

Abstract
Intestinal inflammation causes tight junction changes and death of epithelial cells, and plays an important role in the development of Crohn*s disease (CD). CD52 monoclonal antibody (CD52 mAb) directly targets the cell surface CD52 and is effective in depleting mature lymphocytes by cytolytic effects in vivo, leading to long-lasting changes in adaptive immunity. The aim of this study was to investigate the therapeutic effect of CD52 mAb on epithelial barrier function in animal models of IBD. Interleukin-10 knockout mice (IL-10−/−) of 16 weeks with established colitis were treated with CD52 mAb once a week for 2 weeks. Severity of colitis, CD4+ lymphocytes and cytokines in the lamina propria, epithelial expression of tight junction proteins, morphology of tight junctions, tumour necrosis factor-α (TNF-α)/TNF receptor 2 (TNFR2) mRNA expression, myosin light chain kinase (MLCK) expression and activity, as well as epithelial apoptosis in proximal colon were measured at the end of the experiment. CD52 mAb treatment effectively attenuated colitis associated with decreased lamina propria CD4+ lymphocytes and interferon-γ/IL-17 responses in colonic mucosa in IL-10−/− mice. After CD52 mAb treatment, attenuation of colonic permeability, increased epithelial expression and correct localization of tight junction proteins (occludin and zona occludens protein-1), as well as ameliorated tight junction morphology were observed in IL-10−/− mice. CD52 mAb treatment also effectively suppressed the epithelial apoptosis, mucosa TNF-α mRNA expression, epithelial expression of long MLCK, TNFR2 and phosphorylation of MLC. Our results indicated that anti-CD52 therapy may inhibit TNF-α/TNFR2-mediated epithelial apoptosis and MLCK-dependent tight junction permeability by depleting activated T cells in the gut mucosa.
Keywords: apoptosis, barrier function, CD52 monoclonal antibody, Crohn's disease, interleukin-10 knockout, tight junction
Introduction
Crohn*s disease (CD) is a segmental inflammatory disease of the intestine characterized by a massive infiltration of CD4+ T cells and macrophages and by intestinal barrier dysfunction.1 The intestinal barrier is organized by interactions among several components, including the antibacterial peptides, adhesive mucous gel layer and intercellular tight junctions (TJ).2 Among these components, TJ constitute the major determinant of the intestinal physical barrier.3 Intestinal barrier defects resulting in the permeation of lumenal inflammatory substances induce an abnormally robust inflammatory response.2,4 Intestinal inflammation causes TJ changes and death of epithelial cells, and plays an important role in the development of CD.5,6 Therefore, the maintenance of the intestinal barrier is imperative for intestinal mucosal homeostasis.
Much of our current understanding of the molecular mechanisms involved in inflammatory bowel diseases (IBD) has come from transgenic, knockout and chemically induced mouse models.7&11 Although mouse models of IBD do not fully recapitulate the human disease, interleukin-10 knockout (IL-10−/−) mice display similar characteristics to those of human CD.12 Because the anti-inflammatory effects of IL-10 are required to regulate T helper type 1 (Th1) and Th17 cytokine production and promote immune homeostasis, loss of IL-10 in mice results in colitis under specific pathogen-free conditions driven by an aberrant immunological response to luminal content, including pathogens, toxins and antigens.7,13
In the gut of patients with IBD, immune and non-immune cells produce large amounts of cytokines that drive the inflammatory process, leading to the tissue damage.14 Cytokine blockers, such as anti-tumour necrosis factor-α (TNF-α), have been used with some success in IBD. However, not all patients respond, and the therapeutic effects wane with time, demonstrating the need for more effective and long-lasting anti-inflammatory strategies.15 CD52 is a cell-surface glycoprotein consisting of a short 12-amino-acid peptide with a C-terminal glycosyl-phosphatidyl-inositol anchor, mainly expressed on lymphocytes. Alemtuzumab (Campath-1H), a humanized monoclonal antibody (mAb), directly targets the cell surface CD52 and is effective in depleting mature lymphocytes by cytolytic effects in vivo, leading to long-lasting changes in adaptive immunity.16 This antibody has also been used in the treatment of a wide range of diseases including rheumatoid arthritis, lymphocytic leukaemia and organ transplantation.17,18 In recent phase 3 clinical studies, alemtuzumab showed efficacy in the treatment of relapsing–remitting multiple sclerosis.19,20 Our previous studies demonstrated that treatment with 20 mg of anti-mouse CD52 mAb once per week for 2 weeks improved histological inflammation scores, nutritional status and iron-deficient anaemia in IL-10−/− mice by attenuating colonic inflammation.21,22 However, the relationship between inflammation and epithelial barrier dysfunction after CD52 mAb treatment is not studied. In the present study, we mainly investigated the relationship between the T-cell-induced colonic inflammation and the changes in epithelial barrier function after CD52 mAb treatment in the IL-10−/− model of colitis.
Materials and methods
Animals
Both IL-10−/− and wild-type mice (16 weeks old at the beginning of the study) on a C57BL/6 background were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice were bred and maintained in specific pathogen-free conditions at the Model Animal Research Centre of Nanjing University (Nanjing, China). Previous experiments have demonstrated that most IL-10−/− mice on the C57BL/6 strain under specific pathogen-free conditions develop spontaneous colitis at 12 weeks of age.23 All animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of Nanjing University (Nanjing, China).
Drug administration protocol
Wild-type and IL-10−/− mice were divided into wild-type, control (IL-10 knockout) and treatment (CD52 mAb) groups, each group contained eight mice, three to five mice were housed in one cage. The IL-10−/− mice in the treatment group received anti-CD52mAb (20 μg diluted in PBS; MBL, Naka-kuNagoya, Japan) treatment once a week for 2 weeks, whereas the mice in wild-type and control groups received the same volume of vehicle (IgG). Four weeks after the final drug administration, the therapeutic effects of CD52 mAb were evaluated.
Histology
After mice were killed, proximal colons were fixed in 10% buffer neutral formalin and embedded in paraffin. Thereafter, 6-μm-thick sections were stained with haematoxylin & eosin. Two independent pathologists blinded to the study design gave an inflammation score to samples taking into account the number of lesions as well as the severity of the disease. Each proximal colon segment was scored from 0 to 4 on the following well-established criteria.24 In brief, grade 0 represented no change from normal tissue; grade 1, one or few multifocal mononuclear cell infiltrates in the lamina propria (LP); grade 2, intestinal lesion involved with several multifocal cellular infiltrates in LP; grade 3, lesions involved moderate inflammation and epithelial hyperplasia; grade 4, inflammation involved most of the colon sections. The summation of scores per mouse provided a total colonic disease score.
Enzyme-linked immunosorbent assay
For cytokine determination in colonic mucosa, protein extracts were obtained by homogenization of colonic segments in homogenization buffer consisting of a protease inhibitor. Cytokines were measured by ELISA as described by the manufacturer*s protocol. Mouse interferon-γ (IFN-γ) and IL-17 were measured by ELISA using DuoSet ELISA development kits (R&D Systems, Minneapolis, MN). Concentrations of cytokines were established in triplicate supernatants by comparison with standard curves generated using the appropriate recombinant cytokine.
Cell isolation
The colons were opened longitudinally and then cut into strips 1 cm in length and stirred in Hanks’ balanced salt solution (Gibco-Invitrogen, Grand Island, NY) containing 2 mm EDTA and 1 mm dithiothreitol at 37° for 30 min. The cells from intestinal LP were isolated as described previously.25 In brief, the LP was isolated by digesting intestinal tissue with 40 U/ml collagenase type II (Sigma-Aldrich, St Louis, MO), 5% fetal bovine serum (Gibco-Invitrogen) and 3 mm CaCl2 in RPMI-1640 (Sigma) for 30 min at 37° with moderate stirring. After each 30-min interval, the released cells were centrifuged, stored in complete medium and mucosal pieces were replaced with fresh collagenase solution at least twice. The LP cells were further purified using a discontinuous Percoll (Sigma) gradient collecting at the 40–70% interface.
Flow cytometry analysis
Cell suspensions from the LP were washed twice in RPMI-1640, and isolated cells were thoroughly suspended in each tube in 100 μl RPMI-1640. For cell surface antigen staining, cells were counted and approximately one million cells were transferred to each flow test tube. These cells were stained with FITC-conjugated anti-CD4 (RM4-5; BD Biosciences, San Diego, CA), phycoerythrin-conjugated anti-CD45 (30-F11; BD Biosciences) or an appropriate negative control. Then, the stained cells were incubated at room temperature for 30 min in the dark. The cells were washed twice with 2 ml RPMI-1640 at room temperature and suspended in 500 μl RPMI-1640, and the cells were evaluated on a FACSCalibur flow cytometer (BD Biosciences). All flow data were analysed using flowjo software.
Ussing chamber studies
Segments of proximal colon were harvested and the mucosa was mounted in Lucite chambers exposing mucosal and serosal surfaces to 10 ml of Ringer*s buffer (115 mm NaCl, 8 mm KCl, 1·25 mm CaCl2, 1·2 mm MgCl2, 2·0 mm KHPO4, 25 mm NaCO3, pH 7·33–7·37). The buffers were maintained at 37° by a heated water jacket and circulated with CO2. The serosal buffer and the mucosal buffer contained 10 mm of glucose that was osmotically balanced. For measurement of basal mannitol fluxes, 1 mm of mannitol with 370 KBOr [H3-mannitol] was added to the mucosal side. The spontaneous transepithelial potential difference (PD, mV) was determined, and the tissue was clamped at zero voltage by continuously introducing an appropriate short-circuit current (Isc, uA/cm2) with an automatic DVC 1000 voltage clamp (World Precision Instruments, Sarasota, FL). Tissue ion resistance (1/G), where G is conductance, was calculated from the PD and Isc according to Ohm*s law.26
Quantification of epithelial apoptosis by TUNEL assay
Detection of apoptosis was performed by terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay using the in situ cell death detection kit (Roche, Basel, Switzerland). Sections were permeabilized with 1% Triton X-100, 0·1% sodium citrate, washed and stained for TUNEL according to the manufacturer*s instructions. Sections were counterstained with DAPI. In the end, after washing with PBS, sections were mounted in 50% glycerol and photographed using confocal microscopy (Olympus, Tokyo, Japan). Six random, non-overlapping pictures of 200 × magnified optical fields of two different colon layers were taken (n = 4), and the number of TUNEL-positive cells per field was counted by a person blinded to the genotype of the sample.
Transmission electron microscopy of TJ
Transmission electron microscopy was performed as described previously.27 In brief, sections of proximal colon (3 mm) were fixed for 2 hr in 4% buffered glutaraldehyde, then the sections were cut into smaller pieces, post-fixed in 1% osmium tetroxide, sequentially dehydrated through graded alcohol series, infiltrated through Epon812 and then embedded in resin. Thin sections were cut and stained with uranyl acetate and lead citrate, and examined with a Hitachi H-600 transmission electron microscope (Hitachi) operated at 75 kV.
Immunofluorescence
Immunostaining was performed as described elsewhere.28 After rinsing with ice-cold PBS, tissues were fixed in 4% paraformaldehyde and embedded in optimal cutting temperature compound (Leica, Wetzlar, Germany) and 6-μm-thick frozen sections of proximal colon were transferred to coated slides, fixed in 1% paraformaldehyde, and washed three times with PBS. Thereafter, non-specific binding was blocked with 5% normal goat serum in PBS. After incubation with mAbs against coccludin (Abcam, Cambridge, UK) and zona occludens protein 1 (ZO-1; Abcam) in PBS with 1% goat serum overnight at 4°, sections were washed and incubated with Alexa 488-conjugated secondary antibodies for 60 min. Images were obtained using confocal microscopy (Olympus).
Western blotting and quantitative real-time polymerase chain reaction
Western blotting of isolated intestinal epithelia was performed as described previously.29 Band intensities were quantified after background subtraction and used to calculate the changes in the relative amounts of the corresponding proteins. Quantification of the blots was achieved densitometrically using Quantity One 1-D analysis software (Bio-Rad, Hercules, CA). Quantitative real-time PCR was performed as described previously,30 with primers specific for TNF-α and TNF receptor 2 (TNFR2). Relative expression was calculated using the 2−▵▵Ct method after normalizing to GAPDH or actin. The sequences of the primers used are: TNF-α: 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ and 5′-TGGGAGTAGACAAGGTACAACCC-3′; TNFR2: 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ and 5-′GGATCTTGGAGACAGGGTGT-3′.
Statistical analysis
spss version 17.0 software (SPSS, Inc., Chicago, IL) was used to perform the statistical analyses. The data were expressed as means with their standard errors (SEM). Single-factor variance analyses of variance were used to evaluate changes in groups. Results were considered statistically significant if P-values were < 0·05.
Result
CD52 mAb treatment ameliorated histological colitis typically associated with decreased CD4+ lymphocytes and cytokines in IL-10−/− mice
We first quantified the protective effects of CD52 mAb treatment on colitis severity. Compared with wild-type mice, IL-10−/− mice with vehicle treatment showed more infiltration of inflammatory cells in colonic mucosa, and the mean histological scores were also significantly higher than those in wild-type mice. The IL-10−/− mice receiving CD52 mAb treatment showed a significant reduction in colonic inflammation, reduced inflammatory cell infiltration, lower mean inflammation scores, and a partially restored glandular and goblet cell architecture when compared with mice in the control group (Fig.1a, b).
Figure 1.

Change in histological characterization and inflammation after CD52 monoclonal antibody (mAb) treatment in interleukin-10 deficient (IL-10−/−) mice 4 weeks after the final drug administration. (a) Histological sections of proximal colons from the wild-type and two groups of IL-10−/− mice at the end of the experiment: colon of wild-type mouse (WT), mice with vehicle treatment (IL-10-KO) showed significant inflammatory cells infiltration, while CD52 mAb-treated mice (CD52 mAb) showed markedly decreased inflammatory cell infiltration. (b) The histological inflammation scores of these three groups in proximal colon are presented. (c, d) Percentage of CD4+ CD45+ lymphocytes in wild-type, IL-10−/− and CD52 mAb treatment mice. (e, f) Protein levels of interferon-γ (IFN-γ) and IL-17, expressed as pg/mg of total protein, were determined in isolated colon tissue samples (n = 5). Representative results of at least three individual experiments in each group were shown. Data were presented as means ± SEM. Bars = 100 µm. *P < 0·05 and **P < 0·01 versus IL-10-KO group.
It has been shown that Th1/Th17 cells differentiated from CD4 mainly mediate chronic inflammation in the colon of IL-10−/− mice.31 Next, we evaluated the effect of CD52 mAb on depleting LP CD4+ lymphocytes. Increased percentage of CD4+ CD45+ lymphocytes, IFN-γ/IL-17 responses were observed in IL-10−/− mice receiving vehicle treatment, while CD52 mAb treatment significantly decreased the percentage of CD4+ CD45+ lymphocytes (Fig.1c, d; P < 0·05) and IFN-γ/IL-17 responses (Fig.1e, f; P < 0·05) in colonic mucosa of IL-10−/− mice, resulting in reduced intestinal inflammation.
CD52 mAb treatment ameliorated colonic permeability, epithelial TJ protein expression and morphology in IL-10−/− mice
Increased intestinal permeability has been recognized as an early feature of CD. In this study, we found that colonic permeability to mannitol was increased in the IL-10−/− mice receiving vehicle treatment with a corresponding decrease in electrical resistance. In the CD52 mAb-treated mice, these changes were largely prevented and showed similar results to wild-type mice (Fig.2a, b). Tight junctions are major determinants in the maintenance of barrier permeability. Furthermore, occludin, a TJ protein strongly regulated by TNF-α, was suggested as a general indicator for TJ integrity. To investigate whether CD52 mAb treatment can alter the expression and localization of TJ, the representative TJ-associated proteins (occludin and ZO-1) were assessed. Compared with wild-type mice, Western blotting analysis confirmed decreased occludin and ZO-1 in IL-10−/− mice receiving vehicle treatment. However, CD52 mAb treatment increased expression of occludin and ZO-1 (Fig.2a, b). In addition, immunofluorescence analysis showed that occludin and ZO-1 were differentially localized in IL-10−/− mice receiving vehicle treatment, especially in regions with infiltrations of inflammatory cells, and the TJ density also appeared to be lower, which was improved by CD52 mAb treatment (Fig.2c, d). Transmission electron microscopy analysis was also performed to further investigate the CD52 mAb treatment on the morphology of TJ. As demonstrated in the Fig.2(e), compared with wild-type mice, TJ ultrastructure in IL-10−/− mice with colitis was altered – characterized by decreased electron-dense materials in the TJ. However, more electron-dense materials were present between the adjoining cells near the brush border in IL-10−/− mice with CD52 treatment, which indicated the amelioration of TJ morphology.
Figure 2.

Effect of CD52 monoclonal antibody (mAb) on intestinal permeability, tight junction (TJ) structure and composition, long myosin light chain kinase (MLCK) expression and activity in the proximal colon of interleukin-deficient (IL-10−/−) mice 4 weeks after the final drug administration. Measurements of colonic mannitol flux (a) and electrical resistance (b) in wild-type (WT), IL-10−/− (IL-10-KO) and CD52 mAb treatment (CD52 mAb) mice (n = 6). (c) Representative Western blots of colonic lysates for occludin and zona occludens 1 (ZO-1) in WT mice, IL-10−/− mice or CD52 mAb-treated mice. (d) Densitometric analysis of occludin and ZO-1 (n = 4 to 6). Representative immunofluorescence (green) of occludin (e) and ZO-1 (f) in the proximal colon of mice in three groups (200 × magnification, n = 3 or 4). Nuclei are blue, and arrows show the staining of TJ proteins. (g) Representative transmission electron micrograph of mucosa in three groups (n = 3 or 4), Arrows indicate TJ. (h) Representative Western blots of long MLCK, pMLC, and total MLC (MLC) of isolated colonic epithelia (n = 3). Data in this figure are representative of at least three independent experiments. Data were presented as means ± SEM. *P < 0·05 and **P < 0·01 versus IL-10-KO group.
Recent studies have shown that increased colonic epithelial long myosin light chain kinase (MLCK) expression and activity are observed in CD and immune-mediated colitis.6,32 As expected, compared with wild-type mice, Western blotting analysis confirmed increased long MLCK expression and activity, measured as increased myosin light chain (MLC) phosphorylation in IL-10−/− mice receiving vehicle treatment. However, CD52 mAb treatment decreased colonic epithelial long MLCK expression and activity (Fig.2f).
CD52 mAb treatment decreased epithelial TNFR2 expression and apoptosis in IL-10−/− mice
The TNF signalling is pivotal to the pathogenesis of CD, and expression of this cytokine is markedly increased in IL-10−/− mice. Signalling through TNF-α/TNFR2 promotes T-cell proliferation and epithelial apoptosis.6,33 One important mechanism by which anti-TNF agents exert their therapeutic effect is TNFR2-dependent induction of LP T-cell apoptosis. In addition, increased T-cell apoptosis after alemtuzumab treatment was seen in patients with multiple sclerosis; however, the mechanism is not fully understood.16 To better understand the potential therapeutic effect of CD52 mAb on the TNF-α/TNFR2 signal pathway, the expression of epithelial TNFR2 and tissue TNF-α mRNA was assessed. Remarkably, increased epithelial expression of TNFR2 and tissue TNF-α mRNA were observed in IL-10−/− mice receiving vehicle treatment, while the relative expression of epithelial TNFR2 and mucosal TNF-α were successfully decreased after CD52 mAb treatment (Fig.3a, b; P < 0·01).
Figure 3.
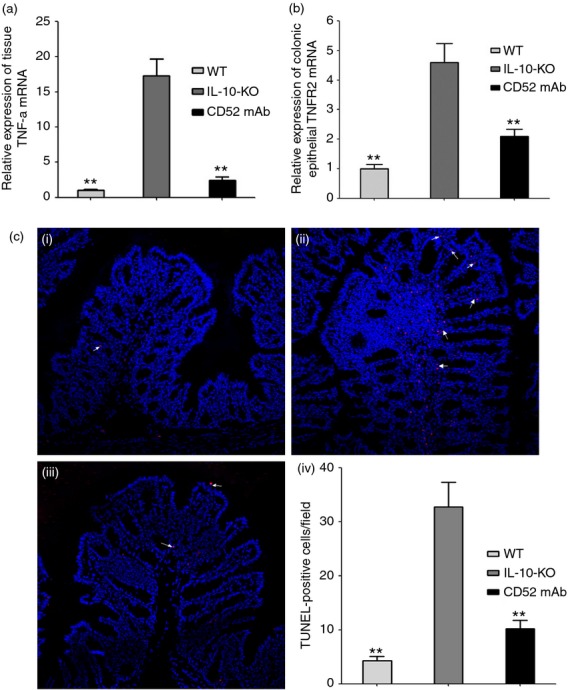
Cytokine RNA expression and apoptotic cells in the proximal colon of interleukin-deficient (IL-10−/−) mice 4 weeks after the final drug administration. Expression of epithelial tumour necrosis factor receptor 2 (TNFR2) and tissue TNF-α in wild-type (WT), IL-10−/− (IL-10-KO) and CD52 monoclonal antibody treatment (CD52 mAb) mice (a and b; n = 6 to 8). (c) Representative TUNEL (white arrow indicates the apoptotic cells with red fluorescence) immunostains with nuclear counterstain (blue fluorescence) in wild-type (i), IL-10−/− (ii) and CD52 mAb treatment (iii) mice (200 × magnification, n = 4), (iv) Morphometric analysis of apoptotic cells in three groups. Data in this figure are representative of at least three independent experiments. Data were presented as means ± SEM. *P < 0·05 and **P < 0·01 versus IL-10-KO group.
Previous reports implicated apoptosis as a potential mechanism of the intestinal barrier loss.4,5 TUNEL staining was used to identify apoptotic cells in proximal colon. The IL-10−/− mice receiving vehicle treatment showed a marked increase in apoptosis (Fig.3c, d; P < 0·05). In contrast, CD52 mAb treatment resulted in far fewer TUNEL-positive cells (Fig.3c, d; P < 0·01).
Discussion
The chronic gastrointestinal inflammation in CD and various experimental models of colitis is associated with a breakdown of immunological unresponsiveness to some food antigens and resident enteric bacteria. This breakdown results in T-cell activation and the production of pro-inflammatory cytokines such as TNF-α, IFN-γ and IL-17.34 Recent studies have revealed that CD is a major burden for society since conventional therapies are neither uniformly effective nor without side effects, novel therapeutic options are always warranted. Our previous reports indicated that CD52 mAb may serve as a potential drug for the treatment of CD.22 Jones et al.16 reported that a single pulse of alemtuzumab treatment resulted in durable T-cell lymphopenia, and regenerated T cells were more susceptible to apoptosis after alemtuzumab treatment in patients with multiple sclerosis characterized by both Th1 and Th17 immune responses. Previous immunological and therapeutic evidence suggests that animal models mimicking colitis that are relevant to human IBD and the pathological process are similar.35 We therefore identified the functional effect of anti-mouse CD52 mAb to abrogate a spontaneous mouse model of chronic colitis, using IL-10−/− mice that were previously reported to spontaneously develop chronic colitis characterized by both Th1 and Th17 polarized inflammation similar to that seen in CD.24 In the current study, we demonstrated that CD52 mAb reversed colitis in IL-10−/− mice, as shown in histopathology and by the reduction in inflammation score. Although many innate leucocyte populations, including natural killer T, natural killer cells and γδ T cells, can secrete Th1 and Th17 cytokines such as IFN-γ and IL-17, the production is predominantly attributed to CD4+ T cells.36 In the present study, we used anti-mouse CD52 mAb to deplete lymphocytes by cytolytic effects. As predicted, Our present data showed that CD52 mAb treatment significantly decreased the percentage of CD4+ CD45+ T cells, as well as IFN-γ/IL-17 levels in colonic LP compared with vehicle treatment in IL-10−/− mice, which could, at least in part, explain the reduction in consequent colonic inflammation score.
Increased intestinal permeability has been linked to a variety of autoimmune and inflammatory disorders. The case is strongest in CD, and reduced barrier function is a marker of impending disease reactivation.3 Despite this association, intestinal barrier loss alone is not sufficient to cause disease in either animal models or human subjects.4 Nevertheless, intestinal barrier defects do accelerate onset and enhance severity of experimental colitis when coupled with disease-inducing stimuli, such as microbes and antigens.6 Intestinal epithelial cells are constantly shed from the tips of villi at an estimated rate of 1400 cells/villus/24 hr.4 The epithelium forms a barrier between the body and the gastrointestinal lumen, so shedding poses a threat to the integrity of the barrier. In healthy individuals, the barrier is maintained by a redistribution of TJ proteins around the shedding cell, which plugs the gap created by the extrusion process.37 However, TJ organization has been shown to be disturbed in active IBD.38 These data suggest that preservation of the TJ barrier can be beneficial in IBD. The influence of pro-inflammatory cytokines on the differential expression of TJ components has been documented in several studies and could explain the observed permeability changes.39 In active CD, occludin and ZO-1 appeared down-regulated and delocalized from tight TJ, whereas electron microscopy analysis showed reduced and discontinuous TJ strands.40&42 In this present study, attenuation of colonic permeability was observed in IL-10−/− mice with CD52 mAb treatment. This suggests that the early evidence of colonic disease, observed in IL-10−/− mice, is prevented by CD52 mAb treatment. Fischer et al.43 demonstrated that adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on TJ proteins in intestinal epithelial cells. In addition, Azuma et al.2 demonstrated that supplemental naringenin protected intestinal barrier function at least partially through protection of the TJ by reducing inflammation in dextran sodium sulphate-induced colitis. To investigate whether an altered abundance of the different TJ proteins can be observed in IL-10−/− mice with CD52 mAb treatment, the expression of TJ-associated proteins and localization of occludin and ZO-1 were assessed in the proximal colon of mice. Western analysis revealed a down-regulation of occludin and ZO-1 expression in IL-10−/− mice with vehicle treatment. Furthermore, immunofluorescence analysis confirmed that occludin and ZO-1 in mice with colitis were differentially localized, especially in the regions with infiltrations of inflammatory cells, these observations are consistent with a previous report.44 However, the changes in TJ proteins were improved by CD52 mAb treatment. Similar ameliorations on the morphology of TJ were also noted in the colon by transmission electron microscopy analysis. Studies in cultured monolayers and animal models have demonstrated that TNF-α, which is central to CD pathogenesis, causes TJ barrier dysfunction via a process that requires MLCK activation.32 Consistent with involvement of this pathway in human disease, MLCK activity, which is measured as increased MLC phosphorylation, is also increased in intestinal epithelia of patients with active IBD.45 In the present study, increased long MLCK and MLC phosphorylation expression were noted in IL-10−/− mice with colitis, whereas CD52 mAb treatment decreased colonic epithelial long MLCK expression and activity. Additionally, increased epithelial expression of TNFR2 and tissue TNF-α mRNA were successfully decreased after CD52 mAb treatment in IL-10−/− mice with colitis. This conclusion, which is consistent with in vitro data showing that TNFR2 signalling mediates TNF-induced long MLCK expression and TJ regulation, might explain the association of TNFR2 polymorphisms with CD.46,47 Furthermore, Su et al.6 demonstrated that signalling through TNFR2 increased long MLCK expression and activity, resulting in TJ dysregulation in immune-mediated inflammatory bowel disease models. The present findings indicated that the depletion of activated T cells by CD52 mAb treatment, resulting in reduced production of the pro-inflammatory cytokines, at least in part contributed to the mechanism of MLCK-mediated TJ changes in the present investigation.
Beyond TJ changes, epithelial apoptosis can also contribute to barrier dysfunction. When the epithelium is intact, intestinal barrier function is largely defined by permeability characteristics of the epithelial TJ. However, epithelial cell death also causes barrier loss, regardless of TJ function. Zeissig et al.41 found that epithelial apoptosis significantly increased in colon samples from patients with CD. Interestingly, levels of apoptosis returned to control levels when patients were treated with anti-TNF agents.41 In the present study, we demonstrated that CD52 mAb treatment effectively suppressed the epithelial apoptosis typically associated with colonic TNF-α mRNA expression compared with IL-10−/− mice receiving vehicle treatment.
In summary, the current study, for the first time, has suggested anti-CD52 therapy may inhibit TNF-α/TNFR2-mediated epithelial apoptosis and MLCK-dependent tight junction permeability by depleting activated T cells in the gut mucosa. Therefore, CD52 mAb may acts as a new biological agent, particularly for patients who do not respond, lose responsiveness, or cannot receive TNF-α blockers. However, further studies are required to determine the exact role that CD52 mAb plays in attenuating activation of the complex immunity and the development of colitis.
Acknowledgments
HW, JD and PS carried out the majority of the biochemical analysis, designed the experiment and contributed to the writing. WZ, JL and NL contributed to the supervision and drafting of the manuscript. JL, YL, LG, JZ, LZ, WZ and JG contributed with technical support, scientific advice and revised the manuscript. This work was supported in part by funding from the National Ministry of Health for the Digestive Disease (Grant 201002020), National Natural Science Foundation of China (Grant 81200263, 81170365 and 81270006) and Jiangsu Provincial Special Programme of Medical Science (BL2012006). The present study was also partly supported by the Model Animal Research Centre, Nanjing University (Nanjing, China). The authors would like to acknowledge the expert technical assistance of Professor Xiang Gao and the members of his laboratory (the Model Animal Research Centre of Nanjing University, China).
Glossary
- CD
Crohn*s disease
- IBD
inflammatory bowel diseases
- IFN-γ
interferon-γ
- IL-10−/−
interleukin-10-knockout
- LP
lamina propria
- mAb
monoclonal antibody
- MLCK
myosin light chain kinase
- Th1
T helper type 1
- TJ
tight junctions
- TNF-α
tumour necrosis factor-α
- TNFR2
tumour necrosis factor receptor 2
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labelling
- ZO-1
zona occludens protein 1
Disclosures
The authors declare that they have no conflict of interest.
References
- Suenaert P, Maerten P, Van Assche G. Effects of T cell-induced colonic inflammation on epithelial barrier function. Inflamm Bowel Dis. 2010;16:1322–31. doi: 10.1002/ibd.21211. [DOI] [PubMed] [Google Scholar]
- Azuma T, Shigeshiro M, Kodama M. Tanabe S, Suzuki T. Supplemental naringenin prevents intestinal barrier defects and inflammation in colitic mice. J Nutr. 2013;143:827–34. doi: 10.3945/jn.113.174508. [DOI] [PubMed] [Google Scholar]
- Juric M, Xiao F, Amasheh S. Increased epithelial permeability is the primary cause for bicarbonate loss in inflamed murine colon. Inflamm Bowel Dis. 2013;19:904–11. doi: 10.1097/MIB.0b013e3182813322. [DOI] [PubMed] [Google Scholar]
- Becker C, Watson AJ, Neurath MF. Complex roles of caspases in the pathogenesis of inflammatory bowel disease. Gastroenterology. 2013;144:283–93. doi: 10.1053/j.gastro.2012.11.035. [DOI] [PubMed] [Google Scholar]
- Schumann M, Gunzel D, Buergel N. Cell polarity-determining proteins Par-3 and PP-1 are involved in epithelial tight junction defects in coeliac disease. Gut. 2012;61:220–8. doi: 10.1136/gutjnl-2011-300123. [DOI] [PubMed] [Google Scholar]
- Su L, Nalle SC, Shen L. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407–15. doi: 10.1053/j.gastro.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D. Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Carter PR, Watts MN, Kosloski-Davidson M, Prasai K, Grisham MB, Harris NR. Iron status, anemia, and plasma erythropoietin levels in acute and chronic mouse models of colitis. Inflamm Bowel Dis. 2013;19:1260–5. doi: 10.1097/MIB.0b013e3182813466. [DOI] [PubMed] [Google Scholar]
- Wang L, Trebicka E, Fu Y. The bone morphogenetic protein–hepcidin axis as a therapeutic target in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:112–9. doi: 10.1002/ibd.21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Li X, Zhang B. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology. 2013;139:494–502. doi: 10.1111/imm.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chen L, Shi J, Liu S, Liu Y. Zheng D. TRAIL receptor deficiency sensitizes mice to dextran sodium sulphate-induced colitis and colitis-associated carcinogenesis. Immunology. 2014;141:211–21. doi: 10.1111/imm.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettel JA, Scott AHM, Olivares-Villagomez D. KSR1 protects from interleukin-10 deficiency-induced colitis in mice by suppressing T-lymphocyte interferon-γ production. Gastroenterology. 2011;140:265–74. doi: 10.1053/j.gastro.2010.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chambrun GP, Body-Malapel M. Frey-Wagner I. Aluminum enhances inflammation and decreases mucosal healing in experimental colitis in mice. Mucosal Immunol. 2014;7:589–601. doi: 10.1038/mi.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di SA, Biancheri P, Rovedatti L. MacDonald TT, Corazza GR. New pathogenic paradigms in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:368–71. doi: 10.1002/ibd.21735. [DOI] [PubMed] [Google Scholar]
- MacDonald TT, Biancheri P, Sarra M, Monteleone G. What*s the next best cytokine target in IBD. Inflamm Bowel Dis. 2012;18:2180–9. doi: 10.1002/ibd.22967. [DOI] [PubMed] [Google Scholar]
- Jones JL, Phuah CL, Cox AL. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H) J Clin Invest. 2009;119:2052–61. doi: 10.1172/JCI37878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SP, Sancho J, Campos-Rivera J. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS One. 2012;7:e39416. doi: 10.1371/journal.pone.0039416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Wu CO, Young NS. Activity of alemtuzumab monotherapy in treatment-naive, relapsed, and refractory severe acquired aplastic anemia. Blood. 2012;119:345–54. doi: 10.1182/blood-2011-05-352328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles AJ, Twyman CL, Arnold DL. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–39. doi: 10.1016/S0140-6736(12)61768-1. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Coles AJ, Arnold DL. Alemtuzumab versus interferon β1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–28. doi: 10.1016/S0140-6736(12)61769-3. [DOI] [PubMed] [Google Scholar]
- Zhu WM, Li Y, Yu C, Li N, Li JS. Antimouse CD52 monoclonal antibody inhibits established spontaneous colitis in IL-10-deficient mice. Inflamm Bowel Dis. 2011;17:E72–3. doi: 10.1002/ibd.21733. [DOI] [PubMed] [Google Scholar]
- Wang H, Dong J, Zuo L. Anti-mouse CD52 monoclonal antibody ameliorates iron-deficient anaemia in IL-10 knockout mice. Br J Nutr. 2014;111:987–95. doi: 10.1017/S0007114513003413. [DOI] [PubMed] [Google Scholar]
- Elliott DE, Setiawan T, Metwali A, Blum A, Urban JF, Jr, Weinstock JV. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. Eur J Immunol. 2004;34:2690–8. doi: 10.1002/eji.200324833. [DOI] [PubMed] [Google Scholar]
- Berg DJ, Davidson N, Kuhn R. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4+ TH1-like responses. J Clin Invest. 1996;98:1010–20. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplugues E, Huber S, Gagliani N. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–8. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looijer-van LM, Hotte N, Dieleman LA, Albert E, Mulder C, Madsen KL. Estrogen receptor-β signaling modulates epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. 2011;300:G621–6. doi: 10.1152/ajpgi.00274.2010. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang W, Zuo L. Bifidobacteria may be beneficial to intestinal microbiota and reduction of bacterial translocation in mice following ischaemia and reperfusion injury. Br J Nutr. 2013;109:1990–8. doi: 10.1017/S0007114512004308. [DOI] [PubMed] [Google Scholar]
- Wu LL, Chiu HD, Peng WH. Epithelial inducible nitric oxide synthase causes bacterial translocation by impairment of enterocytic tight junctions via intracellular signals of Rho-associated kinase and protein kinase C ζ. Crit Care Med. 2011;39:2087–98. doi: 10.1097/CCM.0b013e31821cb40e. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang W, Zuo L. Intestinal dysbacteriosis contributes to decreased intestinal mucosal barrier function and increased bacterial translocation. Lett Appl Microbiol. 2014;58:384–92. doi: 10.1111/lam.12201. [DOI] [PubMed] [Google Scholar]
- Geremia A, Arancibia-Carcamo CV, Fleming MP. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208:1127–33. doi: 10.1084/jem.20101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Finotto S. Translating inflammatory bowel disease research into clinical medicine. Immunity. 2009;31:357–61. doi: 10.1016/j.immuni.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Weber CR, Raleigh DR, Su L. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–46. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Gong J, Zhu J. The suppressive effect of triptolide on chronic colitis and TNF-α/TNFR2 signal pathway in interleukin-10 deficient mice. Clin Immunol. 2008;129:211–8. doi: 10.1016/j.clim.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Dong J, Wang H, Wu G. Oral treatment with SEW2871, a sphingosine-1-phosphate type 1 receptor agonist, ameliorates experimental colitis in interleukin-10 gene deficient mice. Clin Exp Immunol. 2014;177:94–101. doi: 10.1111/cei.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Watson AJ, Chu S, Sieck L. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902–12. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, Dejonckheere E, Van Hauwermeiren F. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol Med. 2013;5:1000–16. doi: 10.1002/emmm.201202100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Clayburgh DR, Mittal N. Epithelial NF-κB enhances transmucosal fluid movement by altering tight junction protein composition after T cell activation. Am J Pathol. 2010;176:158–67. doi: 10.2353/ajpath.2010.090548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering NA, Fromm M, Schulzke JD. Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J Physiol. 2012;590:1035–44. doi: 10.1113/jphysiol.2011.224568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeissig S, Burgel N, Gunzel D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn*s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivinus-Nebot M, Frin-Mathy G, Bzioueche H. Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut. 2014;63:744–52. doi: 10.1136/gutjnl-2012-304066. [DOI] [PubMed] [Google Scholar]
- Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;304:G970–9. doi: 10.1152/ajpgi.00183.2012. [DOI] [PubMed] [Google Scholar]
- Poritz LS, Harris LR, 3rd, Kelly AA, Koltun WA. Increase in the tight junction protein claudin-1 in intestinal inflammation. Dig Dis Sci. 2011;56:2802–9. doi: 10.1007/s10620-011-1688-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- Wang F, Schwarz BT, Graham WV. IFN-γ-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131:1153–63. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschke KA, Villani AC, Vermeire S. Tumor necrosis factor receptor gene polymorphisms in Crohn*s disease: association with clinical phenotypes. Am J Gastroenterol. 2005;100:1126–33. doi: 10.1111/j.1572-0241.2005.40534.x. [DOI] [PubMed] [Google Scholar]