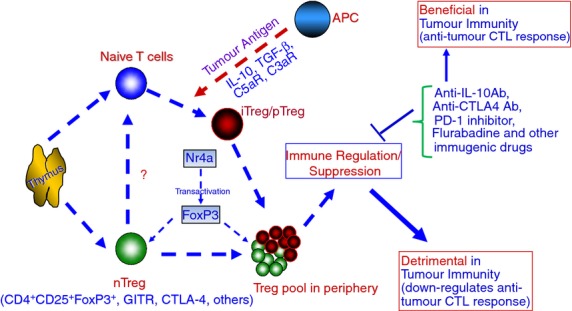
A general schema displaying the derivation of regulatory T (Treg) cells from the thymus and periphery and their roles in inhibiting the anti-tumour response. The roles of Treg cells differ in that natural Treg cells (tTreg) are derived from the thymus, activated by antigen-presenting cells (APC), and modulated by various cellular regulatory molecules (CTLA-4, FoxP3 and GITR), while induced Treg cells are derived in the periphery (pTreg), activated by APC in addition to co-stimulation, and maintained by interleukin-10 (IL-10) and transforming growth factor-β (TFG-β). Both categories of Treg cells inhibit the anti-tumour cytotoxic T lymphocyte (CTL) response and/or expansion. Hence the molecular targeted therapies that inhibit Treg cells (natural or induced to be termed as tTreg or pTreg and not nTreg or iTreg) and enhance CTL-mediated anti-tumour response are important. ? = It is not known how, nTreg/tTreg influence naive CD4 cells to become iTreg/pTreg.
The possibilities to circumvent Treg cell-based constraints
Given that tTreg cells (CD4+ CD25+ T cells) seem to exercise a negative role in tumour immunity, the most obvious strategy to circumvent their negative effects will be to physically remove and/or inactivate them by one mechanism or another. North42 initiated this research by showing that cyclophosphamide could facilitate adoptive immunotherapy of an established tumour by eliminating the CD4+ T cells that induce CD8+ T-cell suppression. Subsequently, North and Awwad43 described the similar effect of a drug-induced elimination of CD4+ suppressor T cells in the regression of an advanced lymphoma. Further research by Berd's group24 employing this strategy in human cancer vaccine therapy and Jaffee's group85 showing that a combination of drugs enhances the anti-tumour immune response of granulocyte–macrophage colony stimulating factor-secreting whole-cell vaccines in mice contributed to the burgeoning information on Treg cell intervention. Jaffee's group has also employed a combination of drugs and vaccine in cancer patients. Preliminary analyses have shown that such an approach could uncover high avidity anti-tumour T cells by inhibiting Treg cells (Laheru and colleagues, personal communication). Other researchers have also used cyclophosphamide as an ‘anti-suppressor cell’ agent in immunotherapy with different forms of cancer vaccines in humans. Important work by Walters et al.86 with single-dose cyclophosphamide and immune responses to the renal cell cancer vaccine IMA901 provides some insight for this particular area. Several investigators worked on the elimination of Treg cells with the immunotoxin-labelled anti-CD25 antibody (ONTAK) in addition to immunotoxin LMB-2, the structure of which contains a fragment of monoclonal antibody against CD25.87 Another important strategy might be to initiate an active immunization approach taking advantage of the weakness of Treg cells. As tTreg cells are less potent when the effector T-cell activation signal is ‘strong,’ their regulatory function might be circumvented by increasing the potency of a vaccine, instituting a ‘strong’ CTL activation/expansion strategy accompanied by the removal/inactivation of the tTreg cells, or active immunization following adoptive transfers of peripheral blood lymphocytes (PBL), depleted of tTreg cells, in a homeostatic expansion mode. Another possible way to ensure the CTL activation/expansion following the inhibition of Treg cells is to block the PD1 ligand, which is up-regulated in many human cancers. PD1 ligands could convert CD8+ T cells into an anergic state in the tumour microenvironment. Therefore, a block in the PD1 ligand-mediated signalling pathway will reinstate the activity of the CD8+ T cells in anti-tumour immunity.27
Similarly, one should also consider the mode of action of pTreg cells and if it might be amenable to circumvention. Indeed, it has been demonstrated that in the presence of anti-IL-10 antibody, a more prolonged anti-tumour CTL response could be obtained through an in vitro CTL generation assay.37 In addition, it was found that in an anti-Mart-127–35-epitope-specific CTL generation model, the regulatory effect of iTreg cells induced from CD4+ CD25− naive cells could be blocked by the presence of antibodies to MHC class II molecules and IL-10R.44 As pTreg (in vivo) iTreg cells (in vitro) act primarily through IL-10, TGF-β or CTLA-4, their action could, therefore, be circumvented by appropriate use of blocking reagents; however, further studies will be needed to answer the question and to test the hypotheses posed here.
We have shown that CD8+ CTL derived from tumours or from vaccine sites decline after initial expansion in in vitro cultures with concomitant rise of CD4+ Treg cells.45 We also showed that PBL derived from patients after successive vaccinations produced increasing amounts of IL-10 on stimulation with IL-2. Further analyses revealed that a fraction of CD4+ T cells from PBL exhibited the essential attributes of Treg cells, i.e. they expressed the CD4+ marker, expressed significantly high levels of CD25, and intracytoplasmic IL-10 was detected in those cells on short-term stimulation with IL-2. These observations have implications in tumour antigen and APC/DC-based cancer vaccines. A better understanding of the mechanism of activation and expansion of such Treg cells in active specific immunotherapy of cancer will be useful.
An interesting observation to derail Treg cell expansion and allowing proliferation of CTL against melanoma tumour antigen in vitro by using a low dose of the anticancer drug fludarabine was demonstrated by Hegde et al.25 In his study, melanoma tumour antigen Mart-127–35-specific CD8+ CTL generated from total PBL maintained their activity and expanded significantly in the presence of low doses of the drug used. A marked inhibition of the expansion of IL-10-producing CD4+ Treg cells was observed concomitantly, which might have allowed the increased activity of antigen-specific CTL.25 This observation shows a desirable effect of the drug halting Treg cell expansion for enhanced anti-tumour immunity without the toxic side effects of high doses of chemotherapy administration. Since fludarabine has already been approved for cancer treatment, the investigation of other commercial chemotherapeutic agents as possible inhibitors of Treg cell expansion should be continued with the hope of further advances in CTL-mediated therapeutic cancer vaccine development.
Regarding current clinical studies in the field of cancer immunotherapy, the usage of ipilimumab as an antibody to CTLA-4 has shown tremendous success in phase III trials for increased survival in metastatic melanoma patients. Because CTLA-4 is constitutively expressed by Treg cells and inhibits the activation of cytotoxic T cells through its signalling effects on the TCR cascade, any block of CTLA-4 to its ligand will interfere with the decline in CTL expansion in an anti-tumour immune response.26 This selective block of CTLA-4 leads to the expansion of the anti-tumour response. That has led to the development of another treatment regimen, tremelimumab. While ipilimumab has been approved since 2011 at a dose of 3 mg/kg for metastatic melanoma in the USA, EU and Australia; tremelimumab has shown positive responses in early phase I and II clinical studies, with melanoma patients, with a premature closure of the phase III study. Use of tremelimumab in a phase II trial for hepatocellular carcinoma, resulted in a decreased tumour burden and disease stabilization. Allison and his co-workers have identified a molecule, inducible co-stimulator (ICOS) as a crucial player in the anti-tumour effects of CTLA-4 blockade.88 ICOS is a T-cell-specific molecule that belongs to the CD28/CTLA-4 family. ICOS expression is up-regulated upon T-cell activation, which is enhanced in the setting of CTLA-4 blockade. Higher frequency of ICOS+ T cells are detected in cancer patients receiving anti-CTLA-4 therapy. The ICOS+ population of CD4+ T cells was found to be tumour-specific that produced interferon-γ, which could mitigate Treg cell activity.88 Overall, the studies into ipilimumab and tremelimumab as CTLA-4 blockers provide hope in the combination of CTLA-4 block and other types of anti-tumour regimens (vaccines, chemotherapy or radiotherapy) for greater efficacy in down-regulating the Treg cells and modulation of the anti-tumour immune response.26
With the promising results from CTLA-4-blocking agents, an investigation into PD1 has also been conducted.27 Studies have shown that expression of PD1 in mouse tumour cells has been shown to inhibit anti-tumour immune responses.27 In addition, PD1 up-regulation in many human cancers has been demonstrated along with PD1 expression on tumour-infiltrating lymphocytes. The most intriguing result was that PD1 expression on CD4+ T effector cells in the PD1 tumour microenvironment actually converts the effector cells into the same Treg cells that dampen the anti-tumour response. With this discovery, agents that block PD1 could inhibit the conversion of effector T cells into Treg cells and allow for the down-regulation of PD1 on tumour-infiltrating lymphocytes.89 Four different agents (MDX-1106, MK-3475, CT-011 and AMP-224) are currently being studied in clinical trials and have shown excellent results in the treatment of advanced melanoma. As in the case of CTLA-4-blocking agents, there is also high hope that PD1 agents in conjunction with other anti-tumour therapies will lead to a better immune response and greater survival rates.27
Several ideas are emerging for future interventions that target Treg cells. These include the antibody against GITR, which could possibly decrease the function of Treg cells in vivo and in vitro, FoxP3 (which has been patented), the anti-vascular endothelial growth factor (VEGF) antibody (bevacizumab) that targets the surface marker VEGFR2, and Toll-like receptor 8 to reverse the suppressive function of different Treg cell populations.7,90 Anti-angiogenic agents that are used to treat solid tumours have effects on tumour endothelial cells as well as on immune cells. Targeting the VEGFA/VEGF receptor 2 (VEGFR2) signalling pathway reduces the proportion of Treg cells in mouse as well as human colorectal cancer because it inhibits tumour-induced Treg cell proliferation.91 Similarly it has been found that the TLR7/8L:CL097 could simultaneously activate CD8+ T cells, B cells and natural killer cells and block Treg cell suppression of T cells and B cells.92
In addition to cyclophosphamide, other drugs that have been shown to inhibit Treg cells include cyclosporine A and tacrolimus, which inhibit IL-2 secretion, and imatinib and dasatinib, which reduce FoxP3 expression.7 Further research into Treg cell intervention involving specific molecular pathways shows promise into targeting the p38 mitogen-activated protein kinase (MAPK) pathway, hypoxia-inducible factor-1α (HIF-1α), the Notch pathway, the OX40 co-stimulatory tumour necrosis factor receptor family molecule, and exosomes. The p38 MAPK pathway and HIF-1α are both highly activated in Treg cells. The Notch pathway and OX40 molecule are involved in FoxP3 expression with OX40 inhibiting FoxP3 expression and Notch signalling involved in cancer initiation. Ruby et al.93 have shown that OX40 stimulation drives all lineages of CD4 T-cell development, including Treg cells, and the plasticity of the response is dependent on local cytokines. Because tumour-derived exosomes have surface-bound TGF-β1, which play a role in FoxP3 expression, the control or elimination of these exosomes could be a promising advancement in immunotherapy in advanced cancer.7
With Derry Ridgway's report on the first 1000 tumour vaccines94 since our first published report of an APC- and peptide-based cancer vaccine study in 1995,34 the collective results on CTL-based immunotherapy remain encouraging. However, the field of cancer vaccine trials with antigen and DC has reached a point that needs lots of innovation. Our earlier observations37,40,45 support the notion that a vaccine-induced activation/expansion of Treg cells has physiological relevance in the peripheral tolerance induction process and suggest that strategies designed to ‘suppress’ or ‘silence’ the suppressors (Treg cells) could be useful.
T-cell tolerance through natural or induced pathways of self-tolerance generation15,33,35,37,44,46,60,87,95 appears to be the most difficult one and this represents a significant challenge to successful cancer immunotherapy. To achieve an effective anti-tumour response, therapeutic vaccines must be capable of overcoming or reversing T-cell tolerance to tumour antigens whether it is via naturally occurring CD4+ CD25+ Treg cells91 or by induced Treg cells (induced from CD4+ CD25− cells).45–47 We have shown that tumour-specific CTL generated in vitro from total PBL declined within 2–3 weeks with a concomitant expansion of suppressor type CD4+ cells.96 We have also shown that even the number of vaccine-induced CTL becomes ineffective in vivo, with a concomitant increase of a class of CD4+ CD25+ cells that demonstrate intracellular IL-10.45 It appears that the peripheral tolerance has to be established before a metastatic cancerous growth takes place. In this regard, it is important to mention the report by Türbachova et al.97 on higher Treg cell ratio in peripheral blood tolerance and metastasis. T-cell-centric cancer therapies face a number of constraints – extrinsic as well as intrinsic to the T cells. A comprehensive understanding of the biology of anti-tumour T cells through a clear insight into their gene expression (transcriptome) combined with its relationship with function and fate will be helpful. The CD4+-induced T regulators play a pertinent role in actively down-regulating specific CTL. To overcome the negative role played by these cells, use of various adjuvants, multiple peptides (class I and class II), viruses, Toll-like receptors,33,35,98–104 Th1 supportive cytokine IL-12, and blocking of Th2-supportive cytokine IL-4 are already being introduced as possible ways to target the immune response.102 More research has to be conducted focusing on the perilous decline in CD8+ CTL numbers accompanying the rise of CD4+ regulatory T cells. By polarizing the PBL with IL-12 and anti-IL-4 antibody in vitro, the life and activity of tumour-specific CTL were increased.96 Although the use of IL-12 was approved for phase I and phase II clinical trials for immunotherapy, no significant clinical trial report is available so far because IL-12 was found to be more toxic to the patients than beneficial. Yang et al.'s104 important work that persistent Toll-like receptor signals can reverse Treg-mediated CD8+ T-cell tolerance has increased the enthusiasm in the field of vaccine therapy for cancer for a possible alternate way of using IL-12. Subsequently, we argued that Th1 conditioning of the total PBL, in vitro, by using a lower dose (than that of previously used in vitro) of IL-12 might help to induce better tumour-specific CTL survivability and activity for a prolonged period in culture.96 We found that preconditioning of PBL toward Th1 and then continuous presence of such conditioning significantly increased the life and activity of tumour antigen-specific CTL in cultures.96 In the future, we hope there would be a new protocol to initiate Th1 conditioning of PBL in vivo in an attempt to circumvent Treg cell induction mechanisms without the hazard of severe toxicity.
Conclusion
Functional characterization of Treg cells in humans has largely been limited to peripheral blood. A detailed analysis of Treg cells with human blood reveals a heterogeneous population composed of resting Treg cells with a ‘naive’ phenotype, ‘activated’ Treg cells with characteristics of memory cells, and FoxP3-expressing cells that secrete effector cytokines and lack suppressive capacity.7 Recently, adoptive cell therapy (ACT) with in vitro expanded populations of T cells engineered to express a set of tumour-epitope-specific TCR is undergoing clinical trials for various malignancies.105 ACT with the melanoma epitope, Mart-1(27–35), specific TCR-engineered T cells has shown encouraging results in metastatic melanoma. Regulatory T cells turned out to be impediments in this form of cancer therapy also. As such, efforts are underway to gain a fuller understanding of the biology (functionality and constraints) of TCR-engineered T cells and the role of Treg cells so as to extract more robust therapeutic effects from ACT. Traditional T-cell-based assays are inadequate for detailed understanding of the entire regulatory process. In this regard, Next Generation RNA-Seq (NGS) has turned out to be a powerful tool to obtain a comprehensive idea of the transcriptome of specific human T cells. Analyses of the genes responsible for T-cell activation, apoptosis, cellular proliferation, cytolytic response and T-cell differentiation towards effector versus regulator could further show the specific points of intervention for active specific blockage of Treg cells.106
In 2002, Shevach postulated that there are ‘more questions than answers’ on the topic of Treg cells.20 Despite the commentary on Treg cells by Cohn,107 it is safe to say that the subject of suppressor cells (in a Treg cell camouflage) is one that demands further exploration. With the current body of research asserting the Treg cells’ dual control over autoimmunity and regulatory function over tumour immunity, it is essential that this delicate balance be exploited in the quest to enhance anti-tumour effector responses. Presently we are armed with a better understanding of tTreg and pTreg cells, their interference against anti-tumour immunity, and the expansive body of research on existing drugs to block Treg cell expansion and CTL decline. As such, further research should continue the advancement of the successful development of therapeutic cancer vaccines with the eventual goal of establishing the ideal schedule of vaccine administration intervals and maintenance. Numerous ideas are continuously generated from the experimental data using animal models, especially transgenic animals. Unlike infectious diseases, cancers grow spontaneously in genetically diverse humans. There are unique mechanisms for each human about how they immunologically handle their cancers. There are significant differences between mice and humans that must be addressed experimentally to refine adoptive immunotherapy. Hence data generated from genetically identical animals with experimentally induced cancers should not be extrapolated to human cancer. Yet, we cannot take a step forward without an appropriate animal model. Despite all the difficulties, tumour immunologists are slowly but steadily gaining ground. We are hopeful that upcoming studies including next-generation of sequencing will show how these therapeutic cancer vaccines will successfully remove Treg cells and engage a fully responsive CTL attack, at perfectly timed intervals with the minimum adverse effect on patients.
Acknowledgments
This work was supported by a grant MO1RR06192 from GCRC and a grant from the Carole and Ray Neag Comprehensive Cancer Center, University of Connecticut Health Center, Farmington, USA. The work was partly supported by Department of Biotechnology, Ministry of Science and Technology, Government of India (Project no: BT/PR13312/GBD/27/247/2009) and by Council of Scientific and Industrial Research (CSIR) (Project No. 37(1542)/12/EMR-II), Ministry of Science and Technology, Government of India. The authors also acknowledge Dr Robert E Cone (Professor, Department of Immunology, UCHC) for his critical reading and valuable suggestions for the manuscript.
Glossary
- ACT
adoptive cell therapy
- APC
antigen-presenting cell
- CTL
cytotoxic T lymphocytes
- CTLA-4
cytotoxic T lymphocyte antigen 4
- DC
dendritic cell
- GITR
glucocorticoid inducible tumour necrosis factor receptor family
- ICOS
inducible co-stimulator
- IL
interleukin
- iTreg (pTreg)
peripherally induced T regulatory cells
- nTreg (tTreg)
thymus-derived regulatory cells
- PBL
peripheral blood lymphocyte
- PD1
programmed cell death 1
- TAA
tumour-associated antigen
- TCR
T-cell receptor
- TGF-β
transforming growth factor-β
- Th17
T helper type 17
- TIL
tumour-infiltrating lymphocyte
- Treg
T regulatory cells
- VGEF
vascular endothelial growth factor
Disclosures
The authors of this article do not have any financial or competing interest.
References
- Bot A, Ahn M, Bosch M. A novel series of conferences tackling the hurdles confronting the translation of novel cancer immunotherapies. J Transl Med. 2012;10:218–22. doi: 10.1186/1479-5876-10-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2009;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–7. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Saxena R. Regulatory T cells: a review. Natl Med J India. 2012;25:341–51. [PubMed] [Google Scholar]
- Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–6. doi: 10.1016/j.coi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi H, Morisaki T, Katano M. Immunotherapy approaches targeting regulatory T-cells. Anticancer Res. 2012;32:997–1003. [PubMed] [Google Scholar]
- Fayyad-Kazan H, Rouas R, Fayyad-Kazan M. MicroRNA profile of circulating CD4-positive regulatory T cells in human adults and impact of differentially expressed microRNAs on expression of two genes essential to their function. J Biol Chem. 2012;287:9910–22. doi: 10.1074/jbc.M111.337154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevac EM, Collins M, Byrne MC. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Suppressor T cells: rebirth, function and homeostasis. Curr Biol. 2000;10:R572–5. doi: 10.1016/s0960-9822(00)00617-5. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–45. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Stephens GL, Shevach EM. Foxp3+ regulatory T cells: selfishness under scrutiny. Immunity. 2007;27:417–9. doi: 10.1016/j.immuni.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4+ CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor β. J Exp Med. 2001;194:629–44. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+ FOXP3− T cells by T-cell receptor stimulation is transforming growth factor-β-dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–90. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cell and tumor immunity. Cancer Immunol Immunother. 2005;54:1153–61. doi: 10.1007/s00262-005-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CS, Deenick EK. Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol. 2014;92:64–71. doi: 10.1038/icb.2013.55. [DOI] [PubMed] [Google Scholar]
- Bach JF. Regulatory T cells under scrutiny. Nat Rev Immunol. 2003;3:189–98. doi: 10.1038/nri1026. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- Lehner T. Special regulatory T cell review: the resurgence of the concept of contra suppression in immunoregulation. Immunology. 2008;123:40–4. doi: 10.1111/j.1365-2567.2007.02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- Germain RN. Special regulatory T-cell review: a rose by any other name: from suppressor T cells to Tregs, approbation to unbridled enthusiasm. Immunology. 2008;123:20–7. doi: 10.1111/j.1365-2567.2007.02779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas A, Benoist C, Bluestone JA. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14:307–8. doi: 10.1038/ni.2554. [DOI] [PubMed] [Google Scholar]
- Shevach EM, Thornton AM. tTregs, pTregs and iTregs. Similarities and differences. Immunol Rev. 2014;259:88–102. doi: 10.1111/imr.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berd D, Mastrangelo MJ. Effect of low dose cyclophosphamide on the immune system of cancer patients: depletion of CD4+, 2H4+ suppressor-inducer T -cells. Cancer Res. 1988;48:1671–5. [PubMed] [Google Scholar]
- Hegde U, Chhabra A, Chattopadhyay S, Das R, Ray S, Chakraborty NG. Presence of low dose of fludarabine in cultures blocks regulatory T cell expansion and maintains tumor-specific cytotoxic T lymphocyte activity generated with peripheral blood lymphocytes. Pathobiology. 2008;75:200–8. doi: 10.1159/000124981. [DOI] [PubMed] [Google Scholar]
- Grosso J, Jure-Kunkel M. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun. 2013;13:5–18. [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Yolcu E, Shirwan H. The promise of PD-1 signaling pathway for cancer immunotherapy. J Clin Cell Immunol. 2012;3:4. [Google Scholar]
- Ehrlich P. Nederlandsch Tijdschrift voor Geneeskunde: ueber den jetzigne Stand Der Karzinomforchung. Weekblad Jaargang Eerst Helft. 1909;5:273. [Google Scholar]
- Srivastava PK, Old LJ. Individually distinct transplantation antigens of chemically induced mouse tumors. Immunol Today. 1988;9:78–83. doi: 10.1016/0167-5699(88)91269-8. [DOI] [PubMed] [Google Scholar]
- Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–9. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175–82. doi: 10.1016/s0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- Anichini A, Vigetti C, Mortini R. The paradox of T cell-mediated antitumor immunity in spite of poor clinical outcome in human melanoma. Cancer Immunol Immunother. 2004;53:855–64. doi: 10.1007/s00262-004-0526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:328–32. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- Mukherji B, Chakraborty NG, Yamasaki S. Induction of antigen specific cytolytic T cells in situ in human melanoma by immunization with synthetic peptide-pulsed autologous antigen presenting cells. Proc Natl Acad Sci. 1995;92:8078–82. doi: 10.1073/pnas.92.17.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayordomo JI, Zorina T, Storkus WJ. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med. 1995;1:1297–302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- Kuwana M. Induction of anergic and regulatory T cells by plasmacytoid dendritic cells and other dendritic cell subsets. Hum Immunol. 2002;63:1156–63. doi: 10.1016/s0198-8859(02)00754-1. [DOI] [PubMed] [Google Scholar]
- Chakraborty NG, Li L, Sporn JR, Kurtzman SH, Ergin MT, Mukherji B. Emergence of Th2 type CD4+ T cell response to repetitive stimulation with antigen and antigen presenting cells, in vitro: implications in designing tumor vaccines. J Immunol. 1999;162:5576–83. [PubMed] [Google Scholar]
- Pawlec G. Tumor escape from the immune response. Cancer Immunol Immunother. 2004;53:843. doi: 10.1007/s00262-004-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad M, Rees RC, Ali SA. Escape from immunotherapy; possible mechanisms that influence tumor regression/progression. Cancer Immunol Immunother. 2004;53:844–54. doi: 10.1007/s00262-004-0540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty NG, Twardzik DR, Sivanandham M, Ergin MT, Hellstrom KE, Mukherji B. Autologous melanoma-induced activation of regulatory T cells that suppress cytolytic response. J Immunol. 1990;145:2359–64. [PubMed] [Google Scholar]
- Mukherji B, Chakraborty NG, Sivanandham M. T cell clones that react against human tumors. Immunol Rev. 1990;116:33–62. doi: 10.1111/j.1600-065x.1990.tb00803.x. [DOI] [PubMed] [Google Scholar]
- North RJ. Cyclophosphamode-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155:1063–74. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ, Awwad M. Elimination of cycling CD4+ suppressor T cells with an anti-mitotic drug releases non-cycling CD8+ T cells to cause regression of an advanced lymphoma. Immunology. 1990;71:90–5. [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Mehrotra S, Chhabra A, Hegde U, Mukherji B, Chakraborty NG. Effect of CD4+CD25+ and CD4+CD25– T regulatory cells on the generation of cytolytic T cell response to a self but human tumor associated epitope in vitro. J Immunol. 2006;176:984–90. doi: 10.4049/jimmunol.176.2.984. [DOI] [PubMed] [Google Scholar]
- Chakraborty NG, Chattopadhyay S, Mehorotra S, Chhabra A, Mukherji B. Regulatory T cell response and vaccine induced CTL response in human melanoma. Hum Immunol. 2004;65:794–802. doi: 10.1016/j.humimm.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–8. [PubMed] [Google Scholar]
- Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, Shevach EM, Wang RF. Tumor specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20:107–18. doi: 10.1016/s1074-7613(03)00359-5. [DOI] [PubMed] [Google Scholar]
- Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukine-2 receptor α) monoclonal antibody. Cancer Res. 1999;59:3128–33. [PubMed] [Google Scholar]
- Gallimore A, Sakaguchi S. Regulation of tumor immunity by CD25+ T cells. Immunology. 2002;107:5–9. doi: 10.1046/j.1365-2567.2002.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Larmonier N, Schmitt E. CD4+CD25+ regulatory T cells suppress tumor immunity sensitive to which allows immunotherapy in established tumors to be curative. Eur J Immunol. 2004;34:336–44. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- Casares N, Arribillaga L, Sarobe P. CD4+/CD25+ regulatory cells inhibit activation of tumor primed CD4+ T cells with IFN-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J Immunol. 2003;171:5931–9. doi: 10.4049/jimmunol.171.11.5931. [DOI] [PubMed] [Google Scholar]
- Wei-Zen W, Morris GP, Kong YC. Antitumor immunity and autoimmunity: a balancing act of regulatory T cells. Cancer Immunol Immunother. 2004;53:73–8. doi: 10.1007/s00262-003-0444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Tanaka J, Jorgen K, Shu S. Depletion of CD4+CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother. 2002;25:207–17. doi: 10.1097/00002371-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Golgher D, Jones E, Powrie F, Elliott T, Gallimore A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine rejection antigens. Eur J Immunol. 2002;32:3267–75. doi: 10.1002/1521-4141(200211)32:11<3267::AID-IMMU3267>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, Kaiser LR, June CH. Cutting edge: regulatory T cells from lung cancer patients inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–6. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Picirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol. 2004;24:366. doi: 10.1002/eji.200324455. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–23. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- Malek TR. The main function of IL-2 is to promote the development of regulatory T cells. J Leukoc Biol. 2003;74:961–5. doi: 10.1189/jlb.0603272. [DOI] [PubMed] [Google Scholar]
- Liyanage UK, Moore TT, Joo HG. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–61. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer. 2003;98:1089–99. doi: 10.1002/cncr.11618. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Viguier M, Lemaître F, Verola O. Foxp3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–53. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- Lin X, Chen M, Liu Y, Guo Z, He X, Brand D, Zheng SG. Advances in distinguishing natural from induced Foxp3+ regulatory T cells. Int J Clin Exp Pathol. 2013;6:116–23. [PMC free article] [PubMed] [Google Scholar]
- Fialova A, Partlova S, Sojka L. Dynamics of T-cell infiltration during the course of ovarian cancer: the gradual shift from a Th17 effector cell response to a predominant infiltration by regulatory T-cells. Int J Cancer. 2013;132:1070–9. doi: 10.1002/ijc.27759. [DOI] [PubMed] [Google Scholar]
- Elkord E, Sharma S, Burt DJ, Hawkins RE. Expanded subpopulation of FoxP3+ T regulatory cells in renal cell carcinoma co-express Helios, indicating they could be derived from natural but not induced Tregs. Clin Immunol. 2011;140:218–22. doi: 10.1016/j.clim.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Elkord E, Al-Ramadi BK. Helios expression in FoxP3+ T regulatory cells. Expert Opin Biol Ther. 2012;12:1423–5. doi: 10.1517/14712598.2012.711310. [DOI] [PubMed] [Google Scholar]
- Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, von Andrian UH. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity. 2006;25:129–41. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Thronton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin-2 production. J Exp Med. 1998;188:287–96. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, Sakaguchi S. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol. 1999;162:5317–26. [PubMed] [Google Scholar]
- Baecher-Allan C, Viglietta V, Hafler DA. Inhibition of human CD4+ CD25+high regulatory T cell function. J Immunol. 2002;169:6210–7. doi: 10.4049/jimmunol.169.11.6210. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger R. Large-scale in vitro expansion of polyclonal human CD4+ CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- Strainic M, Shevach E, An F, Lin F, Medof F. Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat Immunol. 2013;14:162–71. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri-Payer E, Cantor H. Differential cytokine requirements for regulation of autoimmune gastritis and colitis by CD4+CD25+ T cells. J Autoimmun. 2001;16:115–23. doi: 10.1006/jaut.2000.0473. [DOI] [PubMed] [Google Scholar]
- Asseman C, Mauze S, Leach M, Coffman R, Powrie F. An essential role for interleukin-10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sachs DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- Read S, Maimstrom S, Powrie F. CTLA-4 plays an essential role in the function of CD4+CD25+ regulatory cells which control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazuki S, Ueda T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Dominant immunologic self-tolerance by CD4+CD25+ regulatory T cells constitutively expressing CTLA-4. J Exp Med. 2000;192:303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. 2013;45:49–57. doi: 10.1016/j.jaut.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Liotta F. Phenotype localization and mechanism of suppression of CD4+CD25+ human thymocytes. J Exp Med. 2002;196:379–87. doi: 10.1084/jem.20020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-β in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129:263–76. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol. 2000;30:1538–43. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol. 2003;4:355–60. doi: 10.1038/ni908. [DOI] [PubMed] [Google Scholar]
- Sekiya T, Kashiwagi I, Yosida R. Nr4 receptors are essential for thymic regulatory regulatory T cell development and homeostasis. Nat Immunol. 2013;14:230–7. doi: 10.1038/ni.2520. [DOI] [PubMed] [Google Scholar]
- Khan MM, Chatterjee S, Dwivedi VP. CD4+ T cell-derived novel peptide Thp5 induces interleukin-4 production in CD4+ T cells to direct T helper 2 cell differentiation. J Biol Chem. 2012;287:2830–5. doi: 10.1074/jbc.M111.319947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels J-PH, Reilly RT, Emens LA, Ercollini AM, Lei RY, Weintraub D, Okoye FI, Jaffee EM. Cyclophosphamide, doxorubicin and paclitaxel enhance the antitumor immmune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001;61:3689–97. [PubMed] [Google Scholar]
- Walter S, Weinschenk T, Stenzl A. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254–61. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- Turturro F. Denileukin diftitox: a biotherapeutic paradigm shift in the treatment of lymphoid-derived disorders. Expert Rev Anticancer Ther. 2007;7:11–7. doi: 10.1586/14737140.7.1.11. [DOI] [PubMed] [Google Scholar]
- Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, Allison JP, Sharma P. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res. 2013;1:229–34. doi: 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25Hi regulatory T cells. Int Immunol. 2009;21:1065–77. doi: 10.1093/intimm/dxp072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui SA, Frigola X, Bonne-Annee S. Tumor-infiltrating Foxp3–CD4+CD25+ T cells predict poor survival in renal cell carcinoma. Clin Cancer Res. 2007;13:2075–81. doi: 10.1158/1078-0432.CCR-06-2139. [DOI] [PubMed] [Google Scholar]
- Terme M, Tartour E, Taieb J. VEGFA/VEGFR2-targeted therapies prevent the VEGFA-induced proliferation of regulatory T cells in cancer. Oncoimmunology. 2013;2:e25156. doi: 10.4161/onci.25156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voo KS, Bover L, Harline ML, Weng J, Sugimoto N, Liu YJ. Targeting of TLRs inhibits CD4+ regulatory T cell function and activates lymphocytes in human peripheral blood mononuclear cells. J Immunol. 2014;193:627–34. doi: 10.4049/jimmunol.1203334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby CE, Yates MA, Hirschhorn-Cymerman D, Chlebeck P, Wolchok JD, Houghton AN, Offner H, Weinberg AD. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J Immunol. 2009;183:4853–7. doi: 10.4049/jimmunol.0901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway D. The first 1000 dendritic cell vaccines. Cancer Invest. 1993;21:873–86. doi: 10.1081/cnv-120025091. [DOI] [PubMed] [Google Scholar]
- Torre-Amione G, Beauchamp RD, Koeppen H, Park BH, Schreiber H, Moses HL, Rowley DA. A highly immunogenic tumor transfected with a murine transforming growth factor type 1 cDNA escapes immune surveillance. Proc Natl Acad Sci USA. 1990;87:1486–90. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Chakraborty NG. Continuous presence of Th1 conditions are necessary for longer lasting tumor antigen specific CTL activity in stimulation culture with PBL. Hum Immunol. 2005;66:884–91. doi: 10.1016/j.humimm.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Türbachova I, Schwachula T, Vasconcelos I. The cellular ratio of immune tolerance (immunoCRIT) is a definite marker for aggressiveness of solid tumors and may explain tumor dissemination patterns. Epigenetics. 2013;8:1226–35. doi: 10.4161/epi.26334. [DOI] [PubMed] [Google Scholar]
- Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mule JJ, Rosenberg SA, Bennink JR. Identification of human cancers deficient in antigen processing. J Exp Med. 1993;177:265–72. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C, Schreiber H, Rowley D. Role of TGF-β in immune-evasion of cancer. Microsc Res Tech. 2001;52:387–95. doi: 10.1002/1097-0029(20010215)52:4<387::AID-JEMT1023>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Salazar-Onfray F. Interleukin-10: a cytokine used by tumors to escape immunosurveillance. Med Oncol. 1999;16:86–94. doi: 10.1007/BF02785841. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Sakaguchi S. Naturally arising CD25+CD4+ regulatory T cells in maintaining immunologic self-tolerance and preventing autoimmune disease. Curr Mol Med. 2003;3:693–706. doi: 10.2174/1566524033479429. [DOI] [PubMed] [Google Scholar]
- Chang CC, Ciubotariu R, Manavalan JS. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol. 2002;3:237–43. doi: 10.1038/ni760. [DOI] [PubMed] [Google Scholar]
- Dabbagh K, Lewis DB. Toll-like receptors and T-helper-1/T-helper-2 responses. Curr Opin Infect Dis. 2003;16:199–204. doi: 10.1097/00001432-200306000-00003. [DOI] [PubMed] [Google Scholar]
- Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5:508–15. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- Yee C. Adoptive T-cell therapy for cancer: boutique therapy or treatment modality? Clin Cancer Res. 2013;19:4550–2. doi: 10.1158/1078-0432.CCR-13-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn M. Whither T-suppressors: if they did not exist would we have to invent them? Cell Immunol. 2004;227:81–92. doi: 10.1016/j.cellimm.2004.02.004. [DOI] [PubMed] [Google Scholar]