

Abstract
The use of DNA sequence data often leads to the recognition of cryptic species within putatively well-known taxa. The opposite case, detecting less diversity than originally described, has, however, far more rarely been documented. Maniola jurtina, the Meadow Brown butterfly, occurs all over Europe, whereas all other six species in the genus Maniola are restricted to the Mediterranean area. Among them, three are island endemics on Sardinia, Cyprus, and Chios, respectively. Maniola species are almost indistinguishable morphologically, and hybridization seems to occur occasionally. To clarify species boundaries and diversification history of the genus, we reconstructed the phylogeography and phylogeny of all seven species within Maniola analyzing 138 individuals from across its range using mitochondrial and nuclear genetic markers. Examination of variation in mitochondrial and nuclear DNA surprisingly revealed a case of taxonomic “oversplitting”. The topology of the recovered phylogenetic tree is not consistent with accepted taxonomy, but rather reveals haplotype clades that are incongruent with nominal species boundaries: instead of seven species, we recognized only two major, yet incompletely segregated, lineages. Our results are consistent with the hypothesis that Maniola originated in Africa. We suggest that one lineage dispersed over the Strait of Gibraltar and the Iberian Peninsula to the west of Europe, while the other lineage spreads eastward through Asia Minor and over the Bosporus to Eastern Europe.
Keywords: Biogeography, DNA barcoding, endemism, expansion routes, phylogeny, speciation, species delimitation
Introduction
Taxonomic mishaps such as “oversplitting” (the misinterpretation of individual variants as distinct specific entities) and “lumping” (erroneously grouping several species into a single one) continue to pose problems in modern taxonomy and systematics (Dayrat 2005). In the last decade, DNA barcoding has become a handy tool for resolving species identifications, especially in clades where morphological characterization of putative taxa is weak or inconsistent (Dupuis et al. 2012; Puillandre et al. 2012; Ratnasingham and Hebert 2013).
As a consequence, supposedly well-established taxonomic systems for many groups of organisms have become subject to frequent, and often drastic, change due to ongoing revisions. Newly described cryptic species and novel insights into relationships between taxa frequently overturn traditional systematics, even in comparatively well-known groups such as butterflies (e.g., Dinca˘ et al. 2011; Talavera et al. 2013). In particular, the use of DNA barcoding approaches has led to a very substantial increase of recognized species numbers during the last decade (Hebert et al. 2004; Hausmann et al. 2011).
Meadow Brown butterflies have been in the focus of studies in evolutionary genetics and ecology since decades (Scali 1971; Brakefield 1982a,b; Goulson 1993). The center of these studies has been the widespread M. jurtina. The other species in the genus, especially the island endemics, have been largely neglected since their description, apart from a few anecdotal taxonomic papers (e.g., Jutzeler et al. 1997), some work on ecological aspects of differentiation (Grill et al. 2006a,b) and work on morphometrics in M. jurtina and M. nurag, particularly in genitalia (Dapporto 2010). Maniola jurtina alone has been studied more extensively, but so far, no conclusive picture of phylogeographic patterns has emerged (Schmitt et al. 2005; Dapporto et al. 2011). As taxonomic uncertainties last due to great morphological variation and overlap in distribution areas, we here investigated species boundaries with molecular markers.
Maniola comprises seven described species with a rather peculiar distribution: Maniola jurtina (Linnaeus, 1758) is widely distributed over much of Europe, northwest Africa, the Canary Islands, and eastward to the Ural mountains and NW Iran (Tshikolovets 2011), whereas the other six species in the genus are restricted to the Mediterranean area (Grill et al. 2006a, 2007). Among them, four species are narrowly endemic to islands and therefore belong to those butterflies of Europe with the smallest ranges: M. cypricola Graves, 1928 on Cyprus, M. chia Thomson, 1987 on the Aegean islands Chios and Inousses, M. halicarnassus Thomson, 1990 on the Aegean island Nisyros and the neighboring Turkish Bodrum peninsula, and M. nurag Ghiliani, 1852 on Sardinia (Grill et al. 2007; Kudrna et al. 2011). The remaining two species inhabit largely overlapping parts of the eastern Mediterranean realm: M. telmessia (Zeller, 1847) is found on many Aegean islands and throughout southern Turkey from the Bosporus eastward to Syria, Lebanon, Israel, Jordan, Iraq, and NW Iran (Hesselbarth et al. 1995; Tshikolovets 2011). The range of M. megala (Oberthür, 1909) is smaller, from the Aegean island Lesbos through southern Turkey as far as the Syrian border (Tshikolovets 2011).
With these unusual distributions, Maniola stands as an example for a genus with one widespread and several regional species, a pattern that is found similarly in a range of Palaearctic animal genera, not only in butterflies but in fact in many groups including vertebrates (e.g., in wall lizards Podarcis: Poulakakis et al. 2005; in birds of the genus Oenanthe: Randler et al. 2012; in butterflies: Dennis et al., 2008; Dapporto and Strumia 2008).
Maniola jurtina,M. telmessia,M. halicarnassus, and M. megala are broadly sympatric in Turkey, yet evidence for differential habitat preferences is vague (Hesselbarth et al. 1995). Despite minor variation in emergence times, hybridization has been recorded, for example between M. halicarnassus and M. telmessia (Hesselbarth et al. 1995) as well as M. jurtina and M. nurag on Sardinia (Grill et al. 2006b). All Maniola species strikingly resemble each other in terms of wing patterns and genitalia morphology, making taxonomy a challenging task even for experts (Thomson 1973; Olivier 1993; Grill et al. 2004). In Table1, a survey of described morphological and ecological differences of the Maniola species is given.
Table 1.
Survey of morphological and ecological characters of Maniola species, collated from Hesselbarth et al. (1995), Olivier (1993), Tshikolovets (2011), Tolman and Lewington (2008) and Makris and John (2003)
| M. jurtina | M. nurag | M. chia | M. megala | M. telmessia | M. halicarnassus | M. cypricola | |
|---|---|---|---|---|---|---|---|
| Wingspan | Male: 36–44 mm; female: 37–46 mm | Male: 36–40 mm, smaller than M. jurtina; female: 36-40 mm | Male: 36–44 mm; female: 37–46 mm | Male: 36–44 mm; female: 37–46 mm, larger than M. jurtina | Male: 36–45 mm, smaller than M. jurtina; female: 37–46 mm | Male: 42–47 mm; female: 42–47 mm, larger than M. telmessia | 40–50 mm |
| Elevation | Up to 2700 m in Caucasus | 500–1500 m | Up to 800 m | Up to 1000 m | Up to 2000 m | Up to 450 m | Up to 1900 m |
| Flight period | May–September | May–late September | May–October | May–October | April–October | May–October | April–November |
| Habitat | Grassy, bushy, often flowery places | Grassy, flowery places among bushes and rocks | Grassy, rocky and bushy places; cultivated ground | Similar to M. telmessia; grassy areas including woody and cultivated areas | Light woodlands, rocky scrublands | Moist and shaded grassy places | Everywhere on Cyprus |
| Ovum | 11–21 longitudinal ribs, regionally variable | 13–14 longitudinal ribs | 19–21 longitudinal ribs | 14–16 longitudinal ribs, very small eggs | 9–18 longitudinal ribs | ||
| Morphological differences | Yellow–orange areas more extensive than in M. jurtina; better developed, more sharply defined in females; conspicuous sex-brand in males | Indistinguishable from M. jurtina; distinct genitalia and differences in allozyme genetics | Hind wing outer margin averagely more undulate; underside markings darker; very short penis and spiny excrescences on basal part of gnathos | Apical ocellus always distinct and bigger than in M. jurtina, yellow orange encircled | Male sex-brand large, triangular; male genitalia distinguishable from M. megala,M. chia and M. jurtina; females more orange than all other Maniola | Male forewings larger, more elongated and hairy than in M. telmessia, triangular sex-brand; female genitalia different from M. telmessia | |
| No. larval instars | 6 | 5 | Variable, 5 or 6 | ||||
| Notes | Morphologically very variable species | Lesbos: smaller fore wings, highest portion of individuals with bipupillary ocelli | Emergence 2-4 weeks earlier than M. jurtina | Emergence 2.5 weeks after M. telmessia, flies at cooler, shadier places than M. telmessia | Larvae feed only on silicate poor grasses |
Available studies on phylogeography within Maniola conducted so far never included all species of the genus and never had a geographically large enough taxon sampling (Schmitt et al. 2005; Grill et al. 2006a; Dapporto et al. 2011). So, this study aims at (1) reconstructing the phylogeny of the genus Maniola, (2) comparing it with the currently accepted taxonomy, (3) testing the usability of DNA barcoding for species identifications in Maniola, and (4) investigating whether molecular data reveal information about the existence of refugia in the Mediterranean region and possible expansion routes that have led to the current distribution of the species. What we find for Maniola may indicate that similar outcomes are to be expected for a number of other genera with similarly peculiar distribution patterns.
Material and Methods
Sample collection, DNA extraction, amplification, sequencing, and alignment
We used 138 Maniola individuals, from sites across Europe, Anatolia, and northern Africa (Table S2) representing all seven taxonomically described species. Specimens had either been dried or put into 99% ethanol after collection. Samples were then stored at −20°C until DNA extraction. The applied mitochondrial and nuclear genetic markers (cytochrome-c-oxidase [COI], cytochrome-B [CytB], wingless [wgl], and elongation factor 1α [EF1a]) were amplified with varying success (Table2), as the specimens had been collected between the years 1980 and 2013; older samples performed often worse in PCR than the more recent ones. We additionally included COI sequences from 51 individuals from NCBI GenBank.
Table 2.
Successfully amplified marker sequences (mtDNA = COI + CytB; combined = all genetic markers used)
| COI | CytB | wingless | EF 1α | mtDNA | Combined | |
|---|---|---|---|---|---|---|
| M. jurtina (n = 51) | 47 | 29 | 30 | 28 | 27 | 22 |
| M. nurag (n = 25) | 19 | 12 | 12 | 11 | 8 | 9 |
| M. chia (n = 18) | 16 | 7 | 14 | 17 | 6 | 6 |
| M. megala (n = 6) | 5 | 5 | 5 | 3 | 5 | 3 |
| M. cypricola (n = 14) | 12 | 14 | 11 | 9 | 12 | 9 |
| M. telmessia (n = 16) | 11 | 16 | 14 | 13 | 11 | 11 |
| M. halicarnassus (n = 8) | 8 | 8 | 5 | 5 | 8 | 5 |
| Total | 118 | 91 | 91 | 86 | 77 | 65 |
As outgroups, we downloaded sequences of closely related satyrine butterfly species from GenBank (see Table S1), from the subtribes Coenonymphina, Erebiina, Maniolina, Melanargiina, Pronophilina, and Satyrina, to improve support of the phylogeny of the genus. Outgroups were selected from Peña et al. (2006), but as they did not use CytB in their study, we additionally sequenced two specimens of Pyronia cecilia that were at hand in our laboratory to have at least one species available with complete gene sampling.
Total genomic DNA was extracted from two legs, respectively, or, if missing, from thoracic muscles following a standard protocol (DNeasy Blood & Tissue Kit, Qiagen Inc., Valencia, CA). DNA samples were extracted and amplified in a separate room exclusively dedicated to DNA extractions. Primer names, references, primer sequences as well as respective annealing temperature and time are shown in Table3.
Table 3.
Primer sequences used in this study (F = forward, R = reverse)
| Gene | Primer name | References | Sequence (5′-3′) | Annealing temp. and time |
|---|---|---|---|---|
| COI | LepF | Hajibabaei et al. (2006) | ATTCAACCAATCATAAAGATATTGG (F) | 44°C – 1 min 30 s; 46°C – 1 min 15 s |
| LepR | TAAACTTCTGGATGTCCAAAAAATCA (R) | |||
| CytB | CB-J-10933 | Simons et al. (1994) | TATGTACTACCATGAGGACAAATATC (F) | 46°C – 1 min 20 s |
| CB-N-11367 | ATTACACCT CCTAATTTATTAGGAAT (R) | |||
| wgl | LepWG1 | Brower and DeSalle (1998) | GARTAYAARTGYCAYGGYATGTCTGG (F) | 48°C – 1 min 30 s |
| LepWG2 | ACTICGCARCACCARTGGAATGTRCA (R) | |||
| Ef 1α | EF51.9 | Monteiro and Pierce (2001) | CARGACGTATACAAAATCGG (F) | 52.5°C – 1 min 30 s |
| EFrcM4 | ACAGCVACKGTYTGYCTCATRTC (R) | |||
| Starsky/M3 | Cho et al. (1995) | CACATYAACATTGTCGTSATYGG (F) | 54°C – 1 min | |
| Luke/rcM51-1 | CATRTTGTCKCCGTGCCAKCC (R) |
Gene fragments of the mitochondrial (COI,CytB) and nuclear DNA (wgl,EF1α) were amplified using polymerase chain reaction (PCR) in a thermal cycler (Eppendorf Mastercycler pro S vapo.protect). Negative (sterile water) and positive (samples with known genotypes) controls were always used. PCRs were performed in 25 μL volumes containing 1 μL of genomic DNA, 22.5 μL of ReddyMix®, 0.5 μL of the respective forward and reverse primer, and 1 μL BSA. PCR conditions were optimized for each primer pair.
PCR products were visualized on an agarose gel to verify amplification success. Afterward, they were analyzed using an ABI capillary sequencer (3730 DNA analyzer, Applied Biosystems). Sequences contained no gaps or stop codons and were aligned and edited in BioEdit 7.1.3.0 (Hall 1999) for each gene separately. All new sequences have been submitted to GenBank, under accession numbers KP032366 - KP032458 (Cytb), KP032241 - KP032365 (COI), KP032552 - KP032639 (EF1a), and KP032459 - KP032551 (wgl). Accession numbers of outgroup species are provided in Table S1.
Phylogenetic analysis
We used Bayesian inference (BI) to reconstruct phylogenetic trees. The combined genes tree (including 65 individuals, for which all genetic markers worked) was based on an alignment of 2427 base pairs. Trees of COI included 118 sequences with 657 bp length. We used the program JModelTest (v. 2.0) on the Phylemon-Server 2.0 (Sánchez et al. 2011) to estimate models of nucleotide substitutions (of three substitution schemes: JC, HKY, and GTR) as judged by the Bayesian information criterion (BIC) for BI. Only the best-fit models were subsequently used for evaluations of tree topology. BI analysis was performed with MrBayes v.3.2, considering the estimated best-fit substitution models (Huelsenbeck and Ronquist 2001). Trees were visualized using Figtree v.1.3.1 (Rambaut 2009).
For BI trees under BIC, the best-fit models were as follows: [GTR + G] for COI, [HKY + I + G] for CytB, [GTR + I + G] for mtDNA and all genes combined, [K80 + I] for Elongation factor and nDNA, and [HKY + I] for wingless.
Genetic variation within described species was estimated as the numbers of variable sites (S), average numbers of nucleotide differences (k), haplotype diversity (h: Nei, M. 1987), and nucleotide diversity (π: Nei and Li 1979) for each gene as well as species, using the software DnaSP v5 (Librado and Rozas 2009). Genetic distances (Kimura-2-distances) were calculated with MEGA 5.1 (Tamura et al. 2011).
Haplotype networks
Additionally, we generated median-joining networks for several datasets using the program Network 4.6.1.1 (http://www.fluxus-engineering.com). The median-joining method is based on a maximum parsimony algorithm that searches for all shortest trees of a particular dataset (Bandelt et al. 1999).
Results
Genetic diversity and species delimitation by DNA barcoding
For COI (657 bp), 118 sequences showed 37 different haplotypes with 48 variable sites. The 91 CytB sequences (432 bp) displayed 33 haplotypes and 43 variable sites. The 87 sequences of Elongation factor (1051 bp) showed 53 haplotypes and 66 variable sites, and wingless (403 bp; 91 sequences) showed 34 haplotypes and 25 variable sites.
For all species and all genetic markers (Table4), estimates of haplotype diversity (h) were rather high, ranging from 0.333 to 0.996, whereas estimates of nucleotide diversity (π) were much lower (especially in nuclear genes), ranging from 0.00112 to 0.02804. Interestingly, the endemic species did not show lower haplotype and nucleotide diversity than their mainland congeners, and M. nurag even showed the highest diversity of all as well as very high average numbers of nucleotide differences (k). The available M. megala specimens all had exactly the same haplotype (see Fig. 4), and M. halicarnassus revealed rather low numbers of variable sites. As two lineages could be detected in the phylogenetic trees (see subsection Phylogeny and phylogeography), they were also investigated for their nucleotide diversity: Lineage A (comprising the taxa M. cypricola,M. telmessia, and M. halicarnassus) generally had lower nucleotide diversity than Lineage B (all other nominal species).
Table 4.
Summary of molecular diversity indices of COI,CytB,wgl,elongation factor 1α genes, mitochondrial DNA (combined COI and CytB), and nuclear DNA (nDNA); sample size (n), number of haplotypes (no.), number of variable sites (S), average number of nucleotide differences (k), haplotype diversity (h), and nucleotide diversity (π) with standard deviation (SD). Nuclear genes have been doubled for analysis by the program DNAsp to avoid ambiguous sites, as DNAsp cannot analyze diploide genetic information
| Gene | Species | n | no. | S | k | h (±SD) | π (±SD) |
|---|---|---|---|---|---|---|---|
| COI | M. jurtina | 47 | 20 | 40 | 6.29 | 0.834 ± 0.048 | 0.01065 ± 0.00185 |
| M. nurag | 19 | 8 | 33 | 10.60 | 0.871 ± 0.044 | 0.01849 ± 0.00213 | |
| M. megala | 5 | 1 | – | – | – | – | |
| M. cypricola | 12 | 6 | 26 | 8.86 | 0.848 ± 0.074 | 0.01497 ± 0.00461 | |
| M. chia | 16 | 5 | 15 | 2.26 | 0.65 ± 0.108 | 0.00474 ± 0.00283 | |
| M. telmessia | 11 | 7 | 30 | 6.47 | 0.873 ± 0.089 | 0.00985 ± 0.00491 | |
| M. halicarnassus | 8 | 4 | 7 | 2.61 | 0.750 ± 0.139 | 0.00397 ± 0.00114 | |
| Lineage A | 31 | 14 | 17 | 3.71 | 0.890 ± 0.036 | 0.00627 ± 0.00059 | |
| Lineage B | 87 | 23 | 42 | 7.27 | 0.864 ± 0.025 | 0.01585 ± 0.00138 | |
| All samples | 118 | 37 | 48 | 8.84 | 0.919 ± 0.015 | 0.01926 ± 0.00099 | |
| CytB | M. jurtina | 29 | 14 | 30 | 5.48 | 0.894 ± 0.040 | 0.01305 ± 0.00404 |
| M. nurag | 12 | 7 | 32 | 11.67 | 0.909 ± 0.056 | 0.02804 ± 0.00382 | |
| M. megala | 5 | 1 | – | – | – | – | |
| M. cypricola | 14 | 8 | 25 | 7.82 | 0.868 ± 0.068 | 0.01867 ± 0.00563 | |
| M. chia | 7 | 4 | 24 | 8.48 | 0.714 ± 0.181 | 0.02173 ± 0.00831 | |
| M. telmessia | 16 | 7 | 24 | 4.00 | 0.792 ± 0.089 | 0.00952 ± 0.00490 | |
| M. halicarnassus | 8 | 5 | 6 | 1.79 | 0.786 ± 0.151 | 0.00413 ± 0.00120 | |
| All samples | 91 | 33 | 43 | 10.54 | 0.906 ± 0.021 | 0.02703 ± 0.00106 | |
| wgl | M. jurtina | 30 (60) | 11 | 8 | 1.47 | 0.774 ± 0.041 | 0.00432 ± 0.00044 |
| M. nurag | 12 (24) | 13 | 11 | 2.57 | 0.906 ± 0.046 | 0.00754 ± 0.00095 | |
| M. megala | 5 (10) | 7 | 8 | 3.62 | 0.933 ± 0.062 | 0.00899 ± 0.00106 | |
| M. cypricola | 11 (22) | 6 | 7 | 1.05 | 0.671 ± 0.077 | 0.00281 ± 0.00066 | |
| M. chia | 14 (28) | 9 | 5 | 1.83 | 0.831 ± 0.051 | 0.00537 ± 0.00051 | |
| M. telmessia | 14 (28) | 6 | 3 | 1.18 | 0.741 ± 0.067 | 0.00348 ± 0.00040 | |
| M. halicarnassus | 5 (10) | 3 | 4 | 0.96 | 0.511 ± 0.164 | 0.00281 ± 0.00137 | |
| All samples | 91 (182) | 34 | 25 | 2.48 | 0.904 ± 0.011 | 0.00731 ± 0.00033 | |
| EF 1α | M. jurtina | 28 (56) | 25 | 24 | 1.85 | 0.881 ± 0.032 | 0.00183 ± 0.00021 |
| M. nurag | 12 (24) | 19 | 52 | 9.65 | 0.975 ± 0.021 | 0.00952 ± 0.00349 | |
| M. megala | 3 (6) | 2 | 1 | 0.33 | 0.333 ± 0.215 | 0.00032 ± 0.00021 | |
| M. cypricola | 9 (18) | 7 | 7 | 1.87 | 0.784 ± 0.085 | 0.00183 ± 0.00035 | |
| M. chia | 17 (34) | 11 | 10 | 1.43 | 0.811 ± 0.052 | 0.00143 ± 0.00020 | |
| M. telmessia | 13 (26) | 7 | 9 | 1.39 | 0.689 ± 0.088 | 0.00134 ± 0.00035 | |
| M. halicarnassus | 5 (10) | 4 | 5 | 1.16 | 0.533 ± 0.180 | 0.00112 ± 0.00046 | |
| All samples | 87 (174) | 53 | 66 | 2.72 | 0.851 ± 0.021 | 0.00285 ± 0.00060 | |
| mtDNA | M. jurtina | 29 | 14 | 38 | 8.21 | 0.899 ± 0.036 | 0.01389 ± 0.00244 |
| M. nurag | 9 | 5 | 26 | 12.61 | 0.861 ± 0.087 | 0.01974 ± 0.00250 | |
| M. cypricola | 12 | 10 | 51 | 17.59 | 0.955 ± 0.057 | 0.01740 ± 0.00519 | |
| M. chia | 6 | 4 | 35 | 12.13 | 0.867 ± 0.129 | 0.01159 ± 0.00688 | |
| M. telmessia | 11 | 7 | 54 | 11.45 | 0.873 ± 0.089 | 0.01052 ± 0.00570 | |
| M. halicarnassus | 8 | 6 | 13 | 4.39 | 0.893 ± 0.111 | 0.00403 ± 0.00106 | |
| Lineage A | 29 | 19 | 29 | 6.13 | 0.921 ± 0.041 | 0.00606 ± 0.00078 | |
| Lineage B | 51 | 17 | 37 | 9.63 | 0.904 ± 0.022 | 0.01776 ± 0.00153 | |
| All samples | 80 | 30 | 43 | 10.81 | 0.946 ± 0.011 | 0.01994 ± 0.00095 | |
| nDNA | M. jurtina | 26 (52) | 33 | 31 | 3.40 | 0.962 ± 0.015 | 0.00248 ± 0.00022 |
| M. nurag | 11 (22) | 21 | 29 | 6.41 | 0.996 ± 0.015 | 0.00457 ± 0.00029 | |
| M. megala | 3 (6) | 4 | 8 | 3.93 | 0.867 ± 0.129 | 0.00276 ± 0.00059 | |
| M. cypricola | 9 (18) | 11 | 13 | 2.93 | 0.908 ± 0.051 | 0.00208 ± 0.00039 | |
| M. chia | 14 (28) | 20 | 14 | 3.33 | 0.974 ± 0.016 | 0.00245 ± 0.00021 | |
| M. telmessia | 13 (26) | 16 | 12 | 2.61 | 0.902 ± 0.049 | 0.00186 ± 0.00030 | |
| M. halicarnassus | 5 (10) | 5 | 9 | 2.11 | 0.756 ± 0.130 | 0.00148 ± 0.00071 | |
| All samples | 81 (162) | 90 | 62 | 4.42 | 0.976 ± 0.005 | 0.00339 ± 0.00014 |
Figure 4.
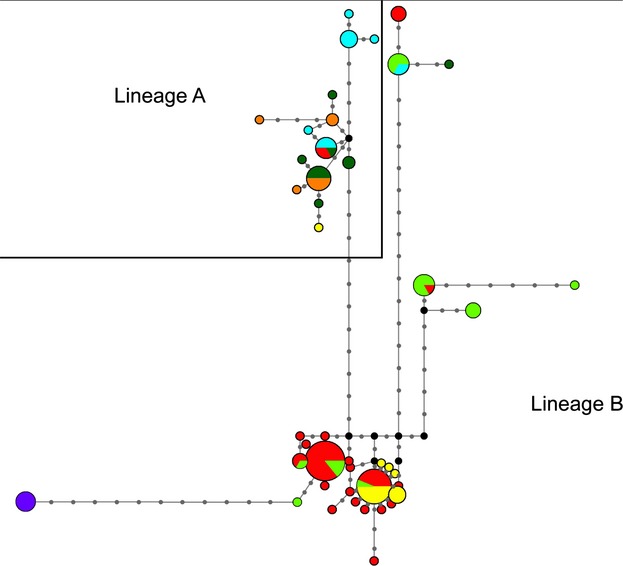
Median-joining network of COI sequences of Meadow Brown butterflies (Maniola). Size of circles is proportional to number of sequences with similar haplotypes; length of lines is proportional to number of mutational steps between haplotype clades. Black dots are missing haplotypes; gray dots are mutational steps. Five different haplotype clades can be recognized. Species color codes: red = M. jurtina, light green = M. nurag, yellow = M. chia, violet = M. megala, blue = M. cypricola, dark green = M. telmessia, orange = M. halicarnassus. Down in the middle two different Maniola jurtina clades or lineages can be seen, to the left of them is the distinct M. megala clade. To the right side, a Maniola nurag clade (with a single M. jurtina from Sardinia) can be found. On the top, there is the M. telmessia,M. halicarnassus, and M. cypricola clade to the left and the mixed island species clade (M. jurtina from Crete, M. nurag,M. cypricola, and one M. telmessia individual from Israel) to the right. Single haplotypes are common to all clades.
To use the barcoding region for species identification, a distinct barcoding gap should exist – separating intra- from interspecific pairwise genetic distances. Among the 118 Maniola COI sequences, no such gap could be found, as intra- and interspecific distances for conventionally delineated species were intermixing. When only the two major genetic lineages (see below) were compared, genetic distances between and within lineages were separated more clearly from each other, although a convincing barcoding gap could still not be found (Fig.1). Approximately 99.8% of pairwise COI sequence comparisons showed two or more percent genetic distance between these two lineages (interlineage), but only 51% of pairwise comparisons showed negligible (0–1% genetic distance) distances within lineages (intralineage). About 26% of sequence comparisons showed high (≥2%) distances within lineages.
Figure 1.
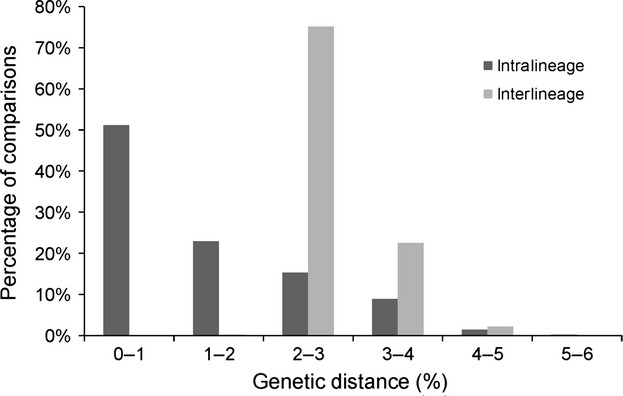
Frequency distribution of pairwise intra- and interlineage Kimura-2-distances of the COI sequences. To ensure the usability of DNA barcoding for species delimitation, a “barcoding gap” should exist between these two data series. In Maniola, however, genetic distances within lineages versus between lineages intermix.
Phylogeny and phylogeography
The topology of the Bayesian Inference tree of all Maniola samples, based on specimens for whom all genetic markers were available plus the selected outgroup species (Fig.2), is not consistent with current taxonomy. Rather, most nominal species form mixed clades. Nevertheless, recurrent clusters can be recognized: there is one lineage of M. jurtina (from various provenances; mixed with M. chia,M. nurag, and M. megala) (Lineage B in the lower part of the tree diagram); another one containing a few M. jurtina (from Crete), M. nurag, two M. cypricola specimens, and one M. telmessia sequence (upper Lineage B); and one M. telmessia (together with all M. halicarnassus and the majority of M. cypricola sequences) lineage (Lineage A). In the BI tree of COI sequences (Fig.3), Lineage B forms a single clade, so the two parts of this lineage are both called Lineage B in the BI tree of the combined genes, as Lineage B can be defined as all specimens that do not belong to Lineage A. The “mixed island species clade” within Lineage B (marked in blue in Fig.2) contains M. nurag,M. jurtina from Crete, M. cypricola, and a single M. telmessia sequence from Israel. All other M. telmessia specimens cluster together in one clade (Lineage A). This also applies to M. halicarnassus, while M. megala forms a distinct clade within Lineage B. Sequences of the island endemics never form coherent clusters. All samples of M. cypricola and M. nurag distribute across several different clades. Most individuals of M. chia cluster with one M. jurtina lineage, but one individual appears in the same M. telmessia/M. halicarnassus cluster that also contains most M. cypricola individuals. Overall, the monophyly of Maniola is very well supported, whereas most clusters within Maniola show low support. Stable, well-supported groupings are only the invariant M. megala samples from Lesbos and the aforementioned “mixed island species clade” within Lineage B.
Figure 2.
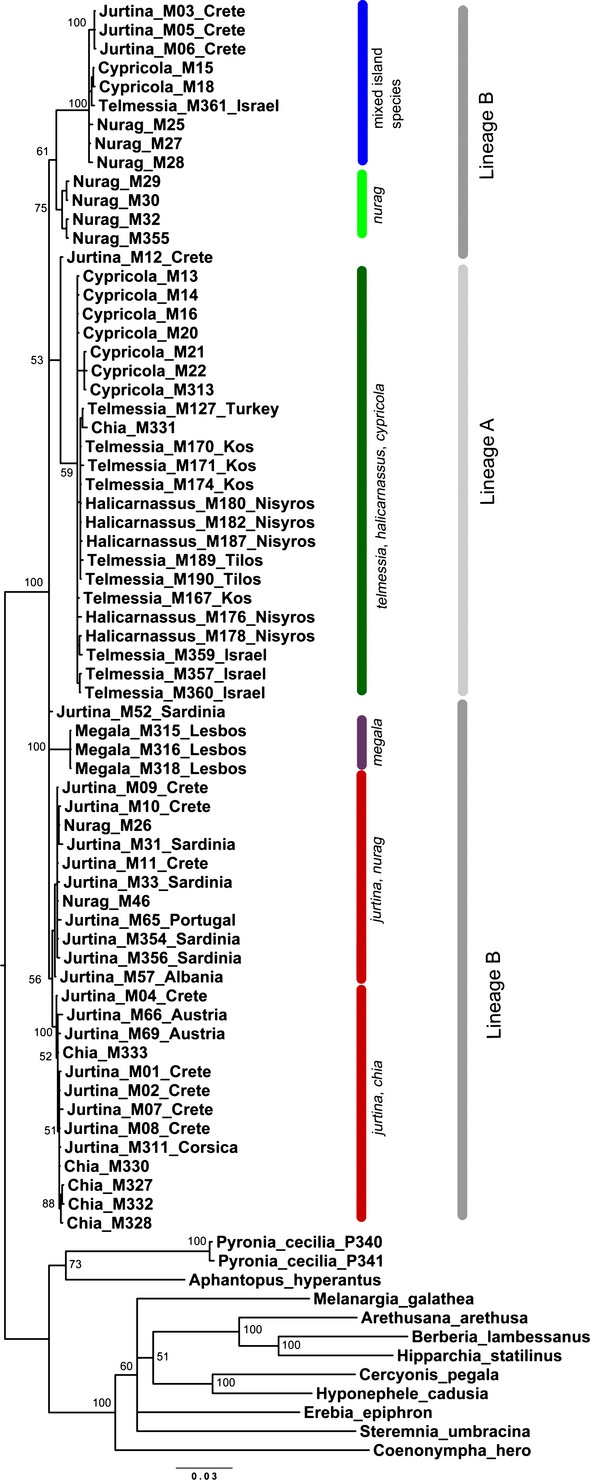
Phylogeny of Maniola butterflies, according to Bayesian inference analysis of combined genes dataset with probability values (%). Nominal species do not form clades according to current taxonomy, but two genetic lineages can be roughly defined: one lineage (A) contains M. telmessia,M. halicarnassus, and M. cypricola (59% prob.), and another lineage (B) contains the remaining species. Of the conventionally accepted species, only M. megala and M. halicarnassus do not occur in several clades across the tree.
Figure 3.
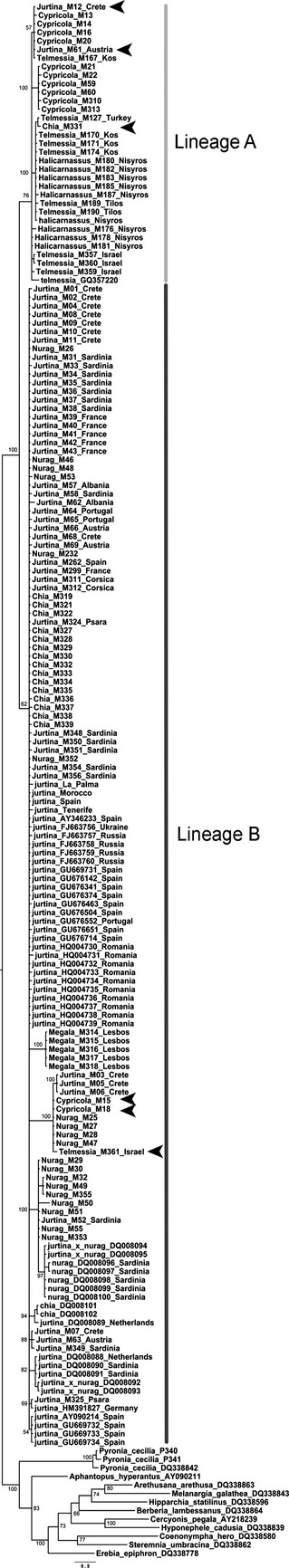
Phylogeny of Maniola butterflies, according to Bayesian inference analysis of COI sequences with support values. Nominal species form intermixed clades that are not consistent with current taxonomy. Also each of the island endemics spreads through several clades. The only monophylum coinciding with a described species is represented by M. megala. The tree shows two main branches (Lineages A and B). Lineage A (light gray) predominantly contains Maniola telmessia,M. halicarnassus, and M. cypricola, whereas (Lineage B; dark gray) contains M. jurtina,M. nurag,M. chia, and M. megala. Lineage A shows a probability value of 76%. Black arrows indicate exceptional sequences which cluster with the “wrong” lineage.
The tree based exclusively on the COI barcoding sequences shows similar groupings (Fig.3), but more clearly emphasizes the split of haplotypes into two main lineages: One lineage (Lineage A; light gray) predominantly contains Maniola telmessia,M. halicarnassus, and M. cypricola, whereas the other contains M. jurtina,M. nurag,M. chia, and M. megala (Lineage B; dark gray), although both with some exceptions (black arrows). Lineage A shows a probability value of 76%.
Median-joining networks were calculated for COI, mtDNA, nDNA, and combined genes and resembled the phylogenetic trees largely; but in comparison, they show a clearer resolution of the relationships between haplotypes. Networks of COI, mtDNA, and combined genes were much alike: they showed the same underlying clustering, but the more genes were used the more single haplotypes and mutational steps between clusters were found. Only the COI network is shown (Fig.4), as it is the most clearly arranged. Groupings are similar as in the phylogenetic trees described above.
Discussion
Our data indeed show one of the – at least up to now – rare cases (cf. Vila et al. 2010), where the differentiation of several nominally described, but rather ambiguously characterized species, did not receive stronger support and higher resolution of “cryptic diversity” by use of genetic methods. Although one might have suspected the existence of even more cryptic species among Maniola because of their geographic distribution around the Mediterranean Sea and its many islands, the opposite was found, viz. according to our data, the whole genus consists of only one, quite variable species.
DNA barcoding, phylogenies, and taxonomic implications
Haplotype as well as nucleotide diversity of the whole Maniola genus is comparable with those of a single widely distributed species (e.g., Jeratthitikul et al. 2013). The high genetic diversity of the island species within Maniola indicates gene flow among the so-called species. Island populations (Frankham 1997), and isolated populations in general (Cassel and Tammaru 2003), typically have less genetic variation than continental populations due to bottlenecks experienced by small founder populations and subsequent inbreeding (Frankham 1998; Keller and Waller 2002). These outcomes are in accordance with earlier results by Grill et al. (2007), who also found no evidence for lower genetic variation in Sardinian populations of Meadow Brown butterflies.
DNA barcoding based on COI sequences cannot be reliably used to split Maniola further into a number of species, in contrast to many cases reported in the recent barcoding literature (Hausmann et al. 2011; Ratnasingham and Hebert 2013). The tree constructed from the barcoding region of the COI gene shows a clear split into only two lineages, which both range across boundaries of all species taxonomically described for this genus. Addition of further sequence markers did not enhance resolution of clades. All individuals (with the exception of M. megala) invariably clustered according to a different pattern than expected from their current taxonomic affiliation.
The results of this study suggest that, instead of accepting seven distinct “good” species, it is more parsimonious to assign all Meadow Browns to one single genetic group. One may distinguish the two lineages A and B as different genetic entities, with some weak further substructure according to haplotypes.
It is understandable that a taxon morphologically as variable as Maniola tempted taxonomists of the 19th and 20th century to describe new species; but according to our genetic data, these descriptions delineate variation, not speciation. For example, the species M. chia and M. halicarnassus (which were proposed as distinct taxa as late as 1987 and 1990, respectively) probably should have been investigated more critically before describing them as new species. If there is endemicity on an island, it is usually observed in a number of unrelated groups of organisms (cf. the high number of endemic species on the Tyrrhenian islands or the Azores). On Chios, as a matter of fact, besides M. chia, only the terrestrial isopod Trachelipus buddelundi is supposed to be endemic (“Only known from its original description. A doubtful species.”; Alexiou and Sfenthourakis 2013). Within the genus Maniola, wing patterns or genitalia morphology are not only determined through the genotype, but also by environmental conditions influencing larval development (Thomson 1973; Goulson 1993; Grill et al. 2004). Hence, even if significant and substantial, differences in phenotypes alone are insufficient to delineate taxonomic entities in that group of butterflies.
Interestingly, a dichotomy – like the two lineages we found – within the “super species” (or species complex) M. jurtina has been postulated multiple times. Starting from Tauber (1970), also Hesselbarth et al. (1995) recognized groupings that were assigned to the more western M. jurtina sensu stricto on the one hand and the eastern M. telmessia on the other hand. Later on, Schmitt et al. (2005) reported the existence of an eastern and a western genetic lineage of Maniola jurtina based on allozyme data, wing patterns, and genitalia morphology as did Dapporto et al. (2011) based on genitalia morphology. Dapporto et al. (2011) rightly called M. jurtina “enigmatic”, as they found contradictory patterns between allozyme and morphological data. Specimens from continental Italy, Sicily, and North Africa shared the same allozyme set, but their genitalia shape was more similar to specimens from the Balkans and some individuals from Eastern and Central Europe. Their explanation for this discrepancy is “recent gene flow in the wake of postglacial range expansions and shifts” (Dapporto et al. 2011). We hypothesize that these contrasting patterns could have come about through by chance-biased sampling from the genetic lineage A for the allozyme study and from lineage B for the genitalia analyses. At any rate, genetic lineages connected to the main Pleistocene glacial refugia, as postulated by Hewitt (1996, 1999), for example, structured according to the three large Mediterranean peninsulas, could not be detected within Maniola.
Hypothetical evolutionary scenario of the genus Maniola and possible expansion routes
Schmitt et al. (2005) hypothesized postglacial expansion routes of an eastern and a western M. jurtina lineage, including a hybrid zone, based on allozyme data. Habel et al. (2009) postulated four postglacial recolonization pathways of Maniola jurtina (three to northern Europe, one to North Africa) out of the Mediterranean peninsulas, also based on allozyme data. Another possible scenario concerning the origin and expansion routes of the whole genus was much earlier suggested by Tauber (1970), who postulated that the western M. jurtina lineage and the eastern M. telmessia species complex diverged from North African ancestors. According to Tauber, the M. jurtina lineage spreads westward over Gibraltar and the Iberian Peninsula, while the M. telmessia complex expanded through Palestine, Lebanon, and Syria to the east. This hypothesis coincides with the fact that M. telmessia is the only Maniola “species” occurring in Israel, Jordan, Lebanon, and Syria (Tshikolovets 2011). According to Tauber's hypothesis, the western M. jurtina complex spreads across Europe and would have met the eastern M. telmessia complex in Asia Minor, giving rise to a sympatric occurrence of both lineages in the eastern Mediterranean nowadays.
Although over 40 years old, Tauber's (1970) “out-of-Africa” hypothesis seems to be a likely scenario for the evolution of the genetic diversity we find in Maniola today. A number of other recent studies revealed similar scenarios for other butterfly species (Weingartner et al. 2006; Kodandaramaiah and Wahlberg 2009; Habel et al. 2010; Husemann et al. 2014). Another fact fitting to an African origin of Maniola is their current distribution: while closely related Satyrinae taxa such as Hyponephele sp. and Aphantopus sp. show trans-Eurasian distributions, the range of Maniola jurtina ends roughly at the Ural Mountains (Tshikolovets 2011).
If the geographic origin of Maniola lay in northern Africa (as it is for other nymphalid butterflies: Aduse-Poku et al. 2009; Kodandaramaiah and Wahlberg 2007), the butterflies could have spread northwestward over Gibraltar and the Iberian Peninsula and then eastward all over Europe and into western Asia. A convincing scenario is exactly what Tauber (1970) hypothesized based on morphological characters and paleo-ecological considerations: a split in the North African stem population, with one part dispersing to the west and the other one to the east. Considering our data, Lineage A (M. cypricola,M. telmessia and M. halicarnassus) could have expanded along an eastern pathway, and Lineage B (M. jurtina,M. nurag,M. chia, and M. megala) could have colonized western and Central Europe through a western migration route (Fig.5). The few individuals that cluster with the “wrong” lineage might indicate a zone of rather recent intermixture in the Aegean Sea and Central Europe. Of course, to further investigate this hypothesis, more haplotypes from this hybrid zone as well as from northern Africa must be examined.
Figure 5.
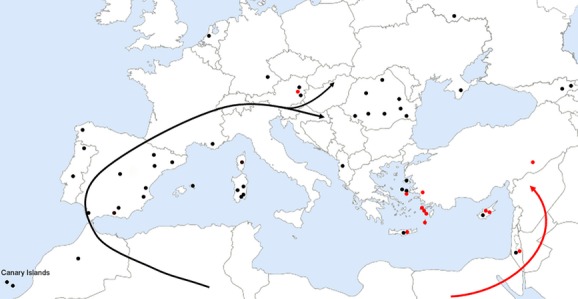
Hypothetical expansion routes of the Maniola lineages A (eastern pathway; red) and B (western pathway; black). Dots show collection sites. (Adapted from User: Madman2001/Wikimedia Commons/CC-BY-SA-3.0.).
Conclusions
During the last decade, phylogenetic studies using sequence data revealed numerous examples for the unexpected discovery of many cryptic species. Our study presents the not so common opposite case: various distinct nominal species melting together to a single species complex. We did not find any proof for the existence of the seven morphologically defined species with the genetic methods used; instead, we found two but moderately distinct genetic lineages. As to the formation of the two lineages, we hypothesize an origin in Africa and two different expansion routes emerging from there, as postulated by Tauber (1970). Thus, we suggest addressing the whole genus as one “super species” M. jurtina. Numerous studies in the past few years have strikingly uncovered similar “out-of-Africa” examples, lending further support to this idea for the satyrine genus Maniola. Future studies, with a larger set of samples from the African continent, will be needed to evaluate our hypothesis. Most importantly, our results raise the controversial question whether oversplitting of species, despite all the contrasting evidence in recent bar code studies, might be more common than expected until now.
Acknowledgments
This research has been financed through the FWF (project V169-B17) and the Department of Botany and Biodiversity Research of the University of Vienna. We express our gratitude to K. Schurian, W. Eckweiler, W. ten Hagen, S. Cuvelier, E. Haeler, S. Schindler, R. Vila, and D. Benyamini for providing butterfly samples. M. Wiemers kindly supplied several COI sequences.
Conflict of Interest
None declared.
Supporting Information
Table S1.Sequences from GenBank with accession numbers used in this study. All sequences from Peña et al. (2006), except CytB sequences.
Table S2. List of specimens used in this study and their collection localities and dates, when available.
References
- Aduse-Poku K, Vingerhoedt E. Wahlberg N. Out-of-Africa again: a phylogenetic hypothesis of the genus Charaxes (Lepidoptera: Nymphalidae) based on five gene regions. Mol. Phylogenet. Evol. 2009;53:463–478. doi: 10.1016/j.ympev.2009.06.021. , and. [DOI] [PubMed] [Google Scholar]
- Alexiou S. Sfenthourakis S. The terrestrial isopods (Isopoda: Oniscidea) of Greece. Parnassiana Arch. 2013;1:3–50. , and. [Google Scholar]
- Bandelt H-J, Forster P. Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. , and. [DOI] [PubMed] [Google Scholar]
- Brakefield PM. Ecological studies on the butterfly Maniola jurtina in Britain. I. Adult behaviour, microdistribution and dispersal. J. Anim. Ecol. 1982a;51:713–726. [Google Scholar]
- Brakefield PM. Ecological studies on the butterfly Maniola jurtina in Britain. II. Population dynamics: the present position. J. Anim. Ecol. 1982b;51:727–738. [Google Scholar]
- Brower A. DeSalle R. Patterns of mitochondrial versus nuclear DNA sequence divergence among nymphalid butterflies: the utility of wingless as a source of characters for phylogenetic inference. Insect Mol. Biol. 1998;7:73–82. doi: 10.1046/j.1365-2583.1998.71052.x. , and. [DOI] [PubMed] [Google Scholar]
- Cassel A. Tammaru T. Allozyme variability in central, peripheral and isolated populations of the scarce heath (Coenonympha hero: Lepidoptera, Nymphalidae); implications for conservation. Conserv. Genet. 2003;4:83–93. , and. [Google Scholar]
- Cho S, Mitchell A, Regier JC, Mitter C, Poole RW, Friedlander TP, et al. A highly conserved nuclear gene for low-level phylogenetics: elongation factor-1 alpha recovers morphology-based tree for Heliothine moths. Mol. Biol. Evol. 1995;12:650–656. doi: 10.1093/oxfordjournals.molbev.a040244. [DOI] [PubMed] [Google Scholar]
- Dapporto L. Satyrinae butterflies from Sardinia and Corsica show a kaleidoscopic intraspecific biogeography (Lepidoptera, Nymphlidae) Biol. J. Linn. Soc. 2010;100:195–212. [Google Scholar]
- Dapporto L. Strumia F. The thorny subject of insular endemic taxonomy: morphometrics reveal no evidence of speciation between Coenonympha corinna and Coenonympha elbana butterflies (Lepidoptera: Nymphalidae) Zootaxa. 2008;1755:47–56. , and. [Google Scholar]
- Dapporto L, Habel JC, Dennis RL. Schmitt T. The biogeography of the western Mediterranean: elucidating contradictory distribution patterns of differentiation in Maniola jurtina (Lepidoptera: Nymphalidae) Biol. J. Linn. Soc. 2011;103:571–577. , and. [Google Scholar]
- Dayrat B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005;85:407–415. [Google Scholar]
- Dennis RLH, Dapporto L, Shreeve TG, John E, Coutsis JG, Kudrna O, Saarinen K, Ryrholm N. Williams WR. Butterflies of European islands: the implications of the geography and ecology of rarity and endemicity for conservation. J. Insect Cons. 2008;12:205–236. , and. [Google Scholar]
- Dinca˘ V, Lukhtanov VA, Talavera G. Vila R. Unexpected layers of cryptic diversity in wood white Leptidea butterflies. Nat. Commun. 2011;2:324. doi: 10.1038/ncomms1329. , and. [DOI] [PubMed] [Google Scholar]
- Dupuis JR, Roe AD. Sperling FAH. Multi-locus species delimitation in closely related animals and fungi: one marker is not enough. Mol. Ecol. 2012;21:4422–4436. doi: 10.1111/j.1365-294X.2012.05642.x. , and. [DOI] [PubMed] [Google Scholar]
- Frankham R. Do island populations have less genetic variation than mainland populations? Heredity. 1997;78:311–327. doi: 10.1038/hdy.1997.46. [DOI] [PubMed] [Google Scholar]
- Frankham R. Inbreeding and extinction: island populations. Conserv. Biol. 1998;12:665–675. [Google Scholar]
- Goulson D. Variation in the genitalia of the butterfly Maniola jurtina (Lepidoptera: Satyrinae) Zool. J. Linn. Soc. 1993;107:65–71. [Google Scholar]
- Grill A, Vos RD. Arkel JV. The shape of endemics: notes on male and female genitalia in the genus Maniola (Schrank, 1801), (Lepidoptera, Nymphalidae, Satyrinae) Bijdragen tot de Dierkunde. 2004;73:293–304. , and. [Google Scholar]
- Grill A, Gkiokia E. Alvarez N. Evolutionary history and patterns of differentiation among European Maniola butterflies (Lepidoptera: Satyrinae) Eur. J. Entomol. 2006a;103:613. , and. [Google Scholar]
- Grill A, Schtickzelle N, Cleary DF, Neve G. Menken SB. Ecological differentiation between the Sardinian endemic Maniola nurag and the pan-European M. jurtina. Biol. J. Linn. Soc. 2006b;89:561–574. , and. [Google Scholar]
- Grill A, Raijmann L, Van Ginkel W, Gkioka E. Menken S. Genetic differentiation and natural hybridization between the Sardinian endemic Maniola nurag and the European Maniola jurtina. J. Evol. Biol. 2007;20:1255–1270. doi: 10.1111/j.1420-9101.2007.01358.x. , and. [DOI] [PubMed] [Google Scholar]
- Habel JC, Dieker P. Schmitt T. Biogeographical connections between the Maghreb and the Mediterranean peninsulas of southern Europe. Biol. J. Linn. Soc. 2009;98:693–703. , and. [Google Scholar]
- Habel JC, Roedder D, Stefano S, Meyer M. Schmitt T. Strong genetic cohesiveness between Italy and North Africa in four butterfly species. Biol. J. Linn. Soc. 2010;99:818–830. , and. [Google Scholar]
- Hajibabaei M, Janzen DH, Burns JM, Hallwachs W. Hebert PD. DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl Acad. Sci. USA. 2006;103:968–971. doi: 10.1073/pnas.0510466103. , and. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Hausmann A, Haszprunar G. Hebert PD. DNA barcoding the geometrid fauna of Bavaria (Lepidoptera): successes, surprises, and questions. PLoS ONE. 2011;6:e17134. doi: 10.1371/journal.pone.0017134. , and. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PD, Penton EH, Burns JM, Janzen DH. Hallwachs W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl Acad. Sci. USA. 2004;101:14812–14817. doi: 10.1073/pnas.0406166101. , and. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselbarth G, Van Oorschot H. Wagener S. Die Tagfalter der Türkei. Bocholt, Germany: Sigbert Wagener; 1995. , and. Vol. 3. [Google Scholar]
- Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 1996;58:247–276. [Google Scholar]
- Hewitt GM. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 1999;68:87–112. [Google Scholar]
- Huelsenbeck JP. Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. , and. [DOI] [PubMed] [Google Scholar]
- Husemann M, Schmitt T, Zachos FE, Ulrich W. Habel JC. Palaearctic biogeography revisited: evidence for the existence of a North African refugium for Western Palaearctic biota. J. Biogeogr. 2014;41:81–94. , and. [Google Scholar]
- Jeratthitikul E, Hara T, Yago M, Itoh T, Wang M, Usami S-I, et al. Phylogeography of Fischer's blue, Tongeia fischeri, in Japan: evidence for introgressive hybridization. Mol. Phylogenet. Evol. 2013;66:316–326. doi: 10.1016/j.ympev.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Jutzeler D, Leigheb G. de Bros E. Écologie, élevage et distribution du Myrtil de Sardaigne Maniola nurag (Ghiliani, 1852) (Lepidoptera: Nymphalidae, Satyrinae) Linneana Belg. 1997;16:143–149. , and. [Google Scholar]
- Keller LF. Waller DM. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002;17:230–241. , and. [Google Scholar]
- Kodandaramaiah U. Wahlberg N. Out-of-Africa origin and dispersal-mediated diversification of the butterfly genus Junonia (Nymphalidae: Nymphalinae) J. Evol. Biol. 2007;20:2181–2191. doi: 10.1111/j.1420-9101.2007.01425.x. , and. [DOI] [PubMed] [Google Scholar]
- Kodandaramaiah U. Wahlberg N. Phylogeny and biogeography of Coenonympha butterflies (Nymphalidae: Satyrinae) – patterns of colonization in the Holarctic. Syst. Entomol. 2009;34:315–323. , and. [Google Scholar]
- Kudrna O, Harpke A, Lux K, Pennerstorfer J, Schweiger O, Settele J, et al. Distributions atlas of butterflies in Europe. Halle: Gesellschaft für Schmetterlingsschutz; 2011. [Google Scholar]
- Librado P. Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. , and. [DOI] [PubMed] [Google Scholar]
- Makris C. John E. Butterflies of Cyprus. Nicosia: Bank of Cyprus Cultural Foundation; 2003. , and. [Google Scholar]
- Monteiro A. Pierce NE. Phylogeny of Bicyclus (Lepidoptera: Nymphalidae) inferred from COI, COII, and EF-1α Gene Sequences. Mol. Phylogenet. Evol. 2001;18:264–281. doi: 10.1006/mpev.2000.0872. , and. [DOI] [PubMed] [Google Scholar]
- Nei M. New York, NY, USA: Columbia University Press; 1987. Molecular Evolutionary Genetics. [Google Scholar]
- Nei M. Li W-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl Acad. Sci. USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. , and. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier A. The Butterflies of the Greek Island of Ródos: Taxonomy, Faunistics, Ecology and Phenology: with a tentative synthesis on the biogeography of the butterflies of Kríti (Crete), Kárpathos, Ródos, the Eastern Aegean Islands and Kípros (Cyprus)-(Lepidoptera: Hesperioidea & Papilionoidea) Antwerpen: Vlaamse Vereinigung voor Entomologie; 1993. [Google Scholar]
- Peña C, Wahlberg N, Weingartner E, Kodandaramaiah U, Nylin S, Freitas AV, et al. Higher level phylogeny of Satyrinae butterflies (Lepidoptera: Nymphalidae) based on DNA sequence data. Mol. Phylogenet. Evol. 2006;40:29–49. doi: 10.1016/j.ympev.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Poulakakis N, Lymberakis P, Valakos E, Pafilis P, Zouros E. Mylonas M. Phylogeography of Balkan wall lizard (Podarcis taurica) and its relatives inferred from mitochondrial DNA sequences. Mol. Ecol. 2005;14:2433–2443. doi: 10.1111/j.1365-294X.2005.02588.x. , and. [DOI] [PubMed] [Google Scholar]
- Puillandre N, Lambert A, Brouillet S. Achaz G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012;21:1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x. , and. [DOI] [PubMed] [Google Scholar]
- Rambaut A. 2009. Computer program and documentation distributed by the authorFigtree v1. 2.2. Available at http://tree.bio.ed.ac.uk/software (accessed 04 June 2013)
- Randler C, Förschler MI, Gonzalez J, Aliabadian M, Bairlein F. Wink M. Phylogeography, pre-zygotic isolation and taxonomic status in the endemic Cyprus Wheatear Oenanthe cypriaca. J. Ornithol. 2012;153:303–312. , and. [Google Scholar]
- Ratnasingham S. Hebert PD. A DNA-based registry for all animal species: the Barcode Index Number (BIN) System. PLoS ONE. 2013;8:e66213. doi: 10.1371/journal.pone.0066213. , and. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez R, Serra F, Tárraga J, Medina I, Carbonell J, Pulido L, et al. Phylemon 2.0: a suite of web-tools for molecular evolution, phylogenetics, phylogenomics and hypotheses testing. Nucleic Acids Res. 2011;39(Suppl. 2):W470–W474. doi: 10.1093/nar/gkr408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scali V. Imaginal diapause and gonadal maturation of Maniola jurtina (Lepidoptera: Satyridae) from Tuscany. J. Anim. Ecol. 1971;40:467–472. [Google Scholar]
- Schmitt T, Röber S. Seitz A. Is the last glaciation the only relevant event for the present genetic population structure of the Meadow Brown butterfly Maniola jurtina (Lepidoptera: Nymphalidae)? Biol. J. Linn. Soc. 2005;85:419–431. , and. [Google Scholar]
- Simons C, Frati F, Beckenbach A, Crespi B, Liu H. Floors P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994;87:651–701. , and. [Google Scholar]
- Talavera G, Lukhtanov VA, Pierce NE. Vila R. Establishing criteria for higher-level classification using molecular data: the systematics of Polyommatus blue butterflies (Lepidoptera, Lycaenidae) Cladistics. 2013;29:166–192. doi: 10.1111/j.1096-0031.2012.00421.x. , and. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M. Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. , and. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber A. Glaziale Reliktformen der Gattung Maniola (Lep. Satyridae) in Vorderasien. Zeitschrift der Arbeitsgemeinschaft Österreichischer Entomologen. 1970;22:101–119. [Google Scholar]
- Thomson G. Geographical variation of Maniola jurtina (L.) (Lepidoptera, Satyridae) Tijdschr. Entomol. 1973;116:185–227. [Google Scholar]
- Tolman T. Lewington R. Collins butterfly guide: the most complete field guide to the butterflies of Britain and Europe. London: Collins; 2008. , and. [Google Scholar]
- Tshikolovets VV. Butterflies of Europe & the Mediterranean area. Kiev: Tshikolovets Publications; 2011. [Google Scholar]
- Vila R, Lukhtanov VA, Talavera G. Pierce NE. How common are dot-like distributions? Taxonomical oversplitting in western European Agrodiaetus (Lepidoptera: Lycaenidae) revealed by chromosomal and molecular markers. Biol. J. Linn. Soc. 2010;101:130–154. , and. [Google Scholar]
- Weingartner E, Wahlberg N. Nylin S. Speciation in Pararge (Satyrinae: Nymphalidae) butterflies – North Africa is the source of ancestral populations of all Pararge species. Syst. Entomol. 2006;31:621–632. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.Sequences from GenBank with accession numbers used in this study. All sequences from Peña et al. (2006), except CytB sequences.
Table S2. List of specimens used in this study and their collection localities and dates, when available.