

Abstract
Quantitative PCR (qPCR) for human T-lymphotropic virus 1 (HTLV-1) is useful for measuring the amount of integrated HTLV-1 proviral DNA in peripheral blood mononuclear cells. Many laboratories in Japan have developed different HTLV-1 qPCR methods. However, when six independent laboratories analyzed the proviral load of the same samples, there was a 5-fold difference in their results. To standardize HTLV-1 qPCR, preparation of a well-defined reference material is needed. We analyzed the integrated HTLV-1 genome and the internal control (IC) genes of TL-Om1, a cell line derived from adult T-cell leukemia, to confirm its suitability as a reference material for HTLV-1 qPCR. Fluorescent in situ hybridization (FISH) showed that HTLV-1 provirus was monoclonally integrated in chromosome 1 at the site of 1p13 in the TL-Om1 genome. HTLV-1 proviral genome was not transferred from TL-Om1 to an uninfected T-cell line, suggesting that the HTLV-1 proviral copy number in TL-Om1 cells is stable. To determine the copy number of HTLV-1 provirus and IC genes in TL-Om1 cells, we used FISH, digital PCR, and qPCR. HTLV-1 copy numbers obtained by these three methods were similar, suggesting that their results were accurate. Also, the ratio of the copy number of HTLV-1 provirus to one of the IC genes, RNase P, was consistent for all three methods. These findings indicate that TL-Om1 cells are an appropriate reference material for HTLV-1 qPCR.
INTRODUCTION
Human T-lymphotropic virus 1 (HTLV-1) was the first retrovirus to be found in humans (1, 2). HTLV-1 is a cause of adult T-cell leukemia (ATL), HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), and HTLV-1-associated uveitis (3). Areas where HTLV-1 is endemic are distributed across several different regions, including southern Japan, the Caribbean, South America, and tropical Africa (4, 5). A recent report has shown that the area affected by this infection has expanded from the southern part of Japan to the entire country, particularly the Tokyo metropolitan area (6). Diagnostic tests for HTLV-1 infection are performed mainly with serological assays, such as enzyme-linked immunoabsorbent assay, particle agglutination assay, and Western blotting. Recently, another diagnostic test has been developed. Quantitation of integrated proviral DNA in peripheral blood (proviral load [PVL]) can be performed by quantitative PCR (qPCR) as a risk assessment for ATL or HAM/TSP (7, 8).
A few studies reported that several samples were positive for viral DNA when tested by PCR even though those samples had been found seroindeterminate for HTLV-1 when tested by Western blotting (9, 10). Their results suggest that HTLV-1 qPCR could be used as an additional test to confirm infection in seroindeterminate samples.
Although many laboratories have developed qPCR methods for HTLV-1 detection in Japan, a wide variety of testing methods are used. For example, the target region, primers and probes, and internal control (IC) genes vary among the laboratories (8, 11–15). These variations lead to significant differences in HTLV-1 PVL when these laboratories measure the same samples (16). As a consequence of these differences, comparison of quantitative data between laboratories will continue to be difficult without standardization.
One possible solution is to establish a reference material, which is indispensable for standardizing multicenter test results. The target material for HTLV-1 qPCR is genomic DNA (gDNA) from peripheral blood mononuclear cells (PBMCs). Therefore, HTLV-1-infected cells would be an ideal source for a reference material. To date, many cell lines from ATL patients have been established, but few of them have been well characterized for the genomic features associated with reference materials for HTLV-1 qPCR.
In this study, we investigated the genomic structure of one of these ATL cell lines, TL-Om1, to establish it as a reference material for HTLV-1 nucleic acid amplification techniques (NATs), namely, HTLV-1 clonality, karyotyping, proviral sequencing, integration sites, and determination of gene copy number of HTLV-1 and cellular genes for IC.
MATERIALS AND METHODS
Cells and gDNA preparation.
Jurkat clone E6-1 cells were obtained from the American Type Culture Collection. HUT102 and SLB-1 cells, which are HTLV-1-infected cell lines, were a kind gift from Masahiro Fujii (Division of Virology, Niigata University Graduate School of Medical and Dental Sciences). PBMCs were kindly provided by the Japanese Red Cross or purchased from AllCells (Alameda, CA, USA). TL-Om1 cells, an ATL-derived cell line established by Sugamura et al. (17), were maintained in RPMI 1640 (Sigma, St. Louis, MO, USA) containing 10% fetal bovine serum (FBS) supplemented with 100 U/ml penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA), 2 mmol/liter l-glutamine, and 10 ng/ml interleukin-2 (PeproTech, London, United Kingdom). Jurkat, HUT102, and SLB-1 cells were maintained in RPMI 1640 containing 10% FBS supplemented with 100 U/ml penicillin-streptomycin and 2 mmol/liter l-glutamine. DNA was extracted using a QIAamp DNA blood mini or maxi kit (Qiagen, Valencia, CA, USA).
Southern blotting.
Southern blotting was performed by SRL Inc. (Tokyo, Japan). DNA was digested with EcoRI and PstI and separated on a 0.8% agarose gel as previously reported (18, 19). DNA was transferred onto nylon membranes (Roche, Mannheim, Germany). The membrane was hybridized with digoxigenin (DIG)-labeled HTLV-1 probe at 42°C overnight. DNA fragments for HTLV-1 probes were obtained from Oncor Inc. (Gaithersburg, MD, USA). Sense and antisense HTLV-1 DNA probes were prepared by random primed labeling using a DIG-High Prime kit (Roche). After the membrane was washed, HTLV-1 probe signals were obtained using a DIG luminescent detection kit (Roche).
FISH analysis.
To stop the cell cycle at M phase, Colcemid (Sigma) was added to the cell culture medium at a concentration of 0.02 μg/ml and incubated for 1 h. Cells were harvested and washed with phosphate-buffered saline (PBS). After treatment with 0.075 M KCl hypotonic solution at 37°C for 1 h, cells were fixed with a solution containing acetic acid and methanol (3:1). Cells were fixed to a glass slide and dried. The complete HTLV-1 genome inserted in pUC18 (15) was used as a probe for provirus, bacterial artificial chromosome (BAC) clone RP11-919G18 was used as a probe for the albumin (ALB) gene, and BAC clones CTD-2326H15 and RP11-203M5 were used as probes for the RNase P (RPPH1) gene. BAC clones were selected from NCBI (http://www.ncbi.nlm.nih.gov/clone/) and were purchased from Advanced Geno Techs Co. (Tsukuba, Japan). The probe for 1q44 was commercially prepared by Chromosome Science Labo Inc. (Sapporo, Japan). For the detection of ALB and RPPH1 genes, the BAC clones were labeled with cyanine 3 (Cy3) and Cy5, respectively. For the detection of provirus, the DIG-labeled probe was prepared by the nick translation method. The probe was hybridized to the sample at 70°C for 5 min, followed by incubation at 37°C overnight. The probe was stained with anti-DIG-Cy3 antibody. Signals were detected by a Leica DMRA2 system and analyzed with Leica CW4000 fluorescent in situ hybridization (FISH) software (Wetzlar, Germany).
Splinkerette PCR analysis.
Splinkerette PCR was performed as previously reported (20). The first-round PCR was performed as indicated in reference 20. The second-round, nested PCR was performed using the HTLV-1 long-terminal-repeat (LTR)-specific primer. The nested PCR product was loaded onto 3% Tris-acetate-EDTA buffer (TAE) agarose gels. Two distinct DNA bands were cut from the agarose gel and purified using a QIAquick gel extraction kit (Qiagen). After thymine and adenine (TA) cloning, each band was sequenced by the Sanger method (21).
Inverse PCR analysis.
TL-Om1 gDNA was digested with BamHI or XbaI. Digested DNA was purified by phenol-chloroform extraction followed by ethanol precipitation. Briefly, 1/10 volume of 3 M sodium acetate and 2.5 volume of 100% ethanol were added to the sample. After centrifugation at 2 × 104 × g for 15 min, the DNA pellet was washed with 70% ethanol and then air dried. Purified DNA was self-ligated using a Ligation-Convenience kit (Nippon Gene, Tokyo, Japan). Ligated DNA was purified again by phenol-chloroform extraction followed by ethanol precipitation. PCR was performed with KOD FX (Toyobo, Osaka, Japan). The PCR mixture contained 20 ng gDNA, 0.4 mM forward and reverse primers, 1 mM deoxynucleoside triphosphate (dNTP), 1× KOD FX buffer, and 0.5 U KOD FX in a total volume of 25 μl, in duplicate. The forward primer sequence was 5′-ACAAATACACCTTGCAATCCTATGG-3′, and the reverse primer sequence was 5′-CGCTTGGGAGACTTCTTGCT-3′. PCR mixtures were denatured at 94°C for 2 min, followed by 34 cycles of 98°C for 10 s and 68°C for 10 min. PCR products were loaded onto 0.8% agarose gels and detected by LAS-3000 (Fujifilm, Tokyo, Japan).
Genomic long PCR.
Genomic long PCRs were performed using KOD FX (Toyobo). Primers are listed in Table S1 in the supplemental material. The conditions for the PCR mixture and thermal cycling program were the same as those for the inverse PCR analysis.
DNA sequencing analysis.
The genomic long PCR and inverse PCR products were purified by a GenElute PCR Clean Up kit (Sigma). Direct sequencing was performed using a BigDye Terminator v3.1 sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequence primers are listed in Table S2 in the supplemental material. Sequences were read and analyzed using a 3120× genetic analyzer (Applied Biosystems).
Synchronized qPCR analysis.
The primers used for the synchronized qPCR amplification are listed in Table 1. The PCR mixture was prepared with SYBR premix Ex Taq II (TaKaRa, Tokyo, Japan) containing 100 ng gDNA and 0.4 mM forward and reverse primers in a total volume of 15 μl, in triplicate. PCR was performed according to the manufacturer's protocol. The ΔCT(RPPH1) value (where CT is threshold cycle) was calculated by the following equation: ΔCT(RPPH1) = average CT of target gene primer results − average CT of RPPH1. The gene copy number was calculated by the following equation: target gene copy number (N) = copy number determined by FISH × 2−ΔCT(RPPH1). Using normal PBMCs or plasmids, the primer correction factor, which can compensate for small differences in amplification efficiency among different primers, was calculated. The correction factor was determined by the difference of each CT value of target gene primers from the average CT value of RPPH1 primers (Table 1). The correction value was calculated as follows: correction CT value = correction factor × actual CT value. By applying the correction factors, we reduced the limits of error of the CT values to 0.1 cycles with normal PBMCs (data not shown).
TABLE 1.
Primers used for qPCR of HTLV-1 and IC genes
| Target gene | Forward name | Forward sequence | Reverse name | Reverse sequence | Size (bp) | Primer correction factor |
|
|---|---|---|---|---|---|---|---|
| Plasmid | gDNA | ||||||
| HTLV-1 gene | LTR202F | ACAATGACCATGAGCCCCAAA | LTR202R | TTAGTCTGGGCCCTGACCT | 101 | 0.9869 | |
| LTR215F | GCTCGCATCTCTCCTTCAC | LTR215R | AGTTCAGGAGGCACCACA | 102 | 0.9942 | ||
| LTR005F | CCTGACCCTGCTTGCTCAAC | LTR005R | TCAGTCGTGAATGAAAGGGAAAG | 99 | 0.9917 | ||
| 056F | TAGTCCCACCCTGTTCGAAATG | 056R | GCCAGGAGAATGTCATCCATGT | 105 | 1.0013 | ||
| 084F | CCTGCCCCGCTTACTATCG | 084R | GGCATCTGTGAGAGCGTTGA | 102 | 0.9922 | ||
| 153F | TTGTCGCGCTACTCCTTCTTG | 153R | AGGGATGACTCAGGGTTTATAAGAGA | 118 | 0.9792 | ||
| pX2-Sa | CGGATACCCAGTCTACGTGTT | pX2-ASa | CAGTAGGGCGTGACGATGTA | 100 | 0.9944 | ||
| RNaseP (RPPH1) gene | RPPH1-05F | TATGCACAATTATGTAATCCCCAAA | RPPH1-05R | CCAGCTCCCTATAACCTGCACTT | 100 | 1.0025 | 1.0012 |
| RPPH1-08F | GCCGGAGCTTGGAACAGA | RPPH1-08R | AATGGGCGGAGGAGAGTAGTCT | 109 | 0.9956 | 0.9937 | |
| RPPH1-12F | AGGAAGCCCACGAAAATTCTAATT | RPPH1-12R | GTCCCCATACTCGGTGATTCTC | 101 | 1.0019 | 1.0052 | |
| Albumin (ALB) gene | ALB-07F | TGCAATGAACACAGGAGAGCTACTA | ALB-07R | CCACCCAGGTAACAAAATTAGCAT | 103 | 0.9971 | 0.9964 |
| ALB-19F | CCTGATGCTTCTCAGCCTGTT | ALB-19R | TCCATTTAAGAGTGTGTGTGGTAGGT | 100 | 1.0019 | 1.0045 | |
| ALB-26F | TGCATTGCCGAAGTGGAAA | ALB-26R | CCTCAGCATAGTTTTTGCAAACA | 100 | 1.0038 | 1.0078 | |
| β-Actin (ACTB) gene | ACTB-06F | TCTGGTGTTTGTCTCTCTGACTAGGT | ACTB-06R | CCGCTTTACACCAGCCTCAT | 100 | 0.9965 | |
| ACTB-12F | TCCTGGGTGAGTGGAGACTGT | ACTB-12R | CCATGCCTGAGAGGGAAATG | 107 | 1.0016 | ||
| ACTB-21F | AGCATCCCCCAAAGTTCACA | ACTB-21R | GGACTTCCTGTAACAACGCATCT | 101 | 1.0106 | ||
| CD81 gene | CD81-01F | GACACATCCCAAGGGTGCTT | CD81-01R | GGACTCAGTTCTCAATGCTTTGC | 107 | 1.0015 | |
| CD81-10F | ACCACGCCTTGCCCTTCT | CD81-10R | GAATCACGCCACTTCCATAACTG | 111 | 1.0021 | ||
| CD81-21F | GGTGCACACAGCATGCATTT | CD81-21R | GTGCGCCTCTGGGTAATCAT | 102 | 1.0009 | ||
| β-Globin (HBB) gene | HBB-11F | TTGGACCCAGAGGTTCTTTGAG | HBB-11R | GGCACCGAGCACTTTCTTG | 103 | 1.0021 | |
| HBB-15F | AGCAGCTACAATCCAGCTACCAT | HBB-15R | GAGGTATGAACATGATTAGCAAAAGG | 105 | 1.0033 | ||
| HBB-24F | CCCACCCCAAATGGAAGTC | HBB-24R | AGCACCATAAGGGACATGATAAGG | 104 | 1.0111 | ||
| RAG-1 gene | RAG1-03F | GCAATCCCATTTGTCCACTTTT | RAG1-03R | TCCCACTGGCCTGCATTACTA | 100 | 1.0045 | |
| RAG1-27F | GAAGTTTAGCAGTGCCCCATGT | RAG1-27R | ACGGGCAGTGTTGCAGATG | 100 | 1.0006 | ||
| RAG1-32F | TCAAAGTCATGGGCAGCTATTGT | RAG1-32R | AGGGAATTCAAGACGCTCAGAA | 100 | 0.9993 | ||
Primer sequences were previously reported in reference 11.
Digital PCR analysis.
Primers and probes for digital PCR analysis of HTLV-1 were previously reported (11, 15). In brief, the primers and probe for HTLV-1 were as follows: forward, 5′-CGGATACCCAGTCTACGTGTT-3′; reverse, 5′-CAGTAGGGCGTGACGATGTA-3′; probe, FAM-5′-CTGTGTACAAGGCGACTGGTGCC-3′-TAMRA (where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine). The primers and probe for albumin were as follows: forward, 5′-TGTCATCTCTTGTGGGCTGT-3′; reverse, 5′-GGTTCTCTTTCACTGACATCTGC-3′; probe, FAM-5′-CCTGTCATGCCCACACAAATCTCTCC-3′-TAMRA. The mixture of primers and probe for RPPH1 was purchased from Applied Biosystems. The PCR mixture was prepared using 2× digital droplet PCR (ddPCR) supermix for probes (Bio-Rad, Hercules, CA, USA). Droplets were prepared on a QX100 droplet generator (Bio-Rad). PCR was performed with a LifePro thermal cycler (Bio-Rad) and detected with a QX100 droplet reader (Bio-Rad). Data were means of triplicate analysis.
In vitro HTLV-1 infectivity test.
Frozen cells were thawed and immediately cultured for a week. Exponentially growing cells were used for the assay. Jurkat, TL-Om1, SLB1, and HUT102 cells were treated with 50 μg/ml mitomycin C (Kyowa Hakko Kirin, Tokyo, Japan) and incubated for 1 h at 37°C. After being washed twice with 2% FBS-PBS, 1 × 105 cells were added to culture medium containing 1 × 106 Jurkat cells. Mitomycin C was used to block the growth of ATL cell lines added to Jurkat cells. Cells were cocultured for 2 weeks and then subjected to qPCR to determine PVL, as described previously (11).
RESULTS
HTLV-1 infectivity in TL-Om1 cells.
We investigated the production potential of infective virus to ascertain the clonal stability of HTLV-1 integration in vitro. Mitomycin C-treated TL-Om1 cells were cocultured with Jurkat cells for 2 weeks. At the end of the 2 weeks, no HTLV-1 integration was observed in the Jurkat cells that were cocultured with TL-Om1 cells, while HTLV-1 integration was observed when Jurkat cells were similarly cocultured with SLB-1 and HUT102 cells (Fig. 1A). These findings suggested that the production of infective HTLV-1 particles from TL-Om1 cells was low or diminished; thus, the increase in copy number over the course of cell culture was thought to be negligible. If TL-Om1 cells had infectious potential, the clonality of HTLV-1 provirus in them would vary because of the mutual HTLV-1 infections between cells. To evaluate the clonality of HTLV-1 provirus in TL-Om1 cells, TL-Om1 gDNA was analyzed by Southern blotting. EcoRI-digested gDNA showed a single band, while PstI digestion produced five DNA bands that contained an HTLV-1 sequence (Fig. 1B). Three of the five DNA bands were HTVL-1 internal sequences. The other two DNA bands contained either 5′ or 3′ HTLV-1 sequences ligated with the host genome (Fig. 1B). These fragment patterns indicated that HTLV-1 provirus integration in TL-Om1 cells was monoclonal.
FIG 1.
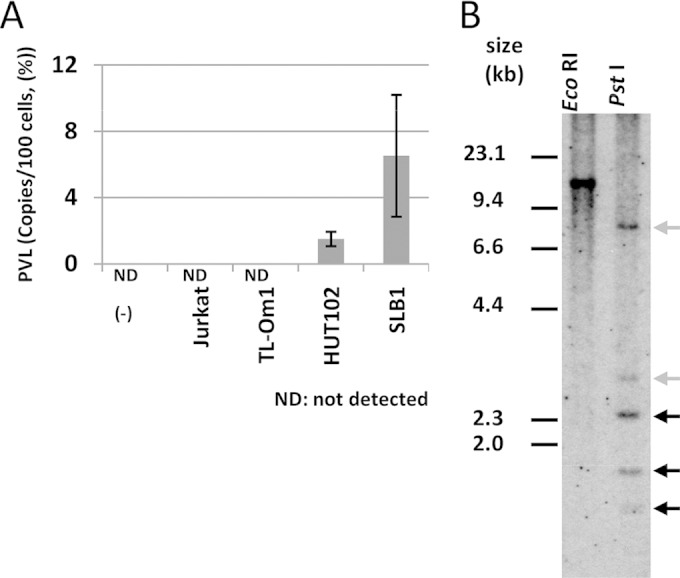
Infectivity and clonality of HTLV-1 provirus in TL-Om1 cells. (A) Mitomycin C-treated Jurkat, TL-Om1, HUT102, and SLB1 cells were cocultured with Jurkat cells. PVL (%) was measured 2 weeks later by qPCR. (B) gDNA from TL-Om1 cells digested with EcoRI or PstI was subjected to Southern blotting probed by the full HTLV-1 genome. Three black arrows show bands for typical HTLV-1 genomic sequences; two gray arrows show bands for host genomic sequences ligated to the HTLV-1 genome. Because the EcoRI site is not included in the HTLV-1 sequence, the number of bands indicates the number of clones in the cells. Detection of two gray bands indicates that there is a pair of 5′ and 3′ HTLV-1 genomes conjugated with the host genome, signifying that the HTLV-1 provirus is monoclonal. On the other hand, detection of more than two gray bands indicates that it is multiclonal.
Determination of copy number and integration site of HTLV-1 provirus by FISH.
To confirm the clonality and copy number of HTLV-1 provirus and of IC genes in detail, we performed a FISH analysis. There were one or two signals of HTLV-1 provirus in the cells. The mean proviral copy number was calculated at 1.8 copies/cell from the count of signals with >250 cells in two independent analyses (Fig. 2Ai and ii). Double-staining of the TL-Om1 genome with both HTLV-1 and 1q44 probes in metaphase showed that all HTLV-1 DNA signals were located on chromosome 1 (Fig. 2Bi). When the number of copies of chromosome 1 was 1, 2, 3, or 4 per cell, the number of HTLV-1 proviruses per cell was 1, 1, 2, and 2, respectively (data not shown). HTLV-1 signals on chromosome 1 were positioned on the band of 1p13 (Fig. 2Bii). These results correlated well with the Southern blotting results that showed monoclonal integration.
FIG 2.

Clonality, copy number, and integration site of HTLV-1 in TL-Om1 cells. (A) HTLV-1 proviral copy number per cell was determined by FISH using an HTLV-1 full-genome probe. (i) Yellow signals indicate the HTLV-1 probe. Lower table shows the counts of HTLV-1 signals per cell. (ii) Vertical axis indicates percentage counts of each fraction in relation to total cells. Data were the results from two independent analyses. (B) Number of HTLV-1 integrated chromosomes was determined in metaphase cells with the HTLV-1 and 1q44 probes. (i, ii) Yellow signals indicate the HTLV-1 probe, and red signals indicate the 1q44 probe. All HTLV-1 signals were located on chromosome 1. HTLV-1 signals on chromosome 1 were positioned at 1p13. (C) Determination of the HTLV-1 integration site in TL-Om1 cells. (i) The 3′ integration site was determined by Splinkerette PCR with an HTLV-1-specific primer. PCR products were subjected to agarose gel electrophoresis. (ii) BamHI- or XbaI-digested TL-Om1 genomes were self-ligated and subjected to inverse PCR with an HTLV-1-specific primer set. PCR products were subjected to agarose gel electrophoresis. (D) 5′ and 3′ HTLV-1 integration sites were determined by a sequencing analysis of DNA fragments from both Splinkerette and inverse PCR. (i) Normal human sequence; (ii) determined HTLV-1 integration site. HTLV-1 was inversely integrated at chromosome 1: NT_077389, 164570 to 164576.
Confirmation of integration site of HTLV-1 in TL-Om1 cells.
To identify the integration site of monoclonal HTLV-1 provirus, Splinkerette PCR was performed with TL-Om1 gDNA. Two specific PCR products were obtained by gel electrophoresis (Fig. 2Ci). The DNA fragments were analyzed by direct sequencing. Sequencing analysis of the lower-molecular-weight DNA fragments (Fig. 2Ci, lower band) showed that they were provirus genomic sequences. Sequencing analysis of the higher-molecular-weight band showed that it contained host gDNA ligated to the 3′ LTR of HTLV-1. We also performed inverse PCR with TL-Om1 gDNA that was digested with BamHI or XbaI followed by self-ligation. Single DNA bands were obtained from both BamHI and XbaI self-ligated templates (Fig. 2Cii). Sequencing analysis demonstrated that both bands contained the same sequences. A BLAST search revealed that the sequence was located on chromosome 1. The integration site was identified, and the HTLV-1 provirus was integrated inversely in between the CATATAT repetitive sequences at the region of NT_077389 from nucleotides (nt) 164570 to 164576 on chromosome 1 (Fig. 2Di and ii).
We determined the full-length sequence of HTLV-1 provirus in TL-Om1 cells by genomic long PCR followed by direct sequencing. The length of HTLV-1 provirus was determined to be 8,941 bp (GenBank accession no. AB979451; see also Text S1 in the supplemental material). The percent identity to the HTLV-1 genomic sequence of the ATK-1 strain (accession no. J02029) was 98.7%. Compared with the full-length HTLV-1 genomic sequence of ATK-1, there was a 93-nt deletion in the env gene. The region that was deleted was equivalent to nt 5547 to 5669 of ATK-1. The deduced amino acid sequence of the deletion was 31 in-frame amino acids (Δ125–155 of Env). The deleted region was located on the receptor binding domain of Env (see Fig. S1 in the supplemental material).
Calculation of chromosome and gene copy numbers of HTLV-1, RPPH1, and ALB genes in TL-Om1 and Jurkat cells.
We counted the chromosome number in TL-Om1 and Jurkat cells by FISH analysis. Jurkat cells were analyzed as one of the control cell lines. The chromosome number differed from 78 to 83 in TL-Om1 cells (Fig. 3A). The mean chromosome number was estimated at 80.2, which indicated that the karyotype of TL-Om1 cells was about 4N. There were 45 or 46 chromosomes in Jurkat cells, indicating that their karyotype is near that of normal human diploid cells (Fig. 3A and B and Table 2).
FIG 3.

Gene copy number of IC cellular genes for HTLV-1 qPCR. (A) The number of chromosomes in TL-Om1 and Jurkat cells at metaphase was counted. Horizontal line indicates the number of chromosomes per cell. (B) Representative FISH images of TL-Om1 and Jurkat cells at metaphase. Yellow and red arrows indicate signals for ALB and RPPH1 probes, respectively. Left panel shows three signals for ALB and four for RPPH1; right panel shows two signals for ALB and two for RPPH1. (C) Determination of the gene copy number of HTLV-1, RPPH1, and ALB genes by digital PCR. gDNA of TL-Om1 and Jurkat cells and of PBMCs from two healthy donors were subjected to digital PCR. Data show the absolute copy number of HTLV-1, RPPH1, and ALB genes per microgram of gDNA. Bars are means from triplicate analyses.
TABLE 2.
Gene copy number of IC genes determined by FISH
| Karyotype | Gene copy no. |
|||
|---|---|---|---|---|
| TL-Om1 (20 analyzed cells) |
Jurkat (20 analyzed cells) |
|||
| RPPH1 gene | ALB gene | RPPH1 gene | ALB gene | |
| 2N | 0 | 0 | 20 | 20 |
| 3N | 1 | 20 | 0 | 0 |
| 4N | 19 | 0 | 0 | 0 |
| Average | 3.95 | 3 | 2 | 2 |
| Ratio to the RPPH1 gene | 1 | 0.76 | 1 | 1 |
The absolute gene copy number of HTLV-1 provirus and IC genes was measured using digital PCR. gDNA from TL-Om1 cells, Jurkat cells, and PBMCs from two healthy donors was subjected to digital PCR and used to calculate the copy numbers of these genes (Fig. 3C). Although the ALB-to-RPPH1 gene copy number ratios in the two PBMC samples were 1.09 and 0.99, the ALB-to-RPPH1 gene copy number ratio in TL-Om1 cells was low (ratio of 0.74) (Table 3). The provirus-to-RPPH1 gene copy number ratio in TL-Om1 cells was 0.51 (Table 3). These results were consistent with the provirus- and ALB-to-RPPH1 gene copy number ratios estimated by FISH, which were 0.46 and 0.76, respectively (Table 3). The usefulness of TL-Om1 as a reference standard is strongly supported by the consistent results from the FISH and digital PCR analyses (Table 4).
TABLE 3.
Summary of ratio of gene copy numbers to the RPPH1 gene
| Method | Cell line | Gene copy no. ratio to the RPPH1 gene |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| RPPH1 gene | ALB gene | ACTB gene | CD81 gene | HBB gene | RAG-1 gene | HTLV-1 gene | LTR gene | ||
| FISH | TL-Om1 | 1.00 | 0.76 | 0.46 | |||||
| Jurkat | 1.00 | 1.00 | |||||||
| Digital PCR | TL-Om1 | 1.00 | 0.74 | 0.51 | |||||
| Jurkat | 1.00 | 0.86 | |||||||
| PBMC1 | 1.00 | 1.09 | |||||||
| PBMC2 | 1.00 | 0.99 | |||||||
| qPCR (plasmid) | TL-Om1 | 1.00 | 0.74 | 0.48 | 1.02 | ||||
| Jurkat | 1.00 | 0.92 | |||||||
| qPCR (gDNA) | TL-Om1 | 1.00 | 0.74 | 1.18 | 0.99 | 0.92 | 0.94 | ||
| Jurkat | 1.00 | 0.95 | 1.07 | 0.99 | 0.90 | 1.08 | |||
| PBMC 1 | 1.00 | 0.99 | 1.00 | 0.98 | 0.99 | 1.00 | |||
| PBMC 2 | 1.00 | 1.01 | 1.01 | 0.99 | 1.00 | 1.01 | |||
TABLE 4.
Absolute gene copy number per microgram gDNA determined by digital PCR
| Cell line | Gene copy no./μg gDNAa |
||
|---|---|---|---|
| HTLV-1 gene | RPPH1 gene | ALB gene | |
| TL-Om1 | 170,171.1 | 335,452.3 | 248,410.8 |
| Jurkat | NT | 434,529.6 | 373,423.9 |
| PBMC1 | NT | 355,116.1 | 388,650.0 |
| PBMC2 | NT | 397,260.3 | 394,520.5 |
Data are means of triplicate analysis. NT, not tested.
Estimation of the gene copy number of HTLV-1 and IC genes by synchronized qPCR.
We previously developed a method to determine inherited allelic deletions by using qPCR with primer sets that can amplify fragments synchronously, even though the target genes are different. The method shows that the difference in CT value determines the difference in gene copy number. We used primer sets for HTLV-1 genes (LTR and coding regions) and ACTB, ALB, CD81, HBB, and RAG-1 IC genes (Table 1). To increase the specificity, we used primer correction factors, which compensate for the slight difference in PCR amplification efficiency between different primers for target genes. As shown in Fig. 4A, TL-Om1 and Jurkat cells did not show the complete synchronized amplifications that were observed in normal PBMCs. By setting the PCR amplification efficiency of all primer sets per cycle to approximately 2-fold, the ratio of the gene copy number against the RPPH1 gene was estimated using the difference in the mean CT scores of the IC gene primer sets from the mean of those for the RPPH1 gene. The ratios of the gene copy number of the ALB gene to that of the RPPH1 gene in TL-Om1 and Jurkat cells were 0.74 and 0.92, respectively (Table 3). When the copy number of the RPPH1 gene in TL-Om1 cells was set at 3.95, which was determined by FISH analysis, the copy number of the IC genes was at least 2.9 (ALB gene) and at most 4.7 (ACTB gene) (Fig. 4B and Table 3).
FIG 4.

Estimation of gene copy number of IC genes in TL-Om1 cells by qPCR. gDNA of TL-Om1 and Jurkat cells and of PBMCs from two healthy donors was tested by qPCR with synchronous amplification primer sets for IC genes. (A) CT scores (cycles) of each primer set for IC genes. Each dot indicates the mean from triplicate analyses. The CT scores in the graph were the results of correction by the factors described in Table 1. (B) Estimated gene copy number of IC genes calculated using the difference in CT scores from RPPH1. The copy numbers of IC genes of TL-Om1 and Jurkat cells were calculated based on FISH analysis for the RPPH1 gene. RPPH1 gene copy number from PBMCs was set as 2N. Equation for the estimation of gene copy number was as follows: gene copy number (N) = RPPH1 gene copy number determined by FISH analysis × 2−ΔCT, ΔCT = CT(target gene) − CT(RPPH1).
Additionally, we tried to determine the HTLV-1 copy number in TL-Om1 cells using a synchronized qPCR method. We prepared a plasmid that had one copy of every target PCR amplicon (Fig. 5A). The plasmid had the same copy number as all the target regions. Using the plasmid as a template, we performed qPCR and confirmed the synchronized amplification of primer sets for HTLV-1, RPPH1, and ALB genes (Fig. 5B). The difference in mean CT scores for the HTLV-1 gene to the RPPH1 gene was 1.05 cycles on average in TL-Om1 cells (Fig. 5C and Table 3). As with the sequencing analysis, use of the synchronized qPCR method also estimated the copy number of the LTR to be 4.01, indicating that TL-Om1 cells have two LTRs (Fig. 5C and Table 3).
FIG 5.

Estimation of the HTLV-1 gene copy number in TL-Om1 cells by synchronized qPCR. gDNA of TL-Om1 and Jurkat cells and of PBMCs from two healthy donors was tested for qPCR with synchronous amplification primer sets for HTLV-1, RPPH1, and ALB genes. (A) Construction of control plasmid with a single copy of each target sequence; (B) data indicate CT scores of HTLV-1, RPPH1, and ALB genes for control plasmid at 1 pg, 100 fg/reaction, and for TL-Om1 and Jurkat cells. qPCR with the plasmid showed synchronous amplification of all primer sets. Each dot indicates the mean from triplicate analyses. The CT scores in the graph are the results of correction by the factors described in Table 1. (C) Estimated HTLV-1 and ALB gene copy number in TL-Om1 and Jurkat cells. Data were estimated using the difference in CT scores between target genes and RPPH1 genes.
Comparison of HTLV-1 copy number from different calculation methods.
We compared the results of HTLV-1 and ALB gene copy number obtained from FISH, digital PCR, and synchronized qPCR. The copy number ratios of the HTLV-1 gene to the RPPH1 gene in TL-Om1 cells were 0.46, 0.51, and 0.48, from FISH, digital PCR, and synchronized qPCR, respectively, and those for the ALB gene were 0.76, 0.74, and 0.74 (Fig. 6 and Table 3). The results from these varied assays strongly support one another, indicating that TL-Om1 cells are suitable for use as a reference material for HTLV-1 qPCR.
FIG 6.

HTLV-1- and ALB-to-RPPH1 gene copy number ratios. (A) Comparison of the HTLV-1- and ALB-to-RPPH1 gene copy number ratios determined by FISH, digital PCR, and qPCR. Data indicate percentages of gene copy number ratio to the RPPH1 gene.
DISCUSSION
Recently, NAT reference materials have been established for the safety of blood and blood products, such as international standards for HIV, hepatitis B virus, and hepatitis C virus (22–24). These materials have been frequently used for the purpose of calibration and validation of test systems, preparation of secondary reference materials, and comparison of multicenter results, which have helped improve the consistency of the results. Most international standards for blood-transmitted viruses use plasma from infected human blood, because the test target is extracted from human plasma. With regard to HTLV-1 NAT, it may be better to use a cell line as a reference material to standardize the qPCR results, because this test uses cells obtained from peripheral blood. An example of NAT reference material using cell lines is reported in a test for quantitation of BCR-ABL mRNA. Panels of K562 cells combined with HL60 cells were set as standards, which have been approved by the WHO Expert Committee of Biological Standardization (25). Although a variety of cell lines harboring HTLV-1 provirus in their genomes has been established, detailed characterization of the candidate cell lines with regard to their suitability as reference materials for HTLV-1 NATs has not yet been performed. Among the HTLV-1 cell lines, TL-Om1 is well known to be latently infected with HTLV-1 and is thought to be stable for HTLV-1 clonality (17, 26). Transcription from HTLV-1 provirus in TL-Om1 cells is blocked by the highly methylated LTR (27).
In this study, we evaluated the distinct genomic properties of HTLV-1 and IC genes in TL-Om1 cells with regard to their suitability as reference materials for HTLV-1 NATs. Precise information about HTLV-1 infectivity, karyotype, and absolute copy number of HTLV-1 and cellular control genes of TL-Om1 is useful for applying TL-Om1 as a reference material for HTLV-1 qPCR. As such, for this use, TL-Om1 has advantages over other cell lines, such as the human ATL cell line MT2 and the rat T-cell line TARL-2. A recent study of HTLV-1 testing in Japanese blood donor screening revealed that virus prevalence is not limited to areas where HTLV-1 is endemic but has shifted to the entire country, especially the Tokyo metropolitan area (6). Nationwide HTLV-1 tests have been performed on pregnant women in Japan since the end of 2010. The frequent occurrence of seroindeterminate results after Western blotting is one weakness of the HTLV-1 antibody tests. HTLV-1 qPCR is thought to be a solution for decreasing the number of seroindeterminate results; therefore, an accurate measurement of HTLV-1 proviral DNA by qPCR is needed. Additionally, a PVL value of >4% in PBMCs is reported to be a risk factor for ATL development from HTLV-1 asymptomatic carriers, which emphasizes the importance of measuring PVL by qPCR (7). PVL monitoring also provides a risk indicator for HAM/TSP (8).
An attempt to minimize the differences between laboratories by using a common plasmid that included the pX region has been reported. When standard curves were constructed by utilizing the common plasmid in all participating laboratories, the differences in median intralaboratory coefficient of variation (CV) could be reduced by about half (16). Although the attempt worked well among participating laboratories with in-house qPCR methods, the transferability of utilizing common plasmids for standard curves to other methods for PVL determination, for example, digital PCR, is uncertain.
To standardize HTLV-1 qPCR, we advocate the use of TL-Om1 cells with finely elucidated HTLV-1 genomic information as reference material. A previous report showed that PVL values of males and females, on average, are 1.39% and 2.10%, respectively (7). Thus, a dilution or a serial dilution of TL-Om1 with PBMCs or Jurkat cells at a PVL value of around 2% would be an appropriate material for the standardization of HTLV-1 qPCR. These kinds of references can be easily prepared, because the absolute gene copy number is determined from the dilution rate of TL-Om1. TL-Om1 cells were also used as a control in a deep-sequencing-based method for the quantification of the clone size of HTLV-1-infected cells in HTLV-1 carrier or ATL patients (28).
We conclude that TL-Om1 cells can be used as a useful reference material for HTLV-1 NATs. By using TL-Om1 cells, researchers will be able to define the exact values of HTLV-1 by quantifying the copy numbers of provirus and IC genes. In the future, we hope that other laboratories will utilize the features of TL-Om1 cells to standardize the HTLV-1 qPCR.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants-in-aid for scientific research and by Health and Labor Sciences research grant H23-shinkou-ippan-016 from the Ministry of Health, Labor and Welfare of Japan.
We thank all the members of the HTLV-1 qPCR standardization group for their useful discussions about this research.
We declare that we do not have any competing interests.
Ethical approval was not required for this study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02254-14.
REFERENCES
- 1.Hinuma Y, Nagata K, Hanaoka M, Nakai M, Matsumoto T, Kinoshita KI, Shirakawa S, Miyoshi I. 1981. Adult T-cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci U S A 78:6476–6480. doi: 10.1073/pnas.78.10.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watanabe T. 1997. HTLV-1-associated diseases. Int J Hematol 66:257–278. doi: 10.1016/S0925-5710(97)00077-7. [DOI] [PubMed] [Google Scholar]
- 4.Gessain A, Cassar O. 2012. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol 3:388. doi: 10.3389/fmicb.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe T. 2011. Current status of HTLV-1 infection. Int J Hematol 94:430–434. doi: 10.1007/s12185-011-0934-4. [DOI] [PubMed] [Google Scholar]
- 6.Satake M, Yamaguchi K, Tadokoro K. 2012. Current prevalence of HTLV-1 in Japan as determined by screening of blood donors. J Med Virol 84:327–335. doi: 10.1002/jmv.23181. [DOI] [PubMed] [Google Scholar]
- 7.Iwanaga M, Watanabe T, Utsunomiya A, Okayama A, Uchimaru K, Koh KR, Ogata M, Kikuchi H, Sagara Y, Uozumi K, Mochizuki M, Tsukasaki K, Saburi Y, Yamamura M, Tanaka J, Moriuchi Y, Hino S, Kamihira S, Yamaguchi K, Joint Study on Predisposing Factors of ATLDi . 2010. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood 116:1211–1219. doi: 10.1182/blood-2009-12-257410. [DOI] [PubMed] [Google Scholar]
- 8.Takenouchi N, Yamano Y, Usuku K, Osame M, Izumo S. 2003. Usefulness of proviral load measurement for monitoring of disease activity in individual patients with human T-lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis. J Neurovirol 9:29–35. doi: 10.1080/13550280390173418. [DOI] [PubMed] [Google Scholar]
- 9.Costa JM, Segurado AC. 2009. Molecular evidence of human T-cell lymphotropic virus types 1 and 2 (HTLV-1 and HTLV-2) infections in HTLV seroindeterminate individuals from Sao Paulo, Brazil. J Clin Virol 44:185–189. doi: 10.1016/j.jcv.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Zanjani DS, Shahabi M, Talaei N, Afzalaghaee M, Tehranian F, Bazargani R. 2011. Molecular analysis of human T cell lymphotropic virus type 1 and 2 (HTLV-1/2) seroindeterminate blood donors from Northeast Iran: evidence of proviral tax, env, and gag sequences. AIDS Res Hum Retroviruses 27:131–135. doi: 10.1089/aid.2010.0017. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe M, Ohsugi T, Shoda M, Ishida T, Aizawa S, Maruyama-Nagai M, Utsunomiya A, Koga S, Yamada Y, Kamihira S, Okayama A, Kikuchi H, Uozumi K, Yamaguchi K, Higashihara M, Umezawa K, Watanabe T, Horie R. 2005. Dual targeting of transformed and untransformed HTLV-1-infected T cells by DHMEQ, a potent and selective inhibitor of NF-kappaB, as a strategy for chemoprevention and therapy of adult T-cell leukemia. Blood 106:2462–2471. doi: 10.1182/blood-2004-09-3646. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka G, Okayama A, Watanabe T, Aizawa S, Stuver S, Mueller N, Hsieh CC, Tsubouchi H. 2005. The clonal expansion of human T lymphotropic virus type 1-infected T cells: a comparison between seroconverters and long-term carriers. J Infect Dis 191:1140–1147. doi: 10.1086/428625. [DOI] [PubMed] [Google Scholar]
- 13.Nagai M, Yamano Y, Brennan MB, Mora CA, Jacobson S. 2001. Increased HTLV-I proviral load and preferential expansion of HTLV-I Tax-specific CD8+ T cells in cerebrospinal fluid from patients with HAM/TSP. Ann Neurol 50:807–812. doi: 10.1002/ana.10065. [DOI] [PubMed] [Google Scholar]
- 14.Kamihira S, Dateki N, Sugahara K, Yamada Y, Tomonaga M, Maeda T, Tahara M. 2000. Real-time polymerase chain reaction for quantification of HTLV-1 proviral load: application for analyzing aberrant integration of the proviral DNA in adult T-cell leukemia. Int J Hematol 72:79–84. [PubMed] [Google Scholar]
- 15.Ueno S, Umeki K, Takajo I, Nagatomo Y, Kusumoto N, Umekita K, Morishita K, Okayama A. 2012. Proviral loads of human T-lymphotropic virus type 1 in asymptomatic carriers with different infection routes. Int J Cancer 130:2318–2326. doi: 10.1002/ijc.26289. [DOI] [PubMed] [Google Scholar]
- 16.Kamihira S, Yamano Y, Iwanaga M, Sasaki D, Satake M, Okayama A, Umeki K, Kubota R, Izumo S, Yamaguchi K, Watanabe T. 2010. Intra- and inter-laboratory variability in human T-cell leukemia virus type-1 proviral load quantification using real-time polymerase chain reaction assays: a multi-center study. Cancer Sci 101:2361–2367. doi: 10.1111/j.1349-7006.2010.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugamura K, Fujii M, Kannagi M, Sakitani M, Takeuchi M, Hinuma Y. 1984. Cell surface phenotypes and expression of viral antigens of various human cell lines carrying human T-cell leukemia virus. Int J Cancer 34:221–228. doi: 10.1002/ijc.2910340213. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. 1984. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc Natl Acad Sci U S A 81:2534–2537. doi: 10.1073/pnas.81.8.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaguchi K, Seiki M, Yoshida M, Nishimura H, Kawano F, Takatsuki K. 1984. The detection of human T cell leukemia virus proviral DNA and its application for classification and diagnosis of T cell malignancy. Blood 63:1235–1240. [PubMed] [Google Scholar]
- 20.Uren AG, Mikkers H, Kool J, van der Weyden L, Lund AH, Wilson CH, Rance R, Jonkers J, van Lohuizen M, Berns A, Adams DJ. 2009. A high-throughput Splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat Protoc 4:789–798. doi: 10.1038/nprot.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.World Health Organization. 2011. International collaborative study to establish the 3rd WHO international standard for HIV-1 NAT assays. WHO ECBS report 2011 WHO/BS/2011.2178. WHO, Geneva, Switzerland. [Google Scholar]
- 23.Fryer JF, Heath A, Wilkinson DE, Minor PD, The Collaborative Study Group . 2011. Collaborative study to evaluate the proposed 3rd WHO international standard for hepatitis B virus (HBV) for nucleic acid amplification technology (NAT)-based assays. WHO ECBS report 2011 WHO/BS/2011.2170. WHO, Geneva, Switzerland. [Google Scholar]
- 24.Fryer JF, Heath A, Wilkinson DE, Minor PD, The Collaborative Study Group . 2011. Collaborative study to evaluate the proposed 4th WHO international standard for hepatitis C virus (HCV) for nucleic acid amplification technology (NAT)-based assays. WHO ECBS report 2011 WHO/BS/2011.2173. WHO, Geneva, Switzerland. [Google Scholar]
- 25.White HE, Matejtschuk P, Rigsby P, Gabert J, Lin F, Lynn Wang Y, Branford S, Muller MC, Beaufils N, Beillard E, Colomer D, Dvorakova D, Ehrencrona H, Goh HG, El Housni H, Jones D, Kairisto V, Kamel-Reid S, Kim DW, Langabeer S, Ma ES, Press RD, Romeo G, Wang L, Zoi K, Hughes T, Saglio G, Hochhaus A, Goldman JM, Metcalfe P, Cross NC. 2010. Establishment of the first World Health Organization international genetic reference panel for quantitation of BCR-ABL mRNA. Blood 116:e111–e117. doi: 10.1182/blood-2010-06-291641. [DOI] [PubMed] [Google Scholar]
- 26.Koiwa T, Hamano-Usami A, Ishida T, Okayama A, Yamaguchi K, Kamihira S, Watanabe T. 2002. 5′-Long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J Virol 76:9389–9397. doi: 10.1128/JVI.76.18.9389-9397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishida T, Hamano A, Koiwa T, Watanabe T. 2006. 5′ Long terminal repeat (LTR)-selective methylation of latently infected HIV-1 provirus that is demethylated by reactivation signals. Retrovirology 3:69. doi: 10.1186/1742-4690-3-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firouzi S, Lopez Y, Suzuki Y, Nakai K, Sugano S, Yamochi T, Watanabe T. 2014. Development and validation of a new high-throughput method to investigate the clonality of HTLV-1-infected cells based on provirus integration sites. Genome Med 6:46. doi: 10.1186/gm568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.