

Abstract
Key clinical studies for HIV coreceptor antagonists have used the phenotyping-based Trofile test. Meanwhile various simpler-to-do genotypic tests have become available that are compatible with standard laboratory equipment and Web-based interpretation tools. However, these systems typically analyze only the most prominent virus sequence in a specimen. We present a new diagnostic HIV tropism test not needing DNA sequencing. The system, XTrack, uses physical properties of DNA duplexes after hybridization of single-stranded HIV-1 env V3 loop probes to the clinical specimen. Resulting “heteroduplexes” possess unique properties driven by sequence relatedness to the reference and resulting in a discrete electrophoretic mobility. A detailed optimization process identified diagnostic probe candidates relating best to a large number of HIV-1 sequences with known tropism. From over 500 V3 sequences representing all main HIV-1 subtypes (Los Alamos database), we obtained a small set of probes to determine the tropism in clinical samples. We found a high concordance with the commercial TrofileES test (84.9%) and the Web-based tool Geno2Pheno (83.0%). Moreover, the new system reveals mixed virus populations, and it was successful on specimens with low virus loads or on provirus from leukocytes. A replicative phenotyping system was used for validation. Our data show that the XTrack test is favorably suitable for routine diagnostics. It detects and dissects mixed virus populations and viral minorities; samples with viral loads (VL) of <200 copies/ml are successfully analyzed. We further expect that the principles of the platform can be adapted also to other sequence-divergent pathogens, such as hepatitis B and C viruses.
INTRODUCTION
The predominant virus variant in early stages of the clinical manifestation of disease, CCR5-tropic HIV, is found in approximately 80% of treatment-naive patients (1, 2). Although this number can vary for the different virus subtypes, the percentage of CXCR4-tropic HIV isolates is generally low and tends to rise with disease progression (3–6). Nevertheless, the fraction of CCR5-tropic viruses in clinical specimens continues to stay at >50% throughout the course of infection (7, 8). As such, the molecular interactions between the viral envelope and the cellular chemokine receptor CCR5 were recognized as potentially attractive targets for drug development and have yielded compounds and drugs able to specifically block CCR5-tropic HIV (9–11). It is this selectivity of the inhibition of one (CCR5) and not the other (CXCR4) viral coreceptor that necessitates tropism testing prior to prescribing drugs of this particular class. Although the chemokine receptor binding site in the HIV envelope is constituted mainly by the V3 loop, the V1/V2 regions, and the C4 conserved region in the HIV protein gp120, coreceptor tropism is dictated predominantly by amino acid sequences of the V3 region (12, 13). But also sequences of other variable env regions can contribute as secondary sites to the viral tropism (14–17).
Initially, all tropism determinations in the key clinical studies during development of CCR5 antagonists, e.g., maraviroc (Celsentri/Selsentry), used several sensitivity versions of the Trofile test, a phenotype-based system developed by Monogram Biosciences. And along with the approval of maraviroc as the first drug in class, the HIV-1 authorities required a mandatory tropism determination prior to any prescription (18). The phenotypic Trofile test, particularly its enhanced-sensitivity version (TrofileES), had proven to represent an excellent tool for determining the tropism of HIV in patients, particularly when the question was to detect with highest sensitivity CXCR4-tropic viruses. At that time, it was crucial for the salvage studies to exclude affected patients from studies in order to minimize therapy failure. However, such highly sophisticated mostly centralized phenotypic testing turned out to be problematic for the everyday settings when introducing this drug class into clinical practice (19). Particularly for requests from Europe, the need for international sample shipment, a limited test sensitivity (>1,000 copies/ml), and long turnaround times were recognized as inacceptable obstacles. As a consequence, various simpler diagnostic tools have been developed and validated (20–23). And this development of genotypic tests created a need for a further refinement of the tools to be able to characterize mixed virus populations, detect CXCR4 tropism in viral minorities, and define predictors of disease progression.
Various methods were developed and evaluated as possible options for simplifying the diagnostic procedures. We, as others, have used amplified HIV-1 env sequences for tropism analysis based on a heteroduplex tracking assay (HTA) (4, 24), Pathway Diagnostics (SensiTrop)-developed commercial HTA-based tests, and later Quest Diagnostics-developed commercial HTA-based tests.
The apparent complexity of the subject is exemplified in a recent publication by Cabral et al., who compared genotypic methods with Trofile as the phenotypic standard and concluded that “composite algorithms may be needed” for predictively assessing the viral tropism when only V3 sequences are analyzed (25). Comparative studies of the commercial genotypic test with the validated Trofile assay found the SensiTrop test to be inferior in identifying CXCR4 tropism in clinical specimens. Hence, the use of the SensiTrop test was suspended and has in the meantime been replaced with sequence-based methods, including sensitive next-generation sequencing (Reflex; Quest Diagnostics). One key element not revealed for most published HTA-based tropism tests is the strategy by which the hybridization oligonucleotide of the test was identified or the methods by which the relative tropism specificity of such oligonucleotides was validated for the commercial test.
This study presents a new approach attempting to improve and simplify genotypic tropism testing. The method presented here is based on the principles of duplex tracking as initially described by Delwart et al. (26); only for a limited number of critical and ambiguous samples (<10%) did it require complementation by sequence information or phenotyping. As is principally typical for homo- and heteroduplex tracking, our system utilizes the analysis of double-stranded hybrids between a patient-derived HIV-1 sequence(s) and a small set of defined synthetic V3 sequences in a standardized capillary assay format. A key element of this development was the primer optimization based on numerous, characterized sequence pairs.
MATERIALS AND METHODS
Clinical specimens were from routine testing in the frame of the Swiss HIV Cohort Study (SHCS). As no preselection of patients or selection during sampling was performed, the genotypic properties were similar to those observed in routine at the Basel center: over 90% of all samples belong to subtype B, with the next most frequent subtype being C (<5%).
Preparation of labeled probe.
Single-stranded (ss) 6-carboxyfluorescein (FAM)-labeled V3 probes were obtained by PCR using commercial FAM-labeled, high-performance liquid chromatography (HPLC)-purified oligonucleotides (FAM-GA ATC TGT AGA AAT TAA TTG TAC AAG AC) in combination with a biotin-tagged oligonucleotide (biotin-TGC TCT ACT AAT GTT ACA ATG TGC TTG TCT TAT) for the opposite strand (Microsynth, Balgach, Switzerland) covering the HIV-1 V3 region. Ten microliters of 5× iProof HF buffer, 1 μl 10 mM deoxynucleoside triphosphate (dNTP) mix, 1 μl of each primer corresponding to 10 pmol, 0.5 μl iProof DNA polymerase (Bio-Rad, Reinach, Switzerland), and 30 ng of DNA template were mixed on ice and water added to 50 μl. After 2 min at 98°C, a standard cycling protocol with 35 cycles (10 s at 98°C, 15 s at 48°C, 10 s at 72°C) was performed with a final extension for 10 min at 72°C.
Single-stranded DNA separation.
PCR products were fixed via a biotin tag onto streptavidin. Forty microliters of Dynabeads M-280 streptavidin suspension (Life Technologies, Zug, Switzerland) was washed three times in 200 μl binding buffer (20 mM Tris-HCl [pH 7.5], 10 mM EDTA, 2 M NaCl) in a 1.5-ml reaction tube (Eppendorf, Hamburg, Germany) by using magnetic separation. Then 50 μl of purified PCR product and beads were mixed in 50 μl binding buffer and incubated for 30 min in the dark. Beads were then separated with the magnet for 2 min, supernatant was removed, and the Dynabead-DNA complex was washed 3 times in 200 μl wash buffer (20 mM Tris-HCl [pH 7.5], 10 mM EDTA, 1 M NaCl). For strand separation, the complex was resuspended in 20 μl 0.2 N NaOH, incubated for 10 min in the dark, and magnetically separated for 2 min. The supernatant containing the single-stranded, labeled DNA was collected in a reaction tube, and 20 μl of 1 M Tris-HCl (pH 7.5) was added. Prior to use, yields were quantified using a NanoDrop ND-1000 photometer (Fisher Scientific, Wohlen, Switzerland). A tetramethylrhodamine (TAMRA)-labeled 80-bp fragment of unrelated bcr-abl DNA served as the molecular weight marker (MWM) for each electrophoretic run. Sample preparation and PCR for XTrack was as follows: for RNA extraction from clinical specimens, the lysis protocol of the Prepito NA body fluid kit was followed (PerkinElmer, Baesweiler, Germany), and a one-step reverse transcription-PCR (RT-PCR) was then performed.
For one-step reverse transcription, primers F-6943 (CAC AGT ACA ATG YAC ACA TGG AAT) and R-7365 (AGT AGA AAA ATT CYC CTC YAC AAT TAA A), each 10 pmol, were mixed with 5 μl RNA template, 25 μl of Herculase II RT-PCR 2× master mix, and 1 μl Affinity Script RT/RNase block (Affinity Script One-Step RT-PCR kit; Agilent Technologies, Basel, Switzerland) according to the manufacturer's instructions in a final volume of 50 μl. Incubation was for 5 min at 45°C, 1 min at 92°C, 40 cycles of 20 s at 92°C, 20 s at 51°C, and 30 s at 68°C, and then 3 min at 68°C.
After the one-step RT-PCR and Illustra Exostar 1-step treatment (GE Healthcare, Opfikon, Switzerland) for 15 min at 37°C and 15 min at 80°C, the 2nd nested PCR step yielded products of ca. 140 bp in length using the following conditions: 5 μl of the one-step RT-PCR product was added to 10 pmol of primer F-7092 (GAA TCT GTA GAA ATT AAT TGT ACA AGA C) and 10 pmol of primer R-7232 (TGC TCT ACT AAT GTT ACA ATG TGC TTG TCT TAT), dNTPs, and 1 μl of PfuUltra II enzyme (Agilent Technologies) in PfuUltra II buffer in a reaction volume of 20 μl. PCR was carried out for 1 min at 92°C, and then 30 cycles of 20 s at 92°C, 20 s at 51°C, and 15 s at 72°C, and then 3 min at 72°C. The product was purified from a 2% agarose gel by excision and recovery of the V3 fragment and quantified. Approximately 20 ng V3 DNA was then mixed with 2 ng single-stranded probe, 2 μl 10× TKE buffer (10 mM Tris-Cl [pH 7.5], 10 mM KCl, 1 mM EDTA), and H2O to a final volume of 10 μl. The mix was denatured at 95°C for 5 min and rapidly cooled on wet ice in the dark for 10 min, after which 10 ng of the bcr-abl molecular weight marker was added. Samples were loaded onto an ABI 310 genetic analyzer fitted with a 47-cm capillary.
Fragment analysis.
The structure-based “genosorting” fragment analysis system uses POP conformational analysis polymer (CAP; ABI-Life Technologies catalog no. 4340379) at 5%, 1× TBE (90 mM Tris, 90 mM boric acid, 2 mM EDTA), and 10% glycerol. Electrophoresis was carried out in 1× TBE and 10% glycerol; samples were injected at 15 kV for 10 s and run for 30 min at 10 kV at a temperature of 30°C.
The resulting electropherograms were used to calculate for each probe the relative run length of a patient-derived HIV-1 sample, compared to the run of the perfect match (equal to a value of 1) and to the prototypic clonal CCR5-tropic reference virus HIVBaL. Based on the validation with phenotypically characterized isolates, this resulted in the reliable rule that values for the sample hybrid/perfect match ratio of below 0.7 were found for X4-tropic viruses, those above 0.79 were found for R5-tropic viruses, and values between 0.7 and 0.79 were called “indeterminate.”
Dideoxy sequencing.
Sequencing was carried out using BigDye Terminator kit v3.1 according to the manufacturer's instructions (Life Technologies, Zug, Switzerland). Reaction products were processed on an ABI3100 sequence analyzer.
rPhenotyping.
Principles and details of the replicative phenotyping protocol have been described elsewhere (27, 28) with the following adaptations for tropism analysis. Amplification of the HIV-1 Env gene isolated from clinical samples was successfully performed when virus loads were greater than 500 copies/ml (29). The HIV-1 envelope was amplified using the AffinityScript One-Step RT-PCR kit (Agilent Technologies, Basel, Switzerland) on a Biometra T3000 cycler (Biometra, Goettingen, Germany); primers matched nucleotide positions as indicated by their numbers: F_5700, GAA ACT TAT GGG GAT ACT TGG; R_8494, AGC TGA AGA GGC ACA GGC TCC. After primer annealing at 65°C for 5 min, 42°C for 2 min, and 25°C for 10 min, reverse transcription was conducted at 45°C for 30 min, 92°C for 1 min, followed by PCR at 40 cycles of 92°C for 20 s, 55°C for 20 s, and 68°C for 3 min, and then extension was at 68°C for 5 min. For the 2nd, nested, PCR for envelope and for introducing the cloning sites MluI and NgoMIV, the Pfu-ultraII Fusion HS DNA polymerase was used (Agilent Technologies, Basel, Switzerland). Primers were F_6435M (CYA CCA ACG CGT GTG TAC CCA C) and R_8319N (TGA RTA TCC CTG CCG GCC TCT ATT YAY TAT AGA AA); cycling conditions were 95°C for 2 min, 35 cycles of 95°C for 20 s, 50°C for 20 s, and 72°C for 1 min and 20 s, followed by an extension at 72°C for 3 min.
Products were cut with MluI and NgoMIV (New England Biolabs; Bioconcept, Allschwil, Switzerland) and purified over a 0.8% agarose gel. Then the respective fragments of 1.9 kbp were ligated into an NL4-3 backbone to reconstitute fully functional proviruses. After transformation of Top10 bacteria (Life Technologies), 4 ml of standard LB broth-Amp was directly inoculated without plating in order to retain viral diversity. Transformed bacteria were grown at 37°C overnight. Plasmid DNA was purified (DNA MiniPrep kit; Macherey-Nagel AG, Oensingen, Switzerland) and directly used for cell transfection. The drug susceptibility assay was performed as described previously (28) by using serial dilutions of the CCR5 antagonist TAK-779 (obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, Bethesda, MD, USA) (30) or the CXCR4 inhibitor AMD3100 (Sigma-Aldrich, St. Louis, MO, USA) (31). Briefly, the susceptible human cells were transfected with recombinant HIV-1 plasmids to produce replication-competent virus. Cultures were maintained in the presence of active concentrations of the respective antagonists. The time window of 4 days permitted up to 4 rounds of viral replication.
For phenotypic comparison, samples were sent to Monogram Biosciences (South San Francisco, CA, USA) for analysis using the TrofileES version of the commercial Trofile test.
Ethics statement.
The SHCS has been approved by the responsible ethical committees of all participating institutions (Ethikkommission Beider Basel; Kantonale Ethikkommission Bern; Comité d'Éthique du Département de Médicine de Hôpitaux Universitaires de Genève; Commission d'Éthique de la Recherche Clinique, Lausanne; Comitato Etico Cantonale, Bellinzona; Ethikkommission des Kanton St. Gallens; and Ethik-Kommission Zürich [all from Switzerland]).
Written informed consent was obtained from and is on file for all study participants. This study has been approved by the scientific review board of the Swiss HIV Cohort Study.
RESULTS
By targeting a host protein, HIV coreceptor antagonists represent a distinct, unique class of HIV inhibitors. As for cell binding, the virus has the principal option to use one of its two main coreceptors, CCR5 or CXCR4; it appears mandatory to assess this preference of the virus prior to prescribing an inhibitor that is restricted to CCR5-tropic virus variants. A test therefore has to provide reliable means for predicting whether or not the virus in the respective patient will respond to the treatment.
Here, we describe a refined genotypic test which is based on sequence hybridization and which does not require knowledge of the genetic envelope sequence.
For an extensive test validation, representative viruses with known tropism were utilized. The env regions of the CXCR4-tropic clonal HIV-1 strain NL4-3 (32), the CCR5-tropic AD87 (33), or the CCR5-tropic BaL (34) were utilized (reviewed in reference 35) and inserted into a viral NL4-3 genome backbone. Virus tropism and replicative fitness of the resulting constructs were assessed by replicative HIV phenotyping. This test, termed PhenXR, permits up to four rounds of virus replication in the presence of inhibitor. Details have been described elsewhere, and the test has been validated for diagnostic drug resistance testing (27). For all above-mentioned env variants, a replicative fitness of >70% compared to that of the wild-type NL4-3 was noted. This confirms that an exchange of larger segments of the env gene is sufficient for a functional tropism determination; it further shows that the exchange retains envelope functionality.
Properties of the “genosorting tropism test” (XTrack).
Although some reports have indicated a tropism-determining role for regions outside the V3 region (36), most tests focus today on V3-derived information (19, 37, 38). The tropism test described here is based on the sequence along with a sequence-implied structure characterized by the high variability of the V3 region of the envelope glycoprotein gp120 of HIV-1. Env sequences of this variable region in clinical samples are hybridized to double strands with an exogenously added sequence probe of known tropism. This step will constitute a more or less perfect double strand. The use of carefully designed, representative probes for diagnostics has successfully been compared against the genotypic method of Geno2Pheno and against a phenotype-based method (39). In the past, different HTA-based approaches have been described by others (40), and also the use of capillary analysis systems has been presented, e.g., by Baumann et al. (41). Principally, a fluorescently labeled single-stranded DNA probe representing the V3 region of known coreceptor tropism is hybridized under nondenaturing conditions to V3 loop sequences which were obtained from the virus in a given clinical sample. The degree of matching between sample and probe will determine a certain electrophoretic mobility that discriminates more perfect duplexes (homoduplex of almost identical strands) from imperfectly matched DNA (heteroduplexes). The principle of the test is to associate the respective mobility areas with a given tropism. This can be accomplished only with the most carefully selected probes that represent a given tropism and cover the various HIV subtypes.
Technical discrimination of R5/X4.
A principal limitation of the HTA approach is that the envelope V3 regions of individual HIV-1 isolates and of the various virus subtypes can differ dramatically from each other. In fact, this is the main reason for a certain inherent limitation of any genotypic prediction of an HIV tropism. Nevertheless, clinical comparisons have clearly demonstrated that for the vast majority of cases, genotyping and phenotypic methods are in quite good agreement, particularly for R5-tropic virus (25, 42). In an HTA approach, the degree of matching between patient-derived virus and the known sequence probes defines discrete migration zones. The capillary running characteristics of the duplexes under semi-denaturing conditions form the technical basis for the definition of these zones. The tropism prediction is based on the ratio between the distance from the start of the sample-probe duplex (“duplex”) and the single-stranded probe termed “1” divided by the distance of the perfectly matched probe-probe duplex, termed “2” (Fig. 1A). The designation of an “area of migration” forms the basis for defining discrete cutoffs between R5- and X4-tropic isolates. The shorter the distance of a sample-probe duplex from the perfect match (PM), the smaller the difference in character or sequence between probe and sample. Hence, the sample is more likely to share the tropism of the probe.
FIG 1.

(A) Typical electropherogram depicting the test principle. Four types of mass peaks are resolved: 80-bp molecular weight marker (MWM); single-stranded V3 probe (ss-probe); hybrid of patient-derived V3 sequence and probe (duplex). Residual double-stranded probe material serves as the ds-control. PM, perfect match; 1 and 2 indicate relative peak distances for tropism calculation. (B) Representative section of the list of V3 loop nucleotide sequences after grouping according to their relatedness to the probe candidate in the top line: HIV-1JR-CSF.
Of note, in earlier studies, which had recruited mostly patients in the U.S. or Europe, the number of non-B-subtype samples was quite small. Comparisons of older and more recent studies thus have the potential limitation that the more divergent non-B samples affect test performance (amplification success) as well as interpretation in a negative way. This is a principal caveat for comparing tests in different settings and time periods.
A major task lies in the broad divergence of HIV-1 sequences, particularly for the variable regions of env. From the literature, only a small amount of information is available for guiding the sequence optimization for a given diagnostic test. Yet it is quite likely that oligonucleotides chosen for amplification or hybridization are responsible for sequence-biased amplification. This will affect the results of the test. In order to systematically approach the design of suitable diagnostic probes, we tried several algorithms and utilized sequences available through the Los Alamos database (43). Its >1,000 env entries formed the basis for designing a larger number of probes. The best-fitting ones were sequentially tested on the same set of specimens.
Probe design strategy 1: direct sequence alignment.
This approach was based on a large sequence alignment with the aim to identify X4- and/or R5-tropic consensus sequences that could serve as representative probes.
For primary amplification of the V3 env target of HIV-1, the primer pair V3_7092F/V3_7232R was used yielding a 140-bp fragment; the primer positions were chosen based on published information (44, 45). Env sequences from 635 patient-derived viruses were obtained from the Los Alamos database (43) and from an in-house set with available subtype and tropism information. All sequences were computed to yield a generic HIV-1 consensus. Using ClustalW (EMBL-EBI, Cambridge, United Kingdom), sequences were aligned, compared to one another, and grouped according to relatedness and homology. Figure 1B shows a representative section of this alignment. Stretches of dots in the sequences indicate polymorphisms and reflect regions, where individual sequences had insertions of the respective length (not shown). The SplitsTree4 software (University of Tubingen, Tubingen, Germany) was then applied to yield a graphical dendrogram (Fig. 2) with discrete branches, largely reflecting the different subtypes of HIV-1. About 67% of the available sequences belonged to R5-tropic isolates (blue) mostly clustering tightly. In contrast, X4-tropic sequences (red) were distributed more toward the periphery. Of note, for subtype D, the available data set contained more X4-tropic sequences, and branches tend to have longer distances than other subtypes from a virtual “best probe” defining the center of the dendrogram. As an interesting correlate with published observations, subtype D associated with the highest rates of dual/mixed (D/M) tropism (2) in a phenotypic test. A distinct set of exclusively R5-tropic sequences (Fig. 2, bottom right) belonged to group O isolates that, in agreement with its phylogeny, possessed the longest distance to our B subtype reference JR-CSF near the center (branch length truncated). Moreover, recent investigations suggest that for CRF01_AE, algorithms for the genotypic tropism prediction have a much higher failure rate than for other subtypes (46). It is therefore interesting to note that the Clustal analysis in Fig. 2 produced a dendrogram that very discretely separates subtype A from CRF01_AE.
FIG 2.

Dendrogram of 655 HIV-1 sequences depicting the maximal relative relationship to one another (Los Alamos database and data from the Swiss HIV Cohort Study). Blue letters, R5-tropic viruses; red letters, X4-tropic isolates. Lobes of related sequences correspond with subtypes/groups (capital letters); the group O branch with 4 isolates is truncated. Yellow arrow indicates arbitrary reference sequence HIV-1JR-CSF.
For a best representation of the different clades, we chose several R5- or X4-tropic V3 sequences as single-stranded candidate DNA probes and tested them on a representative subset of 20 HIV-1 samples with known coreceptor tropism to verify a correct assignment. FAM-919 produced for 17 samples an interpretable result and yielded the highest number of distinct results (R5, X4, or mix) with 88% concordance with the TrofileES test. In comparison, the reference FAM-JRCSF yielded 19/20 (95%) interpretable results but tended to be too high on the R5 side (85%), which led to 10 false predictions; only 9 samples (47%) were in agreement with TrofileES. And FAM-JRCSF failed to dissect mixed virus populations.
Based on its favorable properties, FAM-919 was kept as one promising probe for the XTrack test system. In order to further minimize ambiguous results, we set out to define a second probe, which was, however, not readily identified with this strategy.
Probe design strategy 2: binding enthalpies linked to phylogenetic relatedness.
This approach considered physical properties of base pairing stability and sequence relatedness. The algorithm, kindly provided by Alex Thielen (Max Planck Institute, Kaiserslautern, Germany), compared each individual candidate R5 probe to every other sequence in the entire sample set of 655 R5 and X4 sequences. Each sample sequence would be sorted in relation to the one chosen to serve as the reference. The degree of relatedness between the two assigns a value to this pair. A value of 0 stands for an identical sequence, and a value of 1 stands for the most distant one in the set. Hence, a closer relatedness of any given sequence to the chosen R5 reference is reflected in a smaller resulting value. The aim of this process was to identify a set of sequences with very low scores that would allow a best separation of R5-tropic from X4-tropic viral sequences. Ten R5-tropic sequences representing the main lobes of the dendrogram of Fig. 2 were chosen, and they were aligned one by one to the entire sequence set from the database. A representative section of the results is shown in Table 1.
TABLE 1.
Binding enthalpies of three R5-specific probes to a selection of R5 or X4 sequencesa

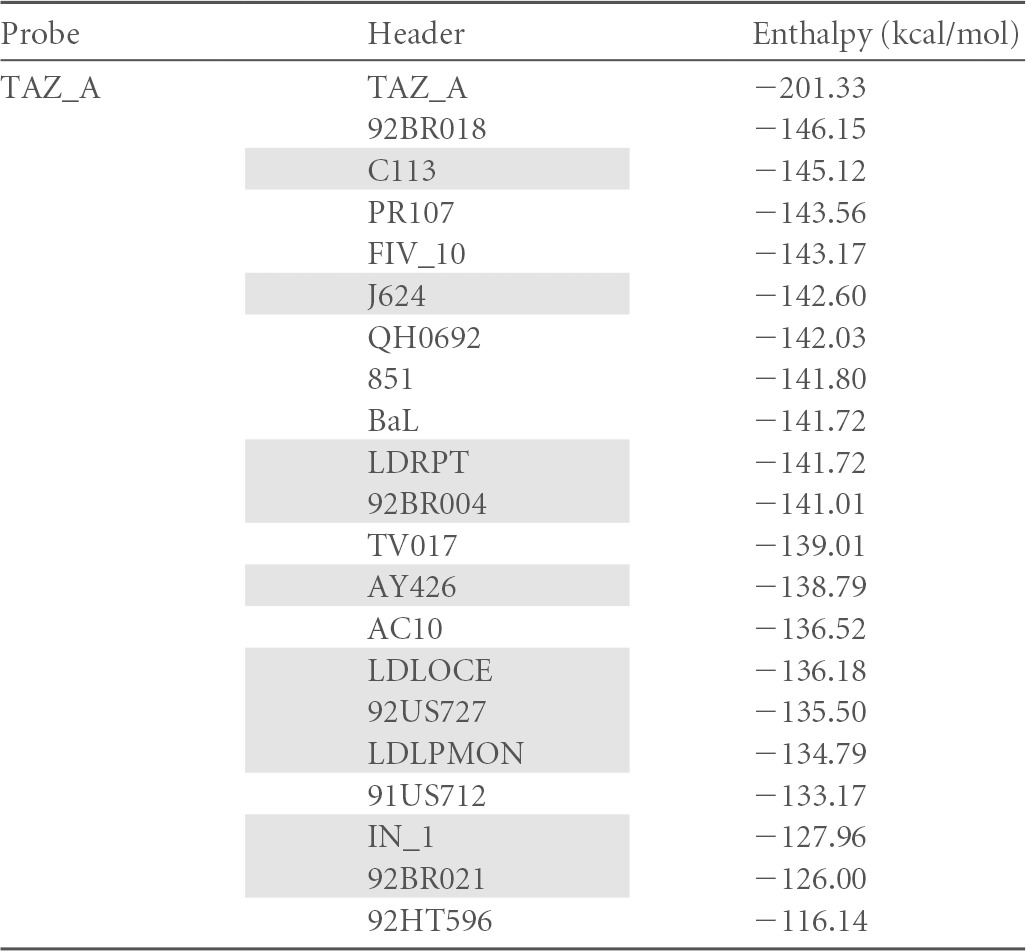
Unshaded, R5 sequences; shaded, X4 sequences. The top value for each probe corresponds to the perfect match of the probe. Values are sorted by decreasing ΔH values.
From the sum of distance values for each sample, a total XR score was calculated for every probe candidate. Table 2 summarizes, e.g., the result for probe candidate 01PB27ZA. This XR algorithm was run against all available isolates, and the obtained values are listed in the “Distance” column. The table is sorted by increasing distance values, with a score of 0 indicating 100% relatedness. In the “1/0 R5” column, a value of 1 is then entered for known R5 tropism according to the Los Alamos database, and a value of 0 for X4-tropic sequences. The “Index” column counts numerically down from 1 for the top sequence (for reference sequence 01PB27ZA) to index 655. A smaller index number thus indicates a closer relatedness to 01PB27ZA. A multiplication of columns “1/0 R5” and “Index” yields the next column, named “Value R5.” It generates a positive value solely for R5-tropic sequences. When the same procedure is now reiterated for every one of the 655 sequences, each reference sequence itself will receive an overall qualifier through the total sum of all “Value R5” scores. This procedure allowed us to define an overall relatedness for all R5 sequences to one another. Simultaneously, it reveals the greatest distance to CXCR4-tropic HIV-1. Subsequently, the same procedure was run with X4-tropic sequences (“1/0 X4” column for X4 viruses). As X4 isolates are underrepresented in the Los Alamos database (only 43% of all entries), we compensated for this by applying the multiplier of 2.3, and cumulative value R5 was divided by cumulative value X4 to yield a final XR score for the winning reference. The top scoring two sequences were used for further experiments. As an example, sequence 01PB27ZA had a final score of 0.93 (shaded in Table 3 ). The lowest overall score (greatest separation of R5 and X4) within the set of 655 samples was 0.46 and the maximal score was 4.9. As the score for a random distribution was 1.23, meaningful sequences have to possess a score below this. Interestingly, the candidate sequences in the top section of the list were contributed mainly by virus subtypes A and C (Table 3).
TABLE 2.
Relative enthalpy-based sequence distance from each V3 sequence to every other one in the list of published isolatesa
| Tropism | Isolate (GenBank accession no.) | Sequence | Distance | 1/0 R5 | Index R5 | Value R5 | 1/0 X4 | Index X4 | Value X4 |
|---|---|---|---|---|---|---|---|---|---|
| R5 | R5-S123 (AF153176) | TGTACAAGAC | 0.021858 | 1 | 3 | 3 | 0 | 3 | 0 |
| R5 | R5-S194 (AF153164) | TGTATAAGAC | 0.021858 | 1 | 4 | 4 | 0 | 4 | 0 |
| R5 | R5-S083 (AF153189) | TGTACAAGAC | 0.027322 | 1 | 5 | 5 | 0 | 5 | 0 |
| R5 | R5-98ZABLM84 (DQ235618) | TGTATAAGAA | 0.027322 | 1 | 6 | 6 | 0 | 6 | 0 |
| X4 | X4-CHN19 | TGTACAAGAC | 0.027322 | 0 | 7 | 0 | 2.3 | 7 | 16.1 |
| R5 | R5-A125 (AY253304) | TGTGTAAGAC | 0.027322 | 1 | 8 | 8 | 0 | 8 | 0 |
| R5 | R5-C054 (AF153155) | TGTACAAGAC | 0.027322 | 1 | 9 | 9 | 0 | 9 | 0 |
| R5 | R5-93MW_965 (AY713413) | TGTACAAGAC | 0.027322 | 1 | 10 | 10 | 0 | 10 | 0 |
| R5 | R5-S071 (AF153162) | TGTACAAGAC | 0.027322 | 1 | 11 | 11 | 0 | 11 | 0 |
| R5 | R5-TV002 (AF254767) | TGTACAAGAC | 0.027322 | 1 | 12 | 12 | 0 | 12 | 0 |
| X4 | X4-CTSC2 (AY043176) | TGTACAAGAC | 0.027322 | 0 | 13 | 0 | 2.3 | 13 | 29.9 |
| R5 | R5-TV019 (AF254783) | TGTACAAGAC | 0.027322 | 1 | 14 | 14 | 0 | 15 | 0 |
| R5 | R5-C022 (AF153186) | TGTACAAGAC | 0.027322 | 1 | 15 | 15 | 0 | 15 | 0 |
| R5 | R5-TV014A (AF391247) | TGCACAAGAC | 0.027322 | 1 | 16 | 16 | 0 | 16 | 0 |
| R5 | R5-99ZASW5 (AY170658) | TGTACAAGAC | 0.027322 | 1 | 17 | 17 | 0 | 17 | 0 |
| R5 | R5-99ZASW38 (AY170667) | TGTACAAGGC | 0.027322 | 1 | 18 | 18 | 0 | 18 | 0 |
| R5 | R5-99ZASW38 (AY505002) | TGTACAAGGC | 0.027332 | 1 | 19 | 19 | 0 | 19 | 0 |
| R5 | R5-99ZATM19 (DQ235629) | TGTACAAGAC | 0.027322 | 1 | 20 | 20 | 0 | 20 | 0 |
| R5 | R5-98ZA502 (AY158534) | TGTACAAGAC | 0.032787 | 1 | 21 | 21 | 0 | 21 | 0 |
| R5 | R5-S147 (AF153169) | TGTACAAGAC | 0.032787 | 1 | 22 | 22 | 0 | 22 | 0 |
| R5 | R5-S171 (AF153150) | TGCACTAGAC | 0.032787 | 1 | 23 | 23 | 0 | 23 | 0 |
| R5 | R5-01PB15ZA (AY510062) | TGTACAAGGC | 0.032787 | 1 | 24 | 24 | 0 | 24 | 0 |
| X4 | X4-C070 (AF153143) | TGTACAAGGC | 0.032787 | 0 | 25 | 0 | 2.3 | 25 | 57.5 |
| R5 | R5-C109 (AF153142) | TGTACAAGAC | 0.032787 | 1 | 26 | 26 | 0 | 26 | 0 |
| R5 | R5-97ZAPET100 (DQ235617) | TGTACAAGAC | 0.032787 | 1 | 27 | 27 | 0 | 27 | 0 |
| R5 | R5-01PB21ZA (AY510065) | TGTACAAGAC | 0.032787 | 1 | 28 | 28 | 0 | 28 | 0 |
| R5 | R5-S059 (AF153151) | TGTACAAGAC | 0.038251 | 1 | 29 | 29 | 0 | 29 | 0 |
| R5 | R5-TV005 (AF254770) | TGTACAAGAC | 0.038251 | 1 | 30 | 30 | 0 | 30 | 0 |
| R5 | R5-TC25 (AY265945) | TGTACAAGAC | 0.038251 | 1 | 31 | 31 | 0 | 31 | 0 |
Highest scores for various HIV-1 isolates to the reference (line 1), with “Distance” value = 0 and sorted by increasing values; columns as detailed in the text.
TABLE 3.
XR scores of relatednessa
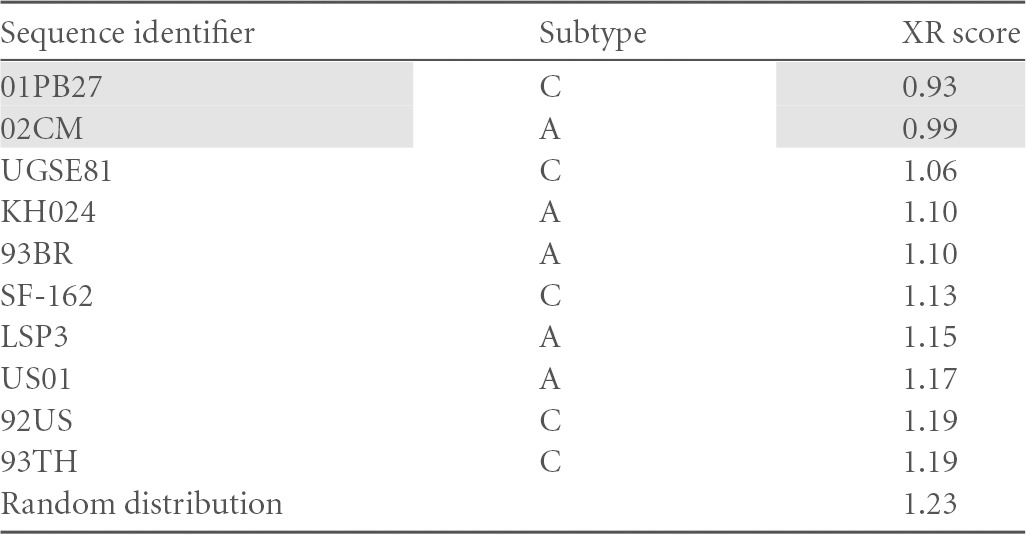
Calculated for the top 10 R5 sequences of HIV-1 subtypes A or C. Random distribution, reference score for a random distribution of R5 and X4 sequences. Shading indicates the top scoring two sequences, which were used for further experiments.
In summary, sequence 01PB27ZA scored favorably with 0.93 and was therefore tested in the XTrack assay.
Probe design strategy 3: free enthalpy (ΔH).
As V3 sequences in clinical specimens typically differ in sequence, a “best match” between sample and probe is expected to yield the most stable hybrid. In this approach, the theoretically best probe candidates were hybridized in silico with each one of the available 655 sequences using the online tool RNAfold (Institute for Theoretical Chemistry, University of Vienna, Vienna, Austria). However, the three probe candidates representing the HIV-1 subtypes B, C, and A (NH45_B, KNH_C, TAZ_A) yielded no consistent ranking. And although X4 sequences principally tended to be closer to the bottom of the rank, there was no clear separation from R5-tropic sequences. We therefore concluded that free enthalpy alone was not suitable for predicting a tropism-specific clustering (data not shown).
In summary, of the three strategies, the highest predictive power was reached with probes targeting subtypes A and C: 01PB27ZA (termed FAM-PB_C) and 02CM (termed FAM-CM_A). By sequence alignment, differences were found at 10 positions, distributed throughout the V3 region (not shown). We added a subtype B candidate, FAM-919. Then the 140-bp DNA segment of each of these V3-probe candidates was generated synthetically by tiling PCR and the gel-purified products cloned into the vector pCR-blunt II-TOPO (Invitrogen).
These probes for XTrack analysis were tried on 20 patient-derived V3 sequences. In parallel, the same patients were analyzed with the TrofileES test. Previous studies have confirmed the good agreement between the phenotypic format of TrofileES and the genotyping tool Geno2Pheno on a retrospective comparison of a large study population in clinical studies (47, 48). As further confirmation, we also performed a parallel analysis with TrofileES on a limited set of 20 clinical samples. Results are summarized in Table 4. For this trial set, the tropism results with FAM-CM_A and FAM-PB_C were identical and concordant with the TrofileES in 13 out of 20 cases. Shorter or longer length than the canonical 105 bp of the V3 loop is likely to affect the hybridization behavior. Among the discordant samples, two had shorter V3 loops (102 bp in length). Related difficulties have also been reported for the genotypic interpretation by G2P (48) and require further attention. Overall, we found for FAM-919 an 88% agreement with the TrofileES results, 71% for FAM-CM_A, and 67% for FAM-PB_C. For test validation and for establishing interpretation rules, we focused on the combination of FAM-CM_A plus FAM-919 due to the superior agreement with the TrofileES test. We chose to accept the results of the clinically validated TrofileES test by default as true. It should be remembered, however, that in clinical studies, TrofileES results were handled quite restrictively in the way that any sign of “non-R5 tropism” or any “uninterpretable result” led to patient exclusion from the respective clinical studies (49). Such policy potentially restricted the number of valid R5 participants by eliminating all less clear cases. A clinical proof for the validity of this exclusion is not available, and tropism changes between screening and baseline in the clinical studies may reflect the test variability or a certain instability of the viral tropism (50–52). Recent deep-sequencing analyses have demonstrated that low proportions of X4-tropic viruses can be found in almost any clinical sample, yet the clinical relevance, e.g., of X4-tropic virus minorities below 2% in a given virus population found in clinical specimens remains unclear (53).
TABLE 4.
XTrack resultsa
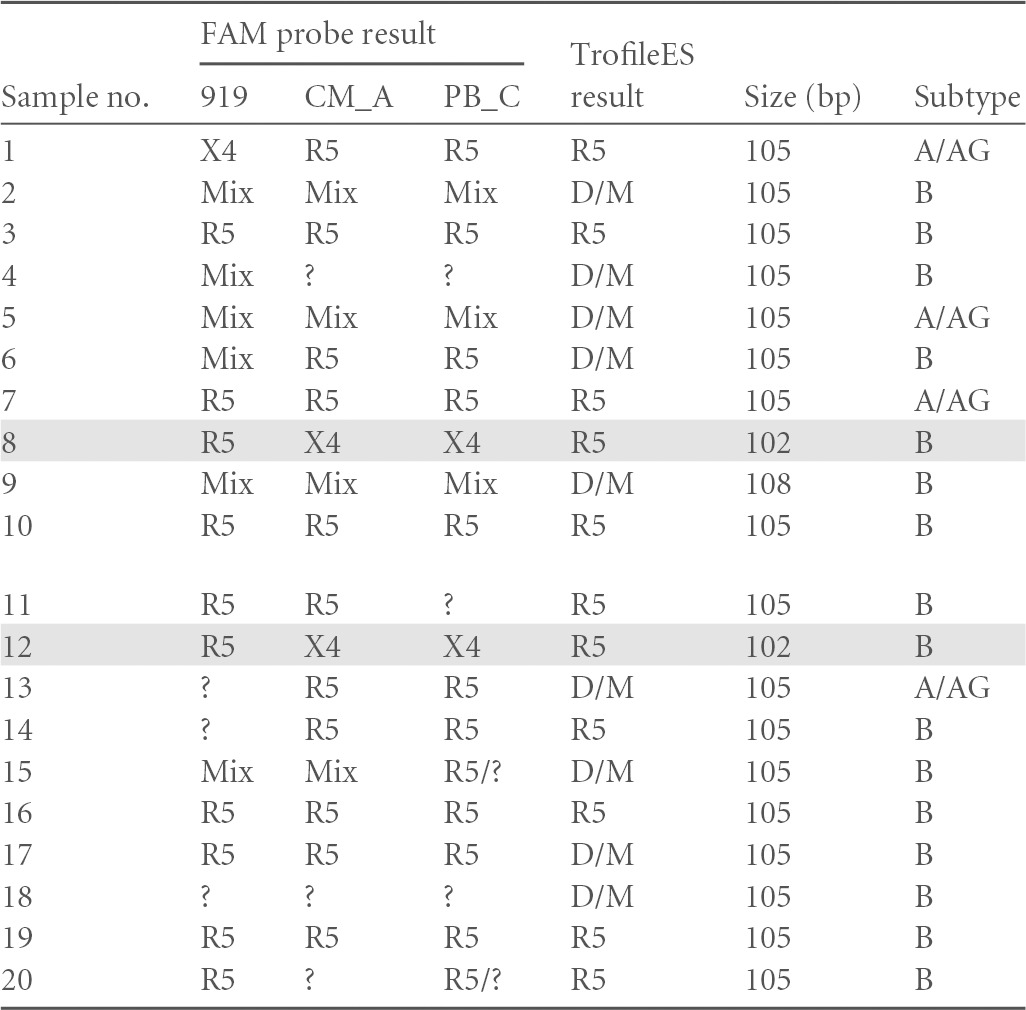
Using probes FAM-919, FAM-CM_A, or FAM-PB_C in columns 2, 3, and 4, respectively, and in comparison to Trofile (column 5). Column 6 lists the V3 loop length and column 7 the subtype for each sample. Samples with shorter V3 loops (no. 8 and 12) are shaded. R5, CCR5-tropic virus; X4, CXCR4-tropic virus; Mix, mixed virus population with both tropisms; D/M, dualtropic or mixed virus; ?, noninterpretable result.
Capillary analysis of duplex species.
A PCR product of about 140 bp containing V3 sequences from HIV-1 in clinical specimens is hybridized to the fluorescently labeled, single-stranded V3 DNA probe of known sequence and tropism. A double-stranded molecular weight marker is mixed with the sample prior to electrophoresis, serving as the migration standard. The relative migration of the products in relation to the marker and to the remaining single-stranded probe allows to determine their relatedness to the homoduplex probe.
In order to assess which of the probes was suitable for analyzing the HIV-1 tropism of clinical samples, we adapted the heteroduplex tracking to a capillary electrophoretic format. A typical electropherogram is depicted in Fig. 3.
FIG 3.

XTrack analysis of one prototypic R5-tropic and one X4-tropic virus at position “sample duplex.” MW, double-stranded molecular weight marker; ss-probe, labeled single-stranded probe; ds-probe, double-stranded probe.
The tropism of sample peaks is calculated based on percent migration, (position of single strand − position of duplex of patient sample and probe)/(ss-probe position − ds-probe [PM] position), as described in Fig. 1 (ds is “double-stranded”). With the help of samples with known tropism in a large validation set, two distinct migration zones corresponding to R5-tropic (80 to 100% migration) and X4-tropic (0 to 69% migration) were defined. Signals mapping to the region between these defined zones (70 to 79%) indicate unassignable samples.
For specimens simultaneously containing several distinct HIV variants, several duplexes will form with different, separable electrophoretic migration, therefore “genosorting.” We have shown that our system will identify, e.g., X4-tropic viral subpopulations that may represent as little as 1% of the total quasispecies (4).
As a potential limitation, the XTrack analysis is restricted to the V3 region of envelope and will therefore not consider other tropism-relevant regions of the HIV genome.
In our study setup with 145 available samples, 139 samples yielded an interpretable result with at least one of the systems. Validation was performed as a side-by-side comparison of XTrack, Geno2Pheno (10% false-positive rate), and TrofileES, and for 57 clinical samples results were obtained with all three systems. For 137/139, such comparison was available between two systems. All analyses were blinded to the interpreting expert so that the link between results from the other systems was not available to the operator. In order to avoid interpretation bias, a second independent operator performed linkage of the results summarized in Table 5. The high number of missing results for the TrofileES system is explained mainly by two factors. As all clinical samples stemmed from the routine laboratory for tropism analysis, either not enough material was available for including the external TrofileES test or the samples were not suitable for Trofile testing due to viral loads being below 1,000 copies/ml (not accepted by the provider). Although the small number is acknowledged as a significant potential limitation of our study, results from earlier work had clearly demonstrated that genotypic techniques correlated very well with the TrofileES results (25). This agreement hence supports the comparative analysis of this study.
TABLE 5.
Side-by-side comparison of XTrack, Geno2Pheno, and Trofile
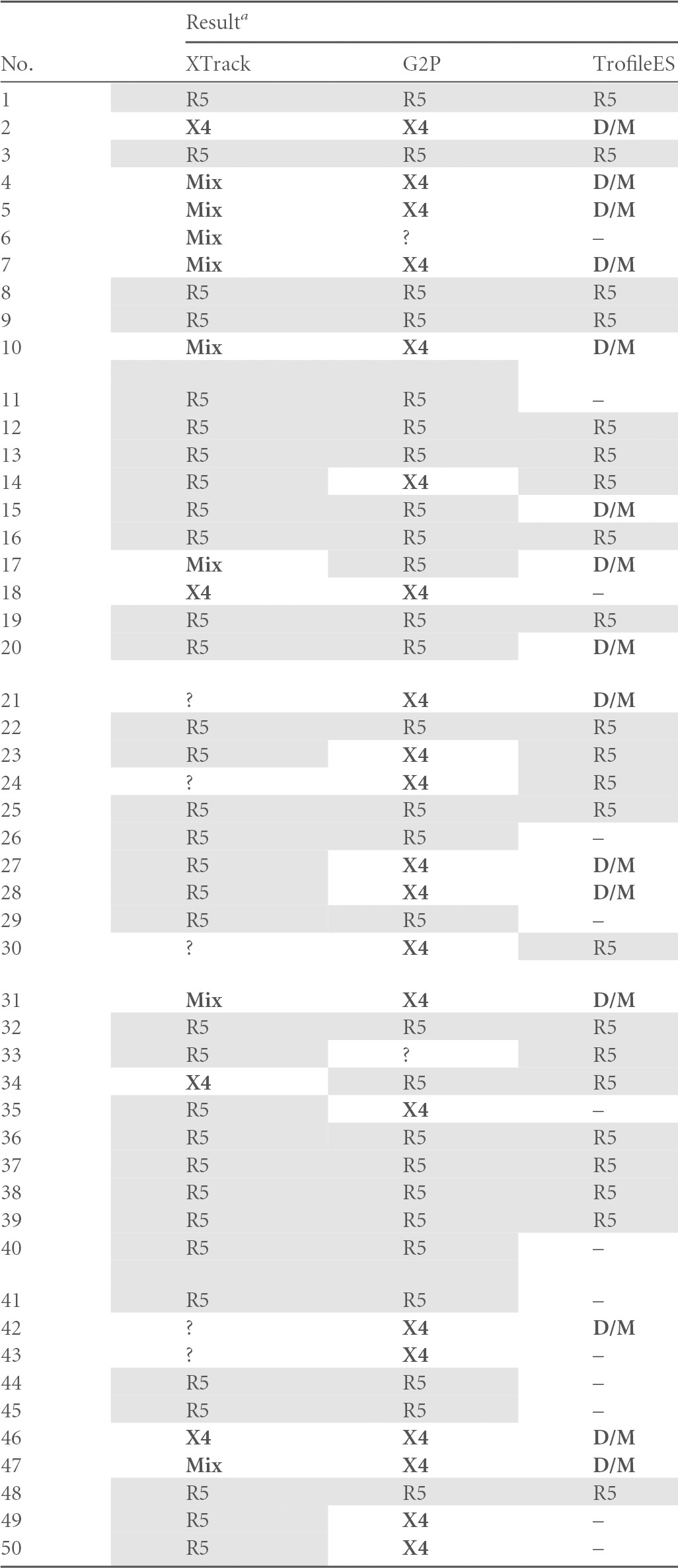
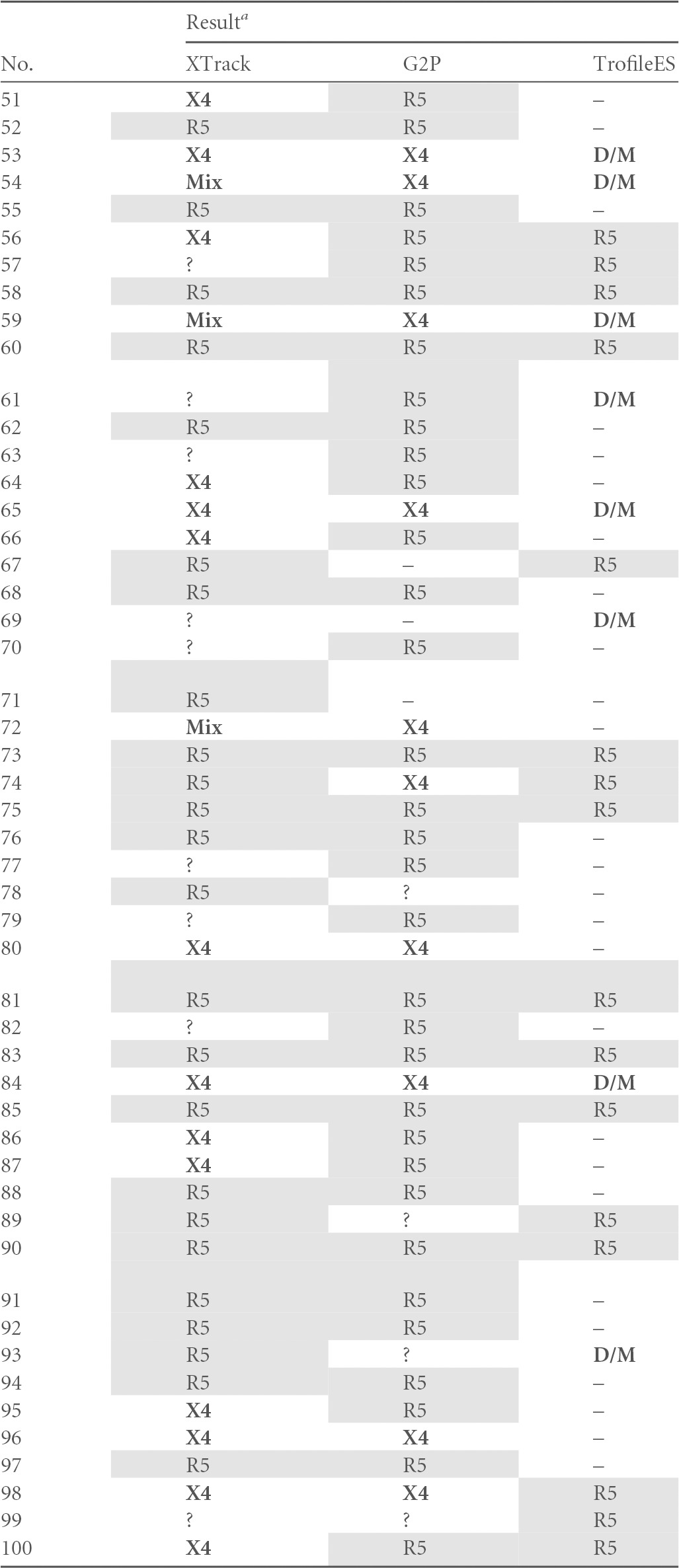
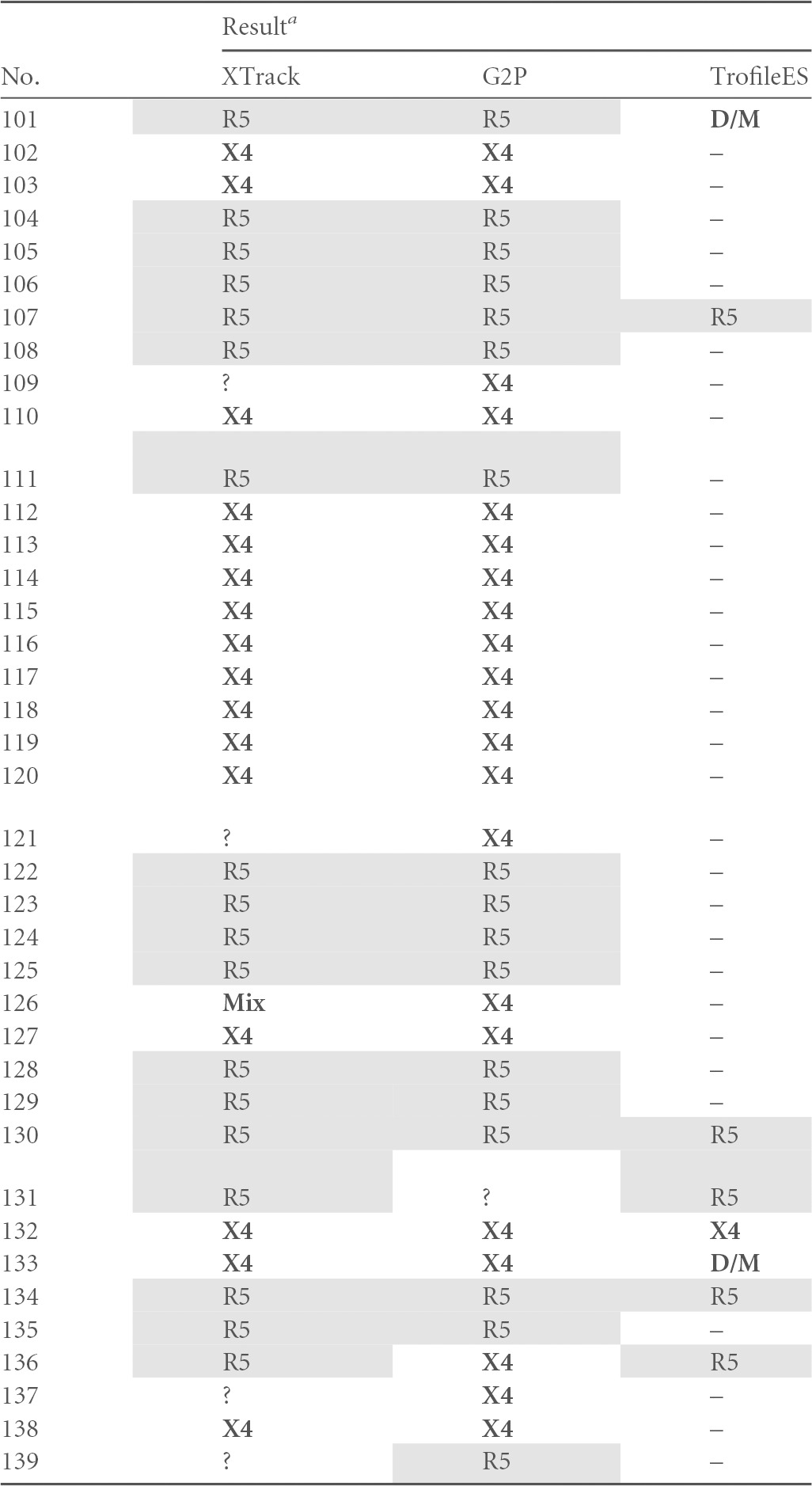
G2P, Geno2Pheno; D/M, dual/mixed tropism; Mix, two viral species with R5 and X4 tropism in the same specimen; ?, no tropism assignable by the respective method (uninterpretable results); –, missing value. X4 viruses, mixes, and D/M are indicated with bold letters, R5 viruses are shaded.
In our analysis, all three systems found R5-tropic virus in 27 samples and X4-tropic HIV or mixes in 15 samples. For 20 samples, TrofileES called D/M, and XTrack identified nine mixed virus populations. All of these correlated with D/M in TrofileES; in contrast, for six of the TrofileES D/M calls, XTrack and Geno2Pheno agreed on X4, and three were called R5. For two samples, XTrack identified an R5 virus and Geno2Pheno an X4 variant. Overall disagreement of at least one system was found in 15 cases (26%).
When assessing the overall performance in six cases, XTrack did not yield a result; in five, no sequence for Geno2Pheno was obtained; and in 56 cases, no TrofileES result was obtained or no such test was performed due to insufficient sample volume or low viral load. Three results were uninterpretable by XTrack due to unusual duplex migration, and for Geno2Pheno four sequences were double sequences that could not be analyzed.
V3 sequence sizes and tentative virus subtypes were determined in order to provide additional information for interpreting discordances. Whereas among the compared samples no link between discordance and subtype was found (not shown), a noncanonical length of the V3 loop (non-105 bp) was more frequently associated with discordances between the tests. With the simultaneous use of probes FAM-CM_A and FAM-919, concordance to TrofileES of over 70% was reached. Less than 5% of XTrack results were noninterpretable.
A statistical analysis produced performance parameters (i) for XTrack versus TrofileES and (ii) for Geno2Pheno versus TrofileES. The sensitivity of determining R as the no. of R5-concordant samples/(no. of R5-concordant samples + no. of X4- and mix-discordant samples) was similar or slightly superior for XTrack in the groups with 89.7% or 81.6%, respectively. The test specificity values for X4 tropism as no. of X4- and mix-concordant samples/(no. of X4- and mix-concordant samples + no. of R5-discordant samples) were 73% and 79%, and the negative predictive values toward X4 tropism as no. of X4- and mix-concordant samples/(no. of X4- and mix-concordant samples + no. of X4- and mix-discordant samples) were 80% and 73%. It has been described that tropism changes can be achieved by exchange of single amino acids in the V3 loop (54), and we set out to investigate whether, as reported earlier in a study for subtype A and D viruses (2, 55), the D/M viruses were dualtropic or alternatively mixed virus populations. In the analysis shown in Table 5, for more than half of the TrofileES D/M results (11/20), the XTrack system did not identify a mixed virus population, and in two samples the V3-based genotypes disagreed. As suggested by Huang et al. (2), a likely explanation is that additional determinants outside the V3 region contributed to the viral tropism. The data of Table 5 are depicted in the histogram of Fig. 4, highlighting the good agreement of 83.0 to 84.9% between the systems, with identical results for all three in 76% of the specimens. The proportions of X4-tropic samples in our patient population were determined to be 38.7% by XTrack, 40.3% by G2P (10% false-positive rate), and 37.7% by TrofileES.
FIG 4.
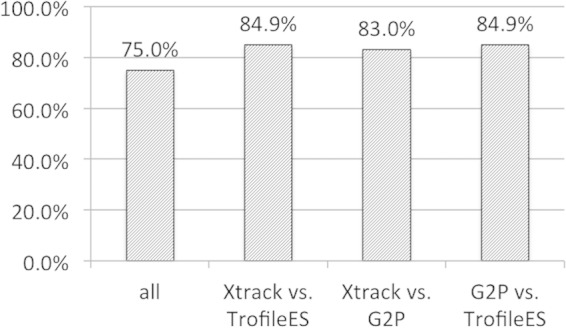
Degree of agreement between the three systems, based on the data in Table 5. Bars indicate either the overall agreement between the three systems (“all”) or between XTrack and TrofileES, or XTrack and Geno2Pheno, or Geno2Pheno and TrofileES, as indicated. Percent agreement is given above each bar.
By including data from ongoing genotypic testing at the Basel center, a total of 256 samples could be analyzed. Full agreement was found in 79.3% of samples, and the proportion of X4-tropic/mixed viruses represented 25% (54) of all analyzed specimens.
In order to address this possibility further, we employed a replicative phenotyping system (deCIPhR), which uses patient-derived full-envelope sequences for recombinantly reconstituting a fully infectious virus, similar to the format described earlier (27). This test allows for the expansion of virus during four replication cycles in the presence of inhibitors, and the virus carries envelopes from clinical specimens. Residual virus after in vitro treatment with, e.g., a CCR5 antagonist can be used for subsequent infections in the presence of a second inhibitor class, e.g., CXCR4 inhibitors. For 23 specimens with available sufficient sample volume, a comparison was conducted for TrofileES and our replicative phenotyping system (PhenXR). All 14 TrofileES analyses with assigned R5 tropism and the only X4 sample were confirmed by PhenXR. TrofileES called eight virus samples D/M, four of which were congruent between both systems and were in our tests exclusively inhibited by the X4 antagonist AMD3100. The remaining four D/M samples by TrofileES were not confirmed by the replicative system PhenXR to contain both tropisms or dualtropic virus. All samples were exclusively inhibited by the CCR5 antagonist in the PhenXR system, and no inhibition or plateau was found with AMD3100; they were thus classified as R5. The four discordant samples were all of subtype B.
The well-characterized CCR5 antagonist TAK-779 was utilized for all studies, as earlier reports had demonstrated its excellent agreement in specificity and potency with maraviroc in cellular systems (56).
Although the PhenXR results were confirmed in repeat experiments (not shown), it cannot be excluded that amplification bias during PCR could have contributed to this discordance between the systems.
Although this report describes the properties of a hybridization-based genotyping principle only for HIV samples, the same physical principles will apply for any genetically divergent pathogen. Hence, an application and similar optimization approaches will be useful for designing suitable test systems also for pathogens such as other highly variable viruses, such as hepatitis B (HBV) and C (HCV) viruses. The same methodology might help to identify optimal probes there, too. This has been suggested already in 1998 by Calvo et al. for genotype 2 of HCV (57).
DISCUSSION
We describe a strategy for complementing and improving the diagnostic genotype-based tropism determination of HIV for clinical use. Sequence-based tropism testing allows a more rapid turnaround time than phenotyping; here, we validated a hybridization-based “genosorting” method, which can further simplify the analysis process by omitting the need for sequencing. In addition, the short PCR fragment required for analysis (<150 bp) allows for the successful application of this system to clinical samples with viral loads below 200 copies/ml, assessed by routine VL testing (Ampliprep-TaqMan; Roche, USA). This poses a major challenge to phenotypic methods that depend on cloning large segments of DNA. Our validation data suggest that by optimization based on enthalpy and sequence relatedness, only 2 or 3 probes are sufficient to predict tropism with precision similar to that of phenotyping or Geno2Pheno (10% false-positive rate). Moreover, genotyping would benefit from additional properties, such as the resolution of mixed virus populations. The simple diagnostic format of XTrack renders the system suitable for diagnostic purposes in routine settings with standard equipment.
The XTrack system confirmed the mixed nature of viral isolates in about 50% of unselected cases of this study, called “dual or mix” by TrofileES. As the other half was assigned to a single tropism, it is likely that this part of the samples is either difficult to judge, truly made up of dualtropic viruses, or misclassified. A functional verification was not available for these samples.
Certain principal shortcomings of genotype-based methods have been identified, particularly for noncanonical V3 loop lengths shorter or longer than 105 bp. The current limitation for those variants is that the respective interpretation rules are still lacking and need to be established. In relation, the suitability for the genetically distant group O variants of HIV-1 could not yet be assessed. Another principal limitation is the fact that the genotyping assays described here restrict the analysis to the Env V3 region. Thereby, this study did not take into consideration the possible contributions to a viral tropism by regions outside this peculiar peptide structure.
It should also be noted that the true value of the term “correct tropism” was not clinically verified for any of the tests. Until today, it remains unclear how meaningful minority viruses of the opposite tropism in clinical specimens are. It also is to be determined whether viruses with unclear tropism assignment (e.g., medium false-positive rate in the Geno2Pheno system or intermediate migration in XTrack) have a higher chance of switching their tropism. For standardization purposes, this study rated TrofileES results as “correct” and set them as the default. Subsequent investigations will have to demonstrate the validity of this relationship using larger panels of molecularly and clinically defined virus isolates.
However, additional validation came from a European ring trial (First European Collaborative Study on HIV Tropism), where various genotypic and phenotypic tropism systems were assessed on blinded identical sets of 12 virus samples by 36 participating laboratories. Results, as reported by Guertler et al. (39), confirmed the very good overall performance of the XTrack and PhenXR systems.
Future work will be needed for connecting XTrack and Geno2Pheno with results from rPhenotyping in order to further improve test performances. By adaptation of the choice of hybridization primers and through primer shortening utilizing minor-groove-binding modifications, this system will also be adaptable to putative emerging new HIV variants.
It is technically likely that the same test principle may also be applicable toward new pathogens, such as hepatitis viruses and other genetically highly variable entities.
ACKNOWLEDGMENTS
We are grateful to Alex Thielen, Max Planck Institute, Kaiserslautern, Germany, for the provision of great expertise in setting up the “RX algorithm” for bulk data analysis of this study and for valuable discussion, and to François Hamy, InPheno AG, Basel, Switzerland, for terrific discussion and suggestions for the algorithm design. We thank Fabian Otte and Isabell Seibert for their excellent technical contribution to refining the test conditions and Kerstin Asal for matching patients and blinded diagnostic results.
TAK-779 was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
This study was financed in the framework of the Swiss HIV Cohort Study, supported by the Swiss National Science Foundation (grant no. SHCS-553).
The members of the Swiss HIV Cohort Study are V. Aubert, M. Battegay, E. Bernasconi, J. Böni, H. C. Bucher, C. Burton-Jeangros, A. Calmy, M. Cavassini, G. Dollenmaier, M. Egger, L. Elzi, J. Fehr, J. Fellay, H. Furrer (Chairman of the Clinical and Laboratory Committee), C. A. Fux, M. Gorgievski, H. Günthard (President of the SHCS), D. Haerry (deputy of the Positive Council), B. Hasse, H. H. Hirsch, M. Hoffmann, I. Hösli, C. Kahlert, L. Kaiser, O. Keiser, T. Klimkait, R. Kouyos, H. Kovari, B. Ledergerber, G. Martinetti, B. Martinez de Tejada, K. Metzner, N. Müller, D. Nadal, D. Nicca, G. Pantaleo, A. Rauch (Chairman of the Scientific Board), S. Regenass, M. Rickenbach (Head of Data Center), C. Rudin (Chairman of the Mother & Child Substudy), F. Schöni-Affolter, P. Schmid, J. Schüpbach, R. Speck, P. Tarr, A. Telenti, A. Trkola, P. Vernazza, R. Weber, S. Yerly.
Footnotes
Citation Edwards S, Stucki H, Bader J, Vidal V, Kaiser R, Battegay M, Klimkait T, the Swiss HIV Cohort Study. 2015. A diagnostic HIV-1 tropism system based on sequence relatedness. J Clin Microbiol 53:597–610. doi:10.1128/JCM.02762-14.
REFERENCES
- 1.Brumme ZL, Goodrich J, Mayer HB, Brumme CJ, Henrick BM, Wynhoven B, Asselin JJ, Cheung PK, Hogg RS, Montaner JS, Harrigan PR. 2005. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J Infect Dis 192:466–474. doi: 10.1086/431519. [DOI] [PubMed] [Google Scholar]
- 2.Huang W, Eshleman SH, Toma J, Fransen S, Stawiski E, Paxinos EE, Whitcomb JM, Young AM, Donnell D, Mmiro F, Musoke P, Guay LA, Jackson JB, Parkin NT, Petropoulos CJ. 2007. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J Virol 81:7885–7893. doi: 10.1128/JVI.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sucupira MC, Sanabani S, Cortes RM, Giret MT, Tomiyama H, Sauer MM, Sabino EC, Janini LM, Kallas EG, Diaz RS. 2012. Faster HIV-1 disease progression among Brazilian individuals recently infected with CXCR4-utilizing strains. PLoS One 7:e30292. doi: 10.1371/journal.pone.0030292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiser B, Philpott S, Klimkait T, Burger H, Kitchen C, Burgisser P, Gorgievski M, Perrin L, Piffaretti JC, Ledergerber B. 2008. HIV-1 coreceptor usage and CXCR4-specific viral load predict clinical disease progression during combination antiretroviral therapy. AIDS 22:469–479. doi: 10.1097/QAD.0b013e3282f4196c. [DOI] [PubMed] [Google Scholar]
- 5.Esbjornsson J, Mansson F, Martinez-Arias W, Vincic E, Biague AJ, da Silva ZJ, Fenyo EM, Norrgren H, Medstrand P. 2010. Frequent CXCR4 tropism of HIV-1 subtype A and CRF02_AG during late-stage disease—indication of an evolving epidemic in West Africa. Retrovirology 7:23. doi: 10.1186/1742-4690-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymond S, Delobel P, Mavigner M, Cazabat M, Encinas S, Souyris C, Bruel P, Sandres-Saune K, Marchou B, Massip P, Izopet J. 2010. CXCR4-using viruses in plasma and peripheral blood mononuclear cells during primary HIV-1 infection and impact on disease progression. AIDS 24:2305–2312. doi: 10.1097/QAD.0b013e32833e50bb. [DOI] [PubMed] [Google Scholar]
- 7.Hunt PW, Harrigan PR, Huang W, Bates M, Williamson DW, McCune JM, Price RW, Spudich SS, Lampiris H, Hoh R, Leigler T, Martin JN, Deeks SG. 2006. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J Infect Dis 194:926–930. doi: 10.1086/507312. [DOI] [PubMed] [Google Scholar]
- 8.Shepherd JC, Jacobson LP, Qiao W, Jamieson BD, Phair JP, Piazza P, Quinn TC, Margolick JB. 2008. Emergence and persistence of CXCR4-tropic HIV-1 in a population of men from the multicenter AIDS cohort study. J Infect Dis 198:1104–1112. doi: 10.1086/591623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao Y, Tagat JR, Cox K, Priestley T, Sorota S, Huang W, Hirsch M, Reyes GR, Baroudy BM. 2005. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother 49:4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin N, Solomon K, Zhou N, Wang KH, Garlapati V, Thomas B, Li Y, Covington M, Baribaud F, Erickson-Viitanen S, Czerniak P, Contel N, Liu P, Burn T, Hollis G, Yeleswaram S, Vaddi K, Xue CB, Metcalf B, Friedman S, Scherle P, Newton R. 2011. Identification and characterization of INCB9471, an allosteric noncompetitive small-molecule antagonist of C-C chemokine receptor 5 with potent inhibitory activity against monocyte migration and HIV-1 infection. J Pharmacol Exp Ther 338:228–239. doi: 10.1124/jpet.111.179531. [DOI] [PubMed] [Google Scholar]
- 11.Marier JF, Trinh M, Pheng LH, Palleja SM, Martin DE. 2011. Pharmacokinetics and pharmacodynamics of TBR-652, a novel CCR5 antagonist, in HIV-1-infected, antiretroviral treatment-experienced, CCR5 antagonist-naive patients. Antimicrob Agents Chemother 55:2768–2774. doi: 10.1128/AAC.00713-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. 1991. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 13.Rosen O, Sharon M, Quadt-Akabayov SR, Anglister J. 2006. Molecular switch for alternative conformations of the HIV-1 V3 region: implications for phenotype conversion. Proc Natl Acad Sci U S A 103:13950–13955. doi: 10.1073/pnas.0606312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groenink M, Andeweg AC, Fouchier RA, Broersen S, van der Jagt RC, Schuitemaker H, de Goede RE, Bosch ML, Huisman HG, Tersmette M. 1992. Phenotype-associated env gene variation among eight related human immunodeficiency virus type 1 clones: evidence for in vivo recombination and determinants of cytotropism outside the V3 domain. J Virol 66:6175–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang W, Toma J, Fransen S, Stawiski E, Reeves JD, Whitcomb JM, Parkin N, Petropoulos CJ. 2008. Coreceptor tropism can be influenced by amino acid substitutions in the gp41 transmembrane subunit of human immunodeficiency virus type 1 envelope protein. J Virol 82:5584–5593. doi: 10.1128/JVI.02676-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monno L, Saracino A, Scudeller L, Punzi G, Brindicci G, Altamura M, Lagioia A, Ladisa N, Angarano G. 2011. Impact of mutations outside the V3 region on coreceptor tropism phenotypically assessed in patients infected with HIV-1 subtype B. Antimicrob Agents Chemother 55:5078–5084. doi: 10.1128/AAC.00743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyd MT, Simpson GR, Cann AJ, Johnson MA, Weiss RA. 1993. A single amino acid substitution in the V1 loop of human immunodeficiency virus type 1 gp120 alters cellular tropism. J Virol 67:3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry CM. 2010. Maraviroc: a review of its use in the management of CCR5-tropic HIV-1 infection. Drugs 70:1189–1213. doi: 10.2165/11203940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19.Vandekerckhove LP, Wensing AM, Kaiser R, Brun-Vezinet F, Clotet B, De Luca A, Dressler S, Garcia F, Geretti AM, Klimkait T, Korn K, Masquelier B, Perno CF, Schapiro JM, Soriano V, Sonnerborg A, Vandamme AM, Verhofstede C, Walter H, Zazzi M, Boucher CA. 2011. European guidelines on the clinical management of HIV-1 tropism testing. Lancet Infect Dis 11:394–407. doi: 10.1016/S1473-3099(10)70319-4. [DOI] [PubMed] [Google Scholar]
- 20.Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R. 2007. Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol 25:1407–1410. doi: 10.1038/nbt1371. [DOI] [PubMed] [Google Scholar]
- 21.Low AJ, Dong W, Chan D, Sing T, Swanstrom R, Jensen M, Pillai S, Good B, Harrigan PR. 2007. Current V3 genotyping algorithms are inadequate for predicting X4 co-receptor usage in clinical isolates. AIDS 21:F17–D24. doi: 10.1097/QAD.0b013e3282ef81ea. [DOI] [PubMed] [Google Scholar]
- 22.Garrido C, Roulet V, Chueca N, Poveda E, Aguilera A, Skrabal K, Zahonero N, Carlos S, Garcia F, Faudon JL, Soriano V, de Mendoza C. 2008. Evaluation of eight different bioinformatics tools to predict viral tropism in different human immunodeficiency virus type 1 subtypes. J Clin Microbiol 46:887–891. doi: 10.1128/JCM.01611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saliou A, Delobel P, Dubois M, Nicot F, Raymond S, Calvez V, Masquelier B, Izopet J. 2008. Concordance between two phenotypic assays and ultradeep pyrosequencing for determining HIV-1 tropism. Antimicrob Agents Chemother 55:2831–2836. doi: 10.1128/AAC.00091-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi B, Weiser B, Styer LM, Kemal K, Brunner C, Anastos K, Burger H. 2012. A novel denaturing heteroduplex tracking assay for genotypic prediction of HIV-1 tropism. J Virol Methods 185:108–117. doi: 10.1016/j.jviromet.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cabral GB, Ferreira JL, Coelho LP, Fonsi M, Estevam DL, Cavalcanti JS, Brigido LF. 2012. Concordance of HIV type 1 tropism phenotype to predictions using Web-based analysis of V3 sequences: composite algorithms may be needed to properly assess viral tropism. AIDS Res Hum Retroviruses 28:734–738. doi: 10.1089/aid.2011.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delwart EL, Busch MP, Kalish ML, Mosley JW, Mullins JI. 1995. Rapid molecular epidemiology of human immunodeficiency virus transmission. AIDS Res Hum Retroviruses 11:1081–1093. doi: 10.1089/aid.1995.11.1081. [DOI] [PubMed] [Google Scholar]
- 27.Fehr J, Glass TR, Louvel S, Hamy F, Hirsch HH, von Wyl V, Boni J, Yerly S, Burgisser P, Cavassini M, Fux CA, Hirschel B, Vernazza P, Martinetti G, Bernasconi E, Gunthard HF, Battegay M, Bucher HC, Klimkait T. 2011. Replicative phenotyping adds value to genotypic resistance testing in heavily pre-treated HIV-infected individuals—the Swiss HIV Cohort Study. J Transl Med 9:14. doi: 10.1186/1479-5876-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vidal V, Potterat O, Louvel S, Hamy F, Mojarrab M, Sanglier JJ, Klimkait T, Hamburger M. 2012. Library-based discovery and characterization of daphnane diterpenes as potent and selective HIV inhibitors in Daphne gnidium. J Nat Prod 75:414–419. doi: 10.1021/np200855d. [DOI] [PubMed] [Google Scholar]
- 29.Kemal KS, Kitchen CM, Burger H, Foley B, Mayers D, Klimkait T, Hamy F, Anastos K, Petrovic K, Minin VN, Suchard MA, Weiser B. 2012. Recombination between variants from genital tract and plasma: evolution of multidrug-resistant HIV type 1. AIDS Res Hum Retroviruses 28:1766–1774. doi: 10.1089/AID.2011.0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, Shiraishi M, Aramaki Y, Okonogi K, Ogawa Y, Meguro K, Fujino M. 1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A 96:5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, Bridger G, Henson GW. 2000. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother 44:1667–1673. doi: 10.1128/AAC.44.6.1667-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Theodore TS, Englund G, Buckler-White A, Buckler CE, Martin MA, Peden KW. 1996. Construction and characterization of a stable full-length macrophage-tropic HIV type 1 molecular clone that directs the production of high titers of progeny virions. AIDS Res Hum Retroviruses 12:191–194. doi: 10.1089/aid.1996.12.191. [DOI] [PubMed] [Google Scholar]
- 34.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. 1986. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233:215–219. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 35.Gorry PR, Sterjovski J, Churchill M, Witlox K, Gray L, Cunningham A, Wesselingh S. 2004. The role of viral coreceptors and enhanced macrophage tropism in human immunodeficiency virus type 1 disease progression. Sex Health 1:23–34. doi: 10.1071/SH03006. [DOI] [PubMed] [Google Scholar]
- 36.Thielen A, Sichtig N, Kaiser R, Lam J, Harrigan PR, Lengauer T. 2010. Improved prediction of HIV-1 coreceptor usage with sequence information from the second hypervariable loop of gp120. J Infect Dis 202:1435–1443. doi: 10.1086/656600. [DOI] [PubMed] [Google Scholar]
- 37.Sing T, Low AJ, Beerenwinkel N, Sander O, Cheung PK, Domingues FS, Buch J, Daumer M, Kaiser R, Lengauer T, Harrigan PR. 2007. Predicting HIV coreceptor usage on the basis of genetic and clinical covariates. Antiviral Ther 12:1097–1106. [PubMed] [Google Scholar]
- 38.Jensen MA, Li FS, van 't Wout AB, Nickle DC, Shriner D, He HX, McLaughlin S, Shankarappa R, Margolick JB, Mullins JI. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J Virol 77:13376–13388. doi: 10.1128/JVI.77.24.13376-13388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guertler L, Kaiser R, Stuermer M. 2012. Evolution of anti-HIV drug resistance—interpretation and its therapeutic progress. Intervirology 55:98–138. doi: 10.1159/000331998. [DOI] [PubMed] [Google Scholar]
- 40.Tressler R, Valdez H, van der Ryst E, James I, Lewis M, Wheeler J. 2008. Comparison of results from the SensiTrop vs Trofile assays on 100 samples from the maraviroc expanded access program, abstr 920a. Abstr 15th Conf Retroviruses Opportunistic Infect, Boston, MA, 3 to 6 February 2008. [Google Scholar]
- 41.Bauman R, Hamdan H, Schwab D, Robins T, Vichayanonda J, Kagan R. 2009. Quest tropism assay: performance of an HIV-1 co-receptor tropism assay utilizing replicate V3 loop sequencing and heteroduplex analysis with capillary electrophoresis, abstr A1-197. Abstr 18th HIV Drug Resist Workshop, Ft. Myers, FL, 9 to 12 June 2009. [Google Scholar]
- 42.McGovern RA, Thielen A, Mo T, Dong W, Woods CK, Chapman D, Lewis M, James I, Heera J, Valdez H, Harrigan PR. 2010. Population-based V3 genotypic tropism assay: a retrospective analysis using screening samples from the A4001029 and MOTIVATE studies. AIDS 24:2517–2525. doi: 10.1097/QAD.0b013e32833e6cfb. [DOI] [PubMed] [Google Scholar]
- 43.Kuiken C, Foley B, Leitner T, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B. 2010. HIV sequence compendium 2010. Theoretical Biology and Biophysics, Los Alamos, NM. [Google Scholar]
- 44.Delwart EL, Gordon CJ. 1997. Tracking changes in HIV-1 envelope quasispecies using DNA heteroduplex analysis. Methods 12:348–354. doi: 10.1006/meth.1997.0489. [DOI] [PubMed] [Google Scholar]
- 45.Nelson JA, Fiscus SA, Swanstrom R. 1997. Evolutionary variants of the human immunodeficiency virus type 1 V3 region characterized by using a heteroduplex tracking assay. J Virol 71:8750–8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulinge M, Lemaire M, Servais JY, Rybicki A, Struck D, da Silva ES, Verhofstede C, Lie Y, Seguin-Devaux C, Schmit JC, Bercoff DP. 2013. HIV-1 tropism determination using a phenotypic Env recombinant viral assay highlights overestimation of CXCR4-usage by genotypic prediction algorithms for CRF01_AE and CRF02_AG [corrected]. PLoS One 8:e60566. doi: 10.1371/journal.pone.0060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prosperi MC, Bracciale L, Fabbiani M, Di Giambenedetto S, Razzolini F, Meini G, Colafigli M, Marzocchetti A, Cauda R, Zazzi M, De Luca A. 2010. Comparative determination of HIV-1 co-receptor tropism by enhanced sensitivity Trofile, gp120 V3-loop RNA and DNA genotyping. Retrovirology 7:56. doi: 10.1186/1742-4690-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seclen E, Soriano V, Gonzalez MM, Gomez S, Thielen A, Poveda E. 2011. High concordance between the position-specific scoring matrix and geno2pheno algorithms for genotypic interpretation of HIV-1 tropism: V3 length as the major cause of disagreement. J Clin Microbiol 49:3380–3382. doi: 10.1128/JCM.00908-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilkin TJ, Goetz MB, Leduc R, Skowron G, Su Z, Chan ES, Heera J, Chapman D, Spritzler J, Reeves JD, Gulick RM, Coakley E. 2011. Reanalysis of coreceptor tropism in HIV-1-infected adults using a phenotypic assay with enhanced sensitivity. Clin Infect Dis 52:925–928. doi: 10.1093/cid/cir072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brumme CJ, Dong W, Chan D, Mo T, Swenson LC, Woods C, Demarest J, Heera J, Valdez H, Harrigan PR. 2010. O123. Abstr 10th Int Congr Drug Ther HIV Infect Glasgow, United Kingdom, 7 to 11 November 2010 J Int AIDS Soc 13(Suppl 4):K13, O11- 51:P11–237. [Google Scholar]
- 51.Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 359:1429–1441. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Genebat M, Ruiz-Mateos E, Gonzalez-Serna A, Pulido I, Munoz-Fernandez MA, Ferrando-Martinez S, Leal M. 2010. Discordance rates between Trofile test and short-term virological response to maraviroc. Antiviral Res 89:182–185. doi: 10.1016/j.antiviral.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 53.Swenson LC, Moores A, Low AJ, Thielen A, Dong W, Woods C, Jensen MA, Wynhoven B, Chan D, Glascock C, Harrigan PR. 2010. Improved detection of CXCR4-using HIV by V3 genotyping: application of population-based and “deep” sequencing to plasma RNA and proviral DNA. J Acquir Immune Defic Syndr 54:506–510. doi: 10.1097/QAI.0b013e3181d0558f. [DOI] [PubMed] [Google Scholar]
- 54.Xiang SH, Pacheco B, Bowder D, Yuan W, Sodroski J. 2013. Characterization of a dual-tropic human immunodeficiency virus (HIV-1) strain derived from the prototypical X4 isolate HXBc2. Virology 438:5–13. doi: 10.1016/j.virol.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.To SW, Chen JH, Wong KH, Chan KC, Chen Z, Yam WC. 2013. Determination of the high prevalence of dual/mixed- or X4-tropism among HIV type 1 CRF01_AE in Hong Kong by genotyping and phenotyping methods. AIDS Res Hum Retroviruses 29:1123–1128. doi: 10.1089/aid.2013.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M. 2008. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharmacol 73:789–800. [DOI] [PubMed] [Google Scholar]
- 57.Calvo PL, Kansopon J, Sra K, Quan S, DiNello R, Guaschino R, Calabrese G, Danielle F, Brunetto MR, Bonino F, Massaro AL, Polito A, Houghton M, Weiner AJ. 1998. Hepatitis C virus heteroduplex tracking assay for genotype determination reveals diverging genotype 2 isolates in Italian hemodialysis patients. J Clin Microbiol 36:227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]