

Abstract
The hypothalamic arcuate nucleus (ARH) is a brain region critical for regulation of food intake and a primary area for the action of leptin in the CNS. In lean mice, the adipokine leptin inhibits neuropeptide Y (NPY) and agouti-related peptide (AgRP) neuronal activity, resulting in decreased food intake. Here we show that diet-induced obesity in mice is associated with persistent activation of NPY neurons and a failure of leptin to reduce the firing rate or hyperpolarize the resting membrane potential. However, the molecular mechanism whereby diet uncouples leptin's effect on neuronal excitability remains to be fully elucidated. In NPY neurons from lean mice, the Kv channel blocker 4-aminopyridine inhibited leptin-induced changes in input resistance and spike rate. Consistent with this, we found that ARH NPY neurons have a large, leptin-sensitive delayed rectifier K+ current and that leptin sensitivity of this current is blunted in neurons from diet-induced obese mice. This current is primarily carried by Kv2-containing channels, as the Kv2 channel inhibitor stromatoxin-1 significantly increased the spontaneous firing rate in NPY neurons from lean mice. In HEK cells, leptin induced a significant hyperpolarizing shift in the voltage dependence of Kv2.1 but had no effect on the function of the closely related channel Kv2.2 when these channels were coexpressed with the long isoform of the leptin receptor LepRb. Our results suggest that dynamic modulation of somatic Kv2.1 channels regulates the intrinsic excitability of NPY neurons to modulate the spontaneous activity and the integration of synaptic input onto these neurons in the ARH.
Keywords: arcuate nucleus, hypothalamus, Kv channels, Kv2.1, leptin, neuropeptide Y
Introduction
The arcuate nucleus of the hypothalamus (ARH) is critical for regulation of food intake and energy balance. The ARH contains a well described microcircuit consisting of orexigenic neurons coexpressing agouti-related peptide (AgRP) and neuropeptide Y (NPY) and anorexigenic neurons expressing pro-opiomelanocortin (POMC). AgRP neurons directly inhibit POMC neurons; however, there is no reciprocal synapse from POMC neurons to AgRP/NPY neurons (Pinto et al., 2004; Atasoy et al., 2012).
AgRP neurons play a critical role in regulating appetite; the activity of these neurons can profoundly alter food intake: direct optogenetic (Aponte et al., 2010; Atasoy et al., 2012) or pharmacogenetic (Krashes et al., 2011) activation of AgRP neurons in vivo rapidly increases food intake. Conversely, inhibition (Krashes et al., 2011) or ablation (Luquet et al., 2005) of AgRP neurons dramatically decreases feeding. AgRP/NPY neurons are activated by peripheral signals associated with hunger (e.g., ghrelin; Cowley et al., 2003; Takahashi and Cone, 2005; Yang et al., 2011; Liu et al., 2012), whereas peripheral satiety signals (e.g., leptin) inhibit their activity (Takahashi and Cone, 2005). The leptin-dependent inhibition of AgRP/NPY neurons is poorly understood, although it may be attributable in part to modulation of K+ channels such as KATP and BK channels (Spanswick et al., 1997, 2000; Cowley et al., 2001; Mirshamsi et al., 2004; Yang et al., 2010). van den Top et al. (2004) described a K+ conductance in ARH NPY neurons sensitive to the voltage-gated K+ (Kv) channel blocker 4-aminopyridine (4-AP), suggesting a critical role for Kv channels in regulating the intrinsic activity of ARH NPY neurons (van den Top et al., 2004).
In obesity, leptin fails to decrease food intake, despite high levels of circulating hormone, because of insensitivity of ARH neurons that regulate energy balance to respond to leptin (Münzberg et al., 2004; Enriori et al., 2007) likely involving defective leptin receptor (LepRb) signaling (Myers et al., 2008). Interestingly, leptin responsiveness can be restored to ARH neurons after weight loss (Enriori et al., 2007), highlighting the plasticity and resilience of the neural circuits controlling energy balance. Nonetheless, there remains relatively little known regarding the leptin-dependent modulation of the ion channels that determine excitability in ARH neurons, including AgRP and POMC neurons. To our knowledge, a role for voltage-gated ion channels in mediating feeding behavior has never been reported, despite the essential role these channels play in regulating neuronal activity.
In this study, we investigated the diet-dependent excitability of ARH NPY neurons from lean and diet-induced obese (DIO) mice. We found that both fasting and diet-induced obesity increase action potential (AP) frequency, but leptin modulated NPY neuronal excitability only in lean mice. The Kv channel blocker 4-AP was sufficient to prevent leptin-dependent inhibition of NPY neurons, suggesting a role for Kv channels in mediating this effect. Consistent with this, we found a large, leptin-sensitive delayed rectifier-type K+ current whose leptin sensitivity is disrupted in DIO mice. Our data suggest that Kv2.1 is a likely molecular correlate of this current, representing a novel target for leptin signaling in ARH NPY neurons.
Materials and Methods
Animal care
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center. Mice were housed at 22–24°C on a 12 h light/dark cycle. A total of 94 male and female mice were used in this study. Most of the experiments described here used transgenic hrGFP-NPY mice in which humanized Renilla green fluorescent protein (hrGFP) is expressed behind the NPY promoter (van den Pol et al., 2009). Quantitative real-time PCR (qPCR) experiments used wild-type C57BL/6J mice (The Jackson Laboratory).
Control, standard diet (SD)-fed mice were fed a standard pelleted mouse chow (Teklad 7912, 17 kcal% fat, 3.1 kcal/g). To generate diet-induced obesity, littermates of the control group were fed a high-fat diet (HFD; D12451, 45 kcal% fat, 4.5 kcal/g, Research Diets) beginning when the mice were weaned (∼3 weeks of age). All mice were weighed weekly; mice that did not exhibit weight gain compared with the control group were removed from the study. For both groups, food and water were available ad libitum. For the experiments using fasted mice, food was removed from the cage 14–16 h before the experiment; water was freely available. In all experiments, slices were prepared between 10:00 and 11:00 A.M. to ensure that all animals were at the same point in their light cycle to control for circadian variation in feeding.
Cell culture
Low-passage-number HEK293 cells were transfected as described previously with cDNAs encoding either GFP-Kv2.1 or GFP-Kv2.2, with or without LepRb (Baver and O'Connell, 2012). The cells were trypsinized and replated at low density 48 h after transfection to obtain single cells for electrophysiology.
Electrophysiology
Slice preparation.
Adult mice (12–16 weeks old) were deeply anesthetized using isoflurane before decapitation and rapid removal of the entire brain. The brain was then immediately submerged in an ice-cold, oxygenated (95% O2/5% CO2) cutting solution (in mm: 80 NaCl, 90 sucrose, 3.5 KCl, 4.5 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 glucose), and the brain was blocked for sectioning. Coronal slices (250–300 μm thick) were cut using a Vibratome (VT1200S, Leica) and incubated in an oxygenated cutting solution at room temperature for at least 1 h before recording.
Slice recording.
Slices were transferred to a recording chamber constantly perfused (∼2 ml/min) with oxygenated aCSF (in mm: 119 NaCl, 2.5 KCl, 1 MgSO4, 2.5 CaCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 glucose). GFP-positive neurons were identified using epifluorescence and standard GFP filters on a fixed-stage Olympus BX-WI microscope equipped with an XM-10IR CCD camera (Olympus). All recordings were performed using a Multiclamp 700B amplifier and Digidata 1440A and controlled using Clampex 10 (Molecular Devices). Data were digitized at 20 kHz and filtered at 5 kHz using the built-in four-pole Bessel filter of the Multiclamp 700B. After the formation of a gigaohm seal, the pipette capacitance was nulled in all experiments.
Recording pipettes were manufactured from filamented thin-wall borosilicate glass (TW150, World Precision Instruments) and had a resistance of 5–7 MΩ when filled with an intracellular solution (in mm: 140 KCl, 0.3 CaCl2, 1 MgCl2, 1 EGTA, 3 MgATP, 0.3 NaGTP, and 10 HEPES, pH 7.35 with KOH). QX-314 (5 μm) was included in the intracellular solution to block voltage-gated Na+ channels. The liquid junction potential (LJP) between the normal aCSF and the intracellular solution was calculated to be +3.5 mV. Because of the relatively small LJP, the command voltage was not corrected. Normal aCSF was supplemented with 100 μm picrotoxin, 10 μm CNQX, and 20 μm d,l-AP-5 to block the GABAA receptor, AMPAR, and NMDAR unless otherwise noted. In some experiments, 1 μm TTX was added to the aCSF to block voltage-gated Na+ channels in the entire slice. Leptin (100 nm) was added to the aCSF and perfused onto the slice for 60–80 s, after which perfusion with normal aCSF resumed.
For current-clamp recordings of resting membrane potential and spike rate, normal aCSF was used as the extracellular solution, and the intracellular solution was modified to contain 130 K-gluconate and 10 KCl. The LJP between the aCSF and this solution was calculated to be 14.2 mV; membrane potential recordings were corrected for the LJP off-line. In current-clamp experiments measuring the effect of leptin (100 nm) on neuronal rheobase, 20 mm 4-aminopyridine was included with the leptin to block Kv channels. In some experiments, spontaneous APs were recorded in the absence and presence of 300 nm stromatoxin-1 (ScTx-1), a more specific Kv channel blocker. All slice recordings were performed at 32–34°C. Because whole-cell recording necessarily disrupts the neuron's internal milieu, in some experiments, we used cell-attached loose-patch recording (in which the membrane beneath the pipette is not ruptured and the intracellular contents are not disturbed) to measure somatic APs to verify that similar rates were obtained using either technique. In this case, the pipette was filled with regular aCSF (under this configuration, the LJP is 0 mV). Since we observed no difference in spike frequency with either approach within each group, all firing rate data were pooled for statistical analysis. Membrane potential and spontaneous action potential firing was recorded for at least 5 min. Neurons that did not exhibit any spontaneous firing activity within 2 min were not included in the analysis.
In voltage-clamp recordings of whole-cell delayed rectifier K+ currents, P/3 on-line leak subtraction was used to compensate for linear leak currents, and series resistance and capacitance compensation were used. Cells with a series resistance of >20 MΩ were not included in the analysis. To elicit delayed rectifier K+ currents, neurons were held at −80 mV, depolarized to +80 mV in +10 mV steps using a 200 ms test pulse, and repolarized to −40 mV to measure tail current deactivation. A 100 ms prepulse to −30 mV was used to inactivate any residual voltage-dependent Na+ current, except in protocols designed to look for A-type K+ currents. The Nernst potential for K+ using the solutions above was calculated to be −104 mV, and this value was used to convert the ionic currents to conductance (G) using Ohm's law. To generate steady-state conductance–voltage (G–V) curves, the normalized conductance was plotted as a function of the test potential and fitted with a standard Boltzmann equation:
 |
where Gmax is the maximal conductance, V1/2 is the midpoint potential for activation, and k is a slope factor.
HEK cell recording.
Delayed rectifier K+ currents from HEK cells expressing either GFP-Kv2.1 or GFP-Kv2.2 were measured as described previously (Baver and O'Connell, 2012). Cells were exposed to 100 nm leptin for 60 s using a fast-step perfusion system (SF-77B Fast-Step Perfusion System, Warner Instruments), and K+ currents were recorded every 2 min for at least 10 min. In some experiments, endogenous Src-family kinases were pharmacologically inhibited by a 30 min pretreatment with 1 μm SrcI1 and 100 nm PP1 (Tocris), after which delayed rectifier currents were measured as described.
Data analysis and statistics
Action potential frequency, waveforms, and the resting membrane potential were measured using Clampfit 10 (Molecular Devices) and MiniAnalysis 6.0 (Synaptosoft). Neither the action potential waveform nor the resting membrane potential can be analyzed from the data from current-clamp experiments using cell-attached recordings of neuronal spiking, so these cells were excluded from those analyses. Group differences were compared using the nonparametric Kruskal–Wallis ANOVA with Dunn's multiple comparisons post hoc test using Prism 6. Steady-state activation curves were generated by plotting the K+ conductance as a function of the test potential and fitting the resulting G–V curve with the Boltzmann distribution (described above). The resulting fit parameters for each group were compared using the paired Student's t test (SigmaPlot 11 and GraphPad Prism 6). All data are presented as the mean ± SEM. For all statistical tests, a value of p < 0.05 was considered significant.
Real-time PCR
The region of the hypothalamus containing the ARH was microdissected from 1-mm-thick coronal brain sections from wild-type C57BL/6J mice and snap frozen in liquid nitrogen. For each experiment, tissues from 10 animals were pooled. The tissues were subsequently homogenized in Trizol reagent (Invitrogen) to isolate total RNA according to the manufacturer's instructions. A total of 1 μg of RNA was used to synthesize cDNA using the GoScript Reverse Transcription system from Promega. Real-time PCR for Kcnb1 and Kcnb2 was performed using the Roche Universal Probe Library system. Primers were designed using the Roche Assay Design Center ProbeFinder algorithm for Kcnb1 (probe 22) and Kcnb2 (probe 67). The ribosomal protein S19 was used as an internal reference gene; all data reported were normalized to S19 levels.
Immunohistochemistry
Three adult (16 weeks old) male hrGFP-NPY mice from each dietary group (SD:Fed, SD:Fast, and HFD) were anesthetized with isoflurane and decapitated. Brains were removed and fixed at least 24 h in 4% paraformaldehyde at 4°C. Coronal sections (50 μm) containing ARH were cut on a vibrating microtome (VT1000S, Leica). Sections were washed (three times for 15 min) in PBS, treated with 1% w/v sodium borohydride for 30 min, washed, and incubated in 5% normal goat serum for 2 h at room temperature. Slices were incubated in rabbit anti-Kv2.1 polyclonal antibody (1:250, APC-012, Alomone Labs) overnight at 4°C and washed, followed by Alexa Fluor 568 goat anti-rabbit secondary antibody (1:200, catalog #A-11011, Invitrogen) for 8 h at 4°C. Sections were mounted on glass slides with polyvinyl alcohol mounting medium with DABCO antifade (Fluka) for imaging.
Sections were imaged at 1024 × 1024 resolution using a Zeiss 710 confocal microscope, and Z-sections were acquired at 0.69 μm optical section thickness. EGFP was excited using a 488 nm laser, and emission was collected using a variable bandpass filter set at 493–569 nm. Alexa Fluor 568 was excited using a 543 nm laser, and emission was collected using a variable bandpass filter set at 568–712 nm. For each section, the detector gain and offset were optimized to use the full 12-bit linear range, and identical settings were used to image primary- and secondary-only control sections. All images were acquired using ZEN software (Zeiss), and off-line image analysis was performed using the ZEN software and NIH ImageJ.
Results
Diet-induced obesity results in electrical remodeling of ARH NPY/AgRP neurons
It has been demonstrated that the firing rate of NPY/AgRP neurons in the ARH correlates well with feeding behavior. Specifically, NPY neurons fire faster in brain slices from food-deprived mice than in slices from satiated (“fed”) animals (Takahashi and Cone, 2005; Yang et al., 2011; Liu et al., 2012). Furthermore, the adipose tissue-derived hormone leptin decreases the firing rate of NPY neurons (Takahashi and Cone, 2005). In dietary models of obesity, leptin-dependent signaling in ARH neurons is impaired, despite elevated plasma leptin levels (Enriori et al., 2007; Kihira et al., 2010). Diet-induced obesity is associated with a persistent increase in the spike rates of ARH NPY neurons (Diano et al., 2011), consistent with a failure of leptin to appropriately decrease the excitability of these neurons. However, the precise mechanism(s) underlying these effects of leptin on NPY firing in both lean and obese mice remains unclear.
To investigate the effect of diet-induced obesity on NPY neuron activity, we fed male hrGFP-NPY mice a HFD comprising 45 kcal% fat beginning at weaning (around 3 weeks of age). Control, age-matched littermates were fed a standard rodent chow diet (SD). Although the mice used in this study express hrGFP behind the NPY promoter to allow visualization of GFP+ NPY neurons, they were similar to wild type and not prone to obesity when fed a standard rodent chow diet (Fig. 1A, Chow). On the other hand, mice fed the HFD began to exhibit significantly higher body weight after 3 weeks on the diet (p < 0.05) and by week 6 were significantly heavier than SD-fed littermates (p < 0.001; Fig. 1A, HFD).
Figure 1.

Diet-induced obesity increases the firing rate and resting membrane potential of arcuate NPY neurons. A, Body-weight curve for male hrGFP-NPY mice fed either a standard chow or a high-fat diet. The arrow indicates the point at which the mean body weight of the HFD:Fed cohort was statistically greater than the SD:Fed cohort. B, Effect of calorie restriction and diet on spontaneous action potential frequency of NPY neurons in acute brain slices from lean satiated (SD:Fed), fasted (SD:Fast), and DIO (HFD:Fed and HFD:Fast) mice. C, D, Summary plots of firing rate (C) and resting membrane potential (D). All data are presented as mean ± SEM. Statistical significance for the firing rate data was determined using a nonparametric Kruskal–Wallis ANOVA with a post hoc Dunn's multiple comparisons test. A standard parametric ANOVA was used to determine significance of the RMP data. The number of GFP-positive NPY neurons in each group is given in parentheses below each column.
We used the whole-cell current-clamp technique to measure AP firing rates in ARH NPY neurons from acute brain slices containing ARH prepared from lean, SD-fed mice (ad libitum access to food); hungry mice (food deprived for 12–14 h before sectioning); or obese, HFD-fed mice (ad libitum access to HFD). Consistent with previous reports, the action potential frequency (fAP) in NPY neurons from SD-fed mice was slower (Fig. 1B,C; SD:Fed fAP = 0.85 ± 0.1 8 s−1, n = 18) than in food-deprived (“fasted”) control mice (Fig. 1B,C; SD:Fast fAP = 3.46 ± 0.4 s−1, n = 19, p < 0.05). We then measured the firing rate of NPY neurons from fed HFD mice and found that these neurons fired faster than control (SD-fed) neurons at 3.4 ± 0.4 s−1 (n = 25, p < 0.001; Fig. 1B,C) but were similar to the fAP measured in NPY neurons from fasted animals. Although these HFD-fed mice had access to a virtually unlimited food supply, chronic consumption of a high-fat diet induced an electrical remodeling in these neurons resulting in firing behavior reminiscent of neurons in the food-deprived (i.e., hungry) SD:Fast mouse. Interestingly, food deprivation of HFD mice did not result in any further significant increase in the firing rate (fAP = 4.1 ± 0.9 s−1, n = 10; Fig. 1C).
Consistent with these diet-related changes in NPY neuron firing rates, we observed a significant depolarization of the resting membrane potential (RMP) between neurons from SD:Fed and SD:Fast animals (Fig. 1D; RMPfed = −59.4 ± 1.7 mV, n = 12; RMPfast = −50.6 ± 1.4 mV, n = 10; p < 0.001), thereby replicating prior work (Liu et al., 2012). We next investigated the impact of chronic consumption of HFD on membrane potential and found that diet-induced obesity is associated with a similar depolarization of the RMP in both fed and fasted obese animals to −53.4 ± 0.8 mV (n = 20; p < 0.001) and −48.6 ± 2.0 (n = 8; p < 0.001), respectively, compared with SD:Fed controls (Fig. 1D).
We next examined the AP waveform from all three groups more closely, as changes in the underlying intrinsic electrical properties that profoundly affect AP spike rate would likely be reflected in the AP waveform. Since food deprivation did not affect either the mean firing rate or the RMP in ARH NPY neurons from DIO mice, we analyzed only waveforms from HFD:Fed mice. Figure 2A shows average AP waveforms from SD:Fed (black), SD:Fast (red), and HFD:Fed (green) neurons (each trace represents an average of 15–20 APs per condition normalized to the peak of the AP). It is apparent that the spikes from both the SD:Fast and HFD:Fed neurons repolarize (decay) more rapidly than the APs from SD:Fed neurons, indicating that changes in the intrinsic conductances underlying the AP waveform are likely to accompany the increased RMP.
Figure 2.
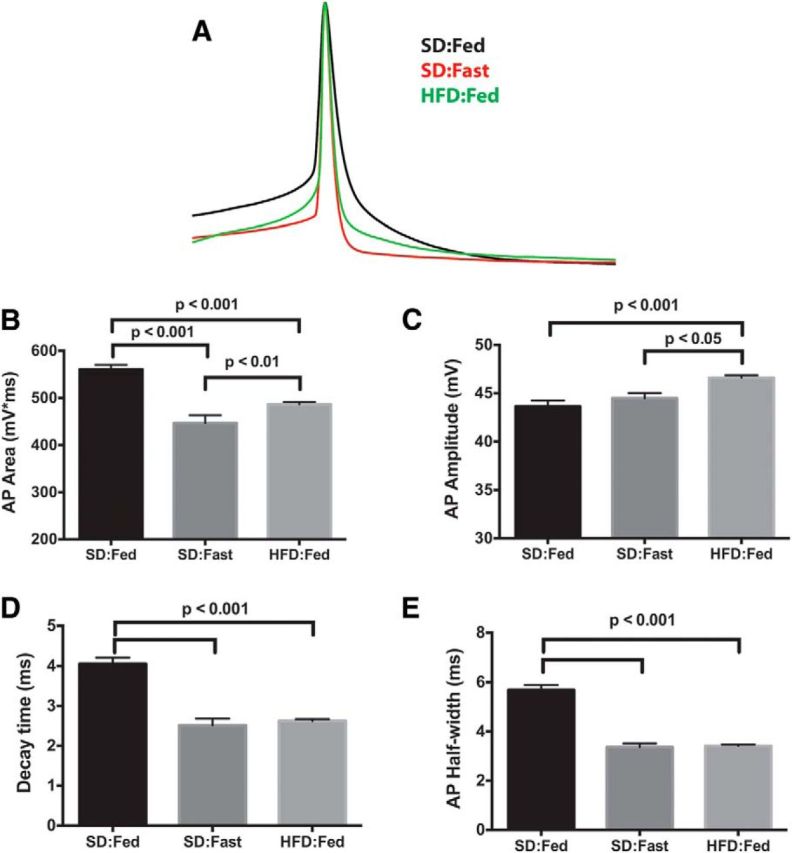
Action potential repolarization is altered in NPY neurons from food-deprived and DIO mice. A, Mean AP waveforms were generated by averaging 15 individual APs from each group. The resulting waveforms were normalized to the peak amplitude and overlaid using the peak for alignment. B, Mean AP area from the point of threshold crossing to return to baseline (positive area only). C, Mean AP amplitude calculated from threshold to peak. D, Mean time for AP to decay e-fold from peak. E, Mean AP width at 50% repolarization.
To quantify these changes, we used the curve-fitting algorithm in Minianalysis 6 to analyze AP waveforms from ∼100 individual APs from the whole-cell current-clamp records of neurons from each group [SD:Fed, 564 events from six cells (three animals); SD:Fast, 384 events from three cells (two animals); HFD:Fed, 1285 events from 13 cells (four animals)]. Consistent with the change in the firing rate (Fig. 1C) and the AP waveform (Fig. 2A), there was a significant decrease in the area of the AP waveform in both the SD:Fast and HFD:Fed neurons compared with the SD:Fed controls (Fig. 2B). This was accompanied by a slight, but statistically significant, decrease in the rise time of the AP in both SD:Fast and HFD:Fed mice (Fig. 2C). Most notably, there was a significant decrease in the decay time (defined as the time required for the AP amplitude to decrease e-fold; Fig. 2D) and the AP half-width (defined as the AP width at half-maximal spike amplitude; Fig. 2E).
Leptin inhibits AgRP/NPY neurons and stimulates anorexigenic POMC neurons, thus promoting increased energy expenditure, decreased food intake, and a negative energy balance. Consistent with previous reports (Takahashi and Cone, 2005), brief application of 100 nm leptin to brain slices from lean, SD:Fast mice resulted in a dramatic decrease in the firing rate (Fig. 3A,C; aCSF alone, 3.2 ± 0.68 s−1; with leptin, 0.38 ± 0.14 s−1; n = 5) as well as a significant hyperpolarization of the RMP (Fig. 3A,D; aCSF alone, −50.5 ± 0.9 mV; with leptin, −64.2 ± 1.9 mV).
Figure 3.

Leptin fails to decrease the spontaneous firing rate or induce hyperpolarization of the RMP in ARH NPY neurons from DIO mice. A, B, Spontaneous APs from NPY neurons in acute brain slices from a lean, fasted mouse (A) or a DIO mouse (B). Leptin (100 nm) was added to the bath solution at the indicated time (arrow). The dotted line is at −65 mV. C, D, Summary bar graph of firing rate (C) and RMP (D). Statistical significance within dietary groups was determined using a paired Student's t test. All data presented as mean ± SEM.
The increased basal firing rate of ARH NPY neurons from DIO mice (Fig. 1B) is consistent with defective leptin signaling, but it is not presently known whether this deficiency in leptin responsiveness extends to the intrinsic electrical activity of NPY neurons. We, therefore, directly assessed the leptin responsiveness of ARH NPY neurons in brain slices from mice fed HFD. As shown in Figure 3, the addition of 100 nm leptin to the bath solution resulted in a significant decrease in the firing rate (Fig. 3A,C) and hyperpolarization of the RMP (Fig. 3D). On the other hand, in brain slices from HFD-fed mice, leptin resulted in a moderate, but not statistically significant, decrease in the spontaneous firing rate of DIO NPY neurons (aCSF alone, 3.8 ± 0.51 s−1; with leptin, 2.0 ± 0.26 s−1, n = 5; p = 0.063; Fig. 3B,C). Notably, this is still approximately twice as fast as the basal spike rate we observed in satiated lean mice (Fig. 1B,C). Interestingly, leptin had no effect on the RMP in NPY neurons from DIO mice (aCSF alone, −54.3 ± 1.6 mV; with leptin, −52.8 ± 0.5 mV; Fig. 3D).
NPY neurons in the ARH exhibit a large delayed rectifier K+ current that is leptin sensitive
Under basal conditions (normal aCSF, −70 mV Vhold), ARH NPY neurons from all dietary groups have similar, relatively high input resistances (SD:Fed, 945.8 ± 131.6 MΩ; SD:Fast, 927.7 ± 189.5 MΩ; HFD:Fed, 1058 ± 133.3 MΩ; HFD:Fast, 797.2 ± 145.7; n.s., p > 0.5, ANOVA). Combined with their relatively small size (Cm = 17.24 ± 0.9 pF; for all groups, no significant difference based on either diet or age was observed), even a small change in input stimulus would be expected to have a large effect on membrane excitability. Consistent with this hypothesis, ARH NPY neurons from lean, fasted animals have a small rheobase. Even small somatic current injections of <10 pA markedly increased the firing rate, with larger depolarizing currents eliciting even faster spike rates (Fig. 4, A,D). In the presence of leptin, a condition under which NPY neurons fire only very slowly, if at all, depolarizing current injection similarly increased the spike rate, but to a lesser extent than under control conditions, and large depolarizing currents elicited only a persistent membrane depolarization, with only one or two spikes at the beginning of the test pulse (Fig. 4, B,D). These data support a model in which leptin activates a hyperpolarizing conductance that effectively “clamps” the membrane potential near EK, thus increasing the rheobase and decreasing the probability of generating an AP.
Figure 4.

Blockade of Kv channels prevents leptin-induced modulation of the intrinsic membrane properties of NPY neurons. A–C, Current-clamp recordings from an ARH NPY neuron in an acute brain slice from a fasted mouse in normal aCSF (A), with 100 nm leptin (B), and with 100 nm leptin and 20 mm 4-AP (C). Black trace, 0 pA; red trace, +10 pA stimulus; green trace, +30 pA stimulus. The dotted line indicates −60 mV. D, Stimulus–response curve for all three conditions. Data are presented as mean ± SEM.
Kv channels exert a profound influence on neuronal firing (Spanswick et al., 1997, 2000; Cowley et al., 2001; Mirshamsi et al., 2004; Cerda and Trimmer, 2010; Yang et al., 2010) and the AP waveform, both of which we observed here. To determine whether Kv channels contribute significantly to the basal firing rate in ARH NPY neurons, we used the Kv channel blocker 4-AP to block K+ flux through Kv channels. Although 4-AP is poorly selective among various Kv channels, it preferentially blocks Kv channels over other nonvoltage-dependent channels, such as KATP channels and other K+ channels (Lesage, 2003; Tamargo, 2004; Hibino et al., 2010). Thus, any effect produced by the addition of 4-AP is most likely attributable to the involvement of Kv channels. When we measured NPY neuronal rheobase in the presence of leptin and 4-AP, we found that 4-AP was able to completely block the effect of leptin on rheobase and restored NPY neuronal firing to levels comparable with that measured in “no leptin” controls (Fig. 4C,D). Strong depolarizing stimuli (>30 pA) tended to decrease the firing rate, most likely because of inhibition of key repolarizing currents in the presence of 4-AP (note the widening of the AP waveform in Fig. 4C). Of note, 4-AP was also able to block the leptin-induced membrane hyperpolarization typically observed in NPY neurons from lean, food-deprived mice (Figs. 3D, 4B), suggesting for the first time that Kv channels also contribute to the leptin-dependent change in membrane potential.
Based on these data, we hypothesized that Kv channels may be a target for LepRb signaling and diet-induced obesity may disrupt this modulation. Therefore, we next investigated the voltage-gated K+ current (IK) in identified NPY neurons from chow- and HFD-fed mice. In acute brain slices from chow-fed mice, NPY neurons had a robust outward IK (Fig. 5A). Although most neurons express multiple Kv channel subunits, their cognate currents can be broadly classified into two main categories based principally on inactivation: (1) rapidly inactivating A-type currents (IA) and (2) slow-inactivating delayed rectifier currents (IDR). As seen in Figure 5A, IK in NPY neurons exhibited biophysical properties consistent with a delayed rectifier-type current: it required relatively strong depolarization to activate (Vthreshold ≈ −25 mV); little to no inactivation occurred during a 200 ms test pulse; and it exhibited slow, voltage-dependent deactivation (τdeact = 22.4 ± 1.4 ms; see tail currents in Fig. 5A,B). Consistent with Roepke et al. (2011), we observed only a small contribution of IA to the total IK in NPY neurons, suggesting that a majority of IK in these cells is likely to be carried by delayed rectifier Kv channel subunits. Unfortunately, there are few pharmacological tools available to dissect voltage-dependent K+ currents; however, IA has a greater sensitivity to 4-AP than does IDR. To confirm this in our system, we used a lower concentration of 4-AP (0.5 mm) that preferentially blocks IA over IDR and measured outward K+ currents before and after the addition of 4-AP. At this lower concentration, we considered the 4-AP-sensitive current to be a rough measure of the total IA in these neurons. As seen in Figure 5C, <50% of the total outward K+ current in NPY neurons is 4-AP sensitive. Note that much of the residual 4-AP-sensitive current exhibits little to no inactivation, suggesting that it is likely to arise from 4-AP-sensitive delayed rectifier-type channels rather than A-type K+ channels. Thus, the bulk of IK in NPY neurons is generated by high-voltage-activated (HVA) delayed rectifier channels.
Figure 5.

Diet-induced obesity mitigates the leptin sensitivity of the delayed rectifier current in NPY neurons. A, B, Representative K+ currents from NPY neurons in brain slices from a lean, SD:Fed mouse (A) and an HFD:Fed mouse (B). C, The relative contribution of delayed rectifier and A-type currents to the total K+ current was ascertained by measurement of voltage-dependent K+ currents before and after bath application of 5 mm 4-AP, followed by off-line subtraction of the 4-AP-sensitive current from the control currents. D, Mean activation midpoints with and without leptin derived from the G–V curves in E–H. The A-type K+ current was measured as the peak current within 10 ms of the onset of depolarization, and the delayed rectifier current was measured as the peak current at the end of the test pulse. Statistical significance within groups was determined by the paired Student's t test, and data are presented as mean ± SEM. The number of GFP-positive NPY neurons in each group is given in parentheses.
Consistent with our hypothesis that IK in NPY neurons is generated by a single population of Kv channels with similar biophysical properties (i.e., principally HVA delayed rectifier channels), the current–voltage (I–V) relationship for IDR in NPY neurons is well described by a single Boltzmann distribution with a membrane potential for half-maximal activation (V1/2) of +9.9 ± 2.6 mV (Fig. 5D,E). A multiple-component fit was not statistically better than a single-component fit. In NPY neurons from lean, food-deprived mice, the voltage dependence of IDR was shifted to more depolarized potentials relative to currents from SD:Fed mice (V1/2 = +15.5 ± 1.7 mV), although this difference was not statistically significant (p = 0.2; Fig. 5D,F).
Although chronic administration of HFD induced obesity and an approximate threefold increase in the firing rate of NPY neurons (Fig. 1B,C), it had no effect on the steady-state parameters of IK in NPY neurons from DIO mice, which exhibited a maximal conductance and voltage dependence similar to those of NPY neurons from SD-fed mice (SD:Fed, Gmax = 180 ± 22 nS vs HFD:Fed, Gmax = 213 ± 14 nS, p = 0.19; Fig. 5B,G). There was no apparent change in the relative contributions of IDR and IA to the total K+ current, as IDR also appeared to comprise a majority of the outward current in cells from obese mice and the I–V relationship was still well described by a single Boltzmann distribution (Fig. 5B,G).
In hippocampal neurons, the modulation of IDR by a variety of stimuli such as glutamate, intracellular Ca2+, and carbachol is an important mechanism for regulating firing rate (Du et al., 2000; Misonou et al., 2004, 2005; Surmeier and Foehring, 2004; Mohapatra et al., 2008, 2009; Baver and O'Connell, 2012). Despite the diversity in the nature of these stimuli, functional modulation occurs by a common mechanism, via induction of a hyperpolarizing shift in the voltage dependence of IDR, which facilitates activation of this key current and decreases somatic excitability. Since leptin potently inhibits spiking in NPY neurons (Fig. 3; Takahashi and Cone, 2005), we investigated whether leptin also modulates IDR in ARH NPY neurons.
As shown in Figure 5, D and E, leptin induced a significant hyperpolarizing shift in the V1/2 for IK within 10 min of administration (V1/2,leptin = −8.1 ± 3.7 mV, p < 0.001 compared with control) in NPY neurons from lean, SD-fed mice. This effect of leptin was persistent; we did not observe any reversal of the shift in V1/2, even up to 1 h after leptin (data not shown). The duration of the shift is not surprising, as the glutamate-induced shift in Ik activation in hippocampal neurons requires nearly 2 h to reverse (Misonou et al., 2004). Importantly, we did not observe a time-dependent shift in the voltage dependence of activation of IDR in control experiments in which only vehicle (PBS) was applied to the slice (data not shown), indicating that this large shift in the voltage dependence of activation of IDR requires leptin. In NPY neurons from lean, food-deprived mice, leptin induced a hyperpolarizing shift of similar magnitude (ΔV1/2,fed = −18 mV; ΔV1/2,fast = −20.5 mV). Note that at steady state, IDR in these neurons was depolarized relative to the SD:Fed controls, thus the activation midpoint after adding leptin was somewhat more positive than observed in neurons from the fed mice (V1/2 = −5.0 ± 1.7 mV).
One consequence of diet-induced obesity is the development of “leptin resistance” in the ARH. We therefore next determined whether leptin could still modulate IDR in NPY neurons from obese mice. Although HFD had no influence on the steady-state parameters of IDR (Fig. 5B–D), the addition of 100 nm leptin to ARH slices from obese mice induced only a slight shift in the activation midpoint of IDR that was not statistically significant (V1/2 = +2.2 ± 1.8 mV, p = 0.07; Fig. 5D,G), indicating that diet-induced obesity blunts, but does not completely eliminate, leptin receptor signaling to delayed rectifier Kv channels in NPY neurons.
Although our data suggest that the total IK in ARH NPY neurons is primarily generated by delayed rectifier-type channels, we also investigated whether leptin modulates IA in NPY neurons. To isolate IA, we recorded currents as described for IDR but omitted the prepulse and used the peak current within 10 ms of the beginning of the test pulse instead of at the end of the test pulse. As shown in Figure 5H, 100 nm leptin had no significant effect on the voltage dependence of IA, with the activation midpoint shifting −5.6 mV from +2.9 ± 2.6 mV to −2.7 ± 4.2 mV. Since it is not possible to pharmacologically dissect IA and IDR, it is possible that contamination from IDR may account for the observed shift.
Kv2.1 channels contribute to the leptin-sensitive delayed rectifier K+ current in NPY neurons
The majority of IK in NPY neurons is a delayed rectifier, HVA current (as shown in Fig. 5), suggesting that the molecular correlate of this current is likely to be a member(s) of the Kv2 or Kv3 families that produces slowly inactivating K+ currents: Kv2.1, Kv2.2, Kv3.1, or Kv3.2 (Kv3.3 and Kv3.4 are A-type channels). Although the currents produced by these four channels have several properties in common, such as slow inactivation and a depolarized voltage dependence of activation, they differ considerably in their deactivation kinetics: Kv2.1 and Kv2.2 deactivate relatively slowly (VanDongen et al., 1990), whereas Kv3.1 and Kv3.2 display ultra-rapid deactivation (Rudy and McBain, 2001). Since the leptin-sensitive IK in NPY neurons deactivates slowly (τdeact = 22.4 ms, compared with <5 ms for Kv3.1 and Kv3.2; Hernández-Pineda et al., 1999), we hypothesized that this current is most likely generated by Kv2.1 or Kv2.2. Unfortunately, the K+ currents generated by Kv2.1 and Kv2.2 are virtually indistinguishable (Kihira et al., 2010), with similar voltage dependences of gating and inactivation as well as nearly identical kinetics for both activation and deactivation. As we and others have previously demonstrated, in HEK293 cells, Kv2.1 is sensitive to many of the same extracellular stimuli the channel is exposed to in the brain (Misonou et al., 2004, 2005; Mohapatra and Trimmer, 2006; Baver and O'Connell, 2012). Kv2.2 has been less well studied, but the channel is robustly expressed in HEK cells (Fig. 6B; Kihira et al., 2010; Baver and O'Connell, 2012), and preliminary studies from our laboratory indicate that in HEK cells, Kv2.2 is also modulated by glutamate in a manner similar to Kv2.1 when coexpressed with the NMDA receptor (data not shown).
Figure 6.

Leptin sensitivity of Kv2-family delayed rectifier channels. A, Top, Representative K+ currents from HEK cells expressing GFP-Kv2.1 with LepRb before and after the addition of 100 nm leptin. Calibration: 5 nA, 50 ms. Bottom, Mean G–V curves for the Kv2.1 current with and without leptin. B, Top, Representative K+ currents from HEK cells expressing GFP-Kv2.2 with LepRb before and after the addition of 100 nm leptin. Calibration: 5 nA, 50 ms. Bottom, Mean G--V curves for the Kv2.2 current with and without leptin. C, Mean G–V curve for Kv2.1 current after Src inhibition by the coaddition of 1 μm SrcI1 and 100 nm PP1 with and without leptin.
As shown in Figure 6, A and B, in HEK cells, both Kv2.1 and Kv2.2 generated large voltage-dependent currents qualitatively similar to IDR in NPY neurons (compare with Fig. 5). Coexpression of Kv2.1 with LepRb had no effect on the voltage dependence of activation of IKv2.1 (V1/2 = +4.0 ± 1.2 mV), but the addition of 100 nm leptin to the bath solution for 60 s induced a significant hyperpolarizing shift in the activation gating of Kv2.1 similar to that observed in NPY neurons (V1/2 = −19.7 ± 2.6, p < 0.001; Fig. 6A). In control cells expressing only Kv2.1 (no LepRb), leptin had no effect on the voltage dependence of Kv2.1 activation (V1/2 = +5.3 ± 1.6 mV). In contrast, in HEK cells coexpressing Kv2.2 and LepRb, 100 nm leptin had no significant effect on the V1/2 of Kv2.2 gating (V1/2 = −3.2 ± 3.6; with leptin, −10.0 ± 2.6; Fig. 6B). These results suggest that Kv2.1 is the most likely molecular correlate of the leptin-sensitive delayed rectifier current in NPY neurons.
Leptin signaling via LepRb is complex, with multiple signal transduction pathways coupled to the activation of LepRb (Villanueva and Myers, 2008). The best characterized is JAK2-dependent phosphorylation of STAT3, the pathway that suppresses expression of the orexigenic peptides Npy and Agrp and promotes expression of the anorexigenic peptide POMC in the ARH (Myers et al., 2008) However, LepRb is coupled to several other signaling pathways, including phosphoinositide-3-kinase (PI3K), ERK, mTOR, and Src (Jiang et al., 2008; Myers et al., 2008; Heida et al., 2010; Heldsinger et al., 2011). Of these known signaling pathways coupled to LepRb, we considered Src to be a likely candidate for LepRb modulation of Kv2.1 as it has recently been established that Kv2.1 is phosphorylated by Src-family kinases (Tiran et al., 2003; Song et al., 2012) and it has been previously demonstrated that PI3K does not regulate Kv2.1 activity (El-Kholy et al., 2003).
To investigate the possible role for Src kinase in leptin-dependent modulation of Kv2.1 activity, we measured voltage-dependent K+ currents in HEK cells coexpressing Kv2.1 and LepRb that were pretreated with the Src inhibitors SrcI1 (1 μm) and PP1 (100 nm) before recording. As shown in Figure 6C, inhibition of Src alone had no significant effect on the voltage dependence of Kv2.1 activation (V1/2 = +9.1 ± 6.9 mV) but completely blocked the hyperpolarization of Kv2.1 activation by LepRb (with leptin, V1/2 = +17.6 ± 7.9 mV). The slight depolarization of the activation midpoint after leptin addition was not statistically significant in this dataset but, nevertheless, may suggest that inhibition of Src activity unmasks some other, as yet unknown LepRb-coupled pathway regulating Kv2.1 activity.
Kv2.1 is highly expressed in the ARH and in NPY neurons
Having established that Kv2.1 is the most likely candidate for the molecular correlate of the leptin-sensitive IDR in ARH NPY neurons, we next set out to verify that Kv2.1 is expressed in these neurons. Previous reports have indicated that Kv2.1 is widely expressed throughout the brain (Hwang et al., 1993), although much less is known about its expression in the hypothalamus, including the ARH. Immunohistochemistry (IHC) performed for Kv2.1 in coronal brain slices from hrGFP-NPY mice indicates that Kv2.1 is highly expressed in the ARH, including in NPY neurons (Fig. 7A). In both cortical and hippocampal neurons, Kv2.1 exhibits a highly restricted distribution to the soma and proximal dendrite, where the channel assembles into high-density clusters (Misonou et al., 2004, 2006; O'Connell et al., 2006; Hermanstyne et al., 2010; Kihira et al., 2010; Baver and O'Connell, 2012) that are recapitulated even in heterologous expression systems such as HEK cells (O'Connell and Tamkun, 2005; Aponte et al., 2010; O'Connell et al., 2010; Baver and O'Connell, 2012). In ARH neurons (including those expressing NPY), Kv2.1 appears to be primarily localized on the soma, with minimal channel expression on the neuritis; however, unlike in cortical neurons (Fig. 7G–I), in the ARH, Kv2.1 does not appear to form the characteristic large clusters typical of this channel. It is instead more evenly distributed over the somatic membrane with smaller clusters than observed in cortical pyramidal neurons (Fig. 7D–F).
Figure 7.

Kv2.1 immunoreactivity in ARH neurons from hrGFP-NPY mice. A–C, Low-magnification view of Kv2.1 (red) in ARH for anatomical reference. Green indicates GFP expression in NPY neurons. Scale bars, 50 μm. D–F, High-magnification image of Kv2.1 alone (red, left) and Kv2.1 with GFP (right). Scale bars, 20 μm. G–I, High-magnification image of Kv2.1 in cortical layer 2/3 pyramidal neurons. Scale bars, 10 μm. J, Relative transcript levels of Kcnb1 (Kv2.1) and Kcnb2 (Kv2.2) in ARH normalized to S19 expression.
Although there are Kv2.2 antibodies commercially available, we were unable to obtain a signal over background when immunostaining for Kv2.2, even after using antigen-retrieval methods (data not shown). It has been reported that, unlike Kv2.1, Kv2.2 expression is more restricted in the mammalian brain (Hermanstyne et al., 2010), raising the possibility that either Kv2.2 is not expressed or not present at high enough levels to be detectable using IHC in the ARH. Nevertheless, to verify this result, we used qPCR of microdissected ARH tissues to probe for mRNA for Kcnb1 and Kcnb2 (the genes for Kv2.1 and Kv2.2, respectively). As shown in Figure 7J, the qPCR signal for Kcnb1 was relatively high, consistent with the IHC results indicating robust expression of Kv2.1 in the ARH. In contrast, the signal for Kcnb2 was barely detectable over the reference gene (S19), consistent with our IHC data suggesting Kv2.2 expression in ARH is low.
Tarantula toxin Stromatoxin-1 significantly increases the basal firing rate of ARH NPY neurons from lean, satiated mice
According to our hypothesis, activation of Kv2.1 by satiety signals such as leptin decreases the basal firing rate of ARH NPY (and thus decreases appetite) by increasing the K+ conductance, opposing depolarization. Therefore, inhibition of Kv2.1 would be predicted to increase the spontaneous firing rate. Although specific blockers of Kv2.1 do not currently exist, there are several spider toxins that have a higher affinity for Kv2- and Kv4-containing channels, with little effect on other K+ channels (Escoubas et al., 2002; Shiau et al., 2003). Stromatoxin-1 (ScTx-1) is a high-affinity (nm) inhibitor of Kv2.1, Kv2.2, and Kv4.2 (Escoubas et al., 2002), but our results here suggest that Kv2.2 and Kv4.2 contribute only minimally to the voltage-dependent K+ current in NPY neurons (Figs. 5C, 6B). Therefore, any effect of ScTx-1 on NPY neuronal firing can be attributed primarily to Kv2.1. As shown in Figure 8, the addition of 300 nm ScTx-1 to acute brain slices from lean, well fed mice resulted in a significant increase in the spontaneous firing rate of NPY neurons, suggesting Kv2.1 plays a key role in setting the firing rate of these neurons.
Figure 8.

Stromatoxin-1 increases the spontaneous firing rate of NPY neurons in brain slices from lean, fed mice. A, B, Representative spontaneous APs recorded in normal aCSF (A) and after bath application of 300 nm ScTx1 (B). C, Summary firing rate data before and after ScTx1 application. Statistical significance was determined using a paired Student's t test. Data are presented as mean ± SEM.
Discussion
In the ARH, AgRP/NPY neurons function as first-order, internal sensory neurons critical for the regulation of food intake and energy balance in mammals (Spanswick et al., 1997; Aponte et al., 2010; Yang et al., 2010; Atasoy et al., 2012). The intrinsic firing rate of these neurons correlates highly with nutritional status (Fig. 1; Takahashi and Cone, 2005; Yang et al., 2011) and is sensitive to peripheral signals of hunger and satiety, such as leptin and ghrelin (Elias et al., 1999; Cowley et al., 2003; Pinto et al., 2004; van den Top et al., 2004; Takahashi and Cone, 2005; Yang et al., 2011). We report here that diet-induced obesity increased the firing rate of ARH NPY neurons from fed mice, consistent with a recent report from Diano et al. (2011). Our data provide a plausible mechanism and a specific molecular target for this effect. Analysis of AP waveforms revealed significant acceleration of repolarization in NPY neurons from obese and food-deprived mice, compared with APs recorded from lean mice, indicative of change in the K+ currents. Indeed, we found a large, leptin-sensitive delayed rectifier K+ current (IDR) in these neurons, which is likely generated by Kv2.1 channels. In parallel with the development of leptin insensitivity in the ARH of obese mice, the leptin responsiveness of IDR was reduced in NPY neurons from DIO mice. Together, our results point to a critical role for somatic Kv2.1 channels in regulating the output of orexigenic AgRP/NPY neurons and thus regulation of energy balance as a whole.
At first glance, it is perplexing that activating a delayed rectifier channel such as Kv2.1 would result in a decrease in the neuronal firing rate. However, key to understanding its unique role in modulating AP frequency is the fact that Kv2.1 is atypical; compared with most other delayed rectifier-type Kv channels, Kv2.1 activates at fairly positive potentials and opens relatively slowly. The net effect of these two properties is that under steady-state conditions, it is unlikely that Kv2.1 opens much at all during a single AP. The threshold for Kv2.1 activation under these conditions is approximately −25 to −20 mV, whereas the voltage-dependent Na+ (Nav) current activates at around −40 mV. Thus, there is more than ample opportunity for sufficient Nav channels (which are responsible for initiation of the AP) to open and depolarize the membrane before the Kv2.1-dependent IDR can act to oppose firing. However, in NPY neurons, leptin shifts the activation of IDR/Kv2.1 such that the threshold for the current is now approximately −50 mV, so stronger depolarizing stimuli are required to reach the threshold for Nav channel activation. In addition, another unique property of Kv2.1 channels is that once open, the channels tend to remain open: Kv2.1 inactivates and deactivates quite slowly, and the resulting persistent K+ efflux acts as a shunt to decrease overall excitability.
The restricted localization of Kv2.1 also plays a part in the importance of the channel in regulating AP frequency. Most, if not all, electrical signals (including those initiating at post-synaptic sites in the dendrites) pass through the soma before integration at the axon initial segment, so the presence or absence of a large somatic K+ conductance can exert a profound influence on the ability of these incoming signals to trigger an AP. At steady state, when Kv2.1 tends to remain closed, the somatic resistance is relatively high, so a small change in somatic current (whether it be from incoming synaptic signals or some intrinsic excitatory stimulus) can result in a comparatively large change in membrane potential, thus readily generating an AP. On the other hand, when IDR/Kv2.1 is active (such as in the presence of leptin), the larger somatic K+ conductance decreases somatic resistance, effectively “shunting” small excitatory stimuli that might otherwise have produced a spike. Such a role for Kv2.1 channels is not surprising; it has been well established that Kv2.1 plays a critical role in shaping the AP and mediating activity-dependent changes in AP frequency (Du et al., 2000; Surmeier and Foehring, 2004; Jacobson et al., 2007; Mohapatra et al., 2009).
Although there are multiple complex signaling pathways coupled to activation of LepRb, we were able to block the leptin-induced hyperpolarizing shift in Kv2.1/IDR activation by inhibiting Src-family kinases, suggesting that Src plays a key role in coupling LepRb and Kv2.1. It has been previously demonstrated that Kv2.1 is phosphorylated by Src (Tiran et al., 2003, 2006; Song et al., 2012), resulting in an increase in current density, with no effect on the voltage dependence of activation. In this study, we observed no change in the maximal K+ conductance, suggesting that LepRb-dependent activation of Src may act as an intermediate step in the modulation of Kv2.1, rather than directly phosphorylating the channel.
Regulation of neuronal excitability is extremely complex, and we acknowledge that Kv2.1 alone is unlikely to be entirely responsible for setting the firing rate of AgRP/NPY neurons. Our results cannot exclude the involvement of other ion channels, including other K+ channels, some of which have been reported to be sensitive to leptin or energy status (Spanswick et al., 1997; Yang et al., 2010; Roepke et al., 2011; Williams et al., 2011). The change we observed in the RMP in fasted and DIO mice may more likely be caused by changes in KATP channel activity rather than Kv channels. That said, the conditions used here (high-glucose, intracellular ATP) were identical between the experimental groups and would likely preclude any significant change in KATP channel activity. Moreover, the addition of 4-AP, which does not efficiently block KATP or other inward rectifier K+ channels, was sufficient to inhibit leptin-induced hyperpolarization of the RMP (Fig. 4). Additionally, the whole-cell current-clamp recordings used slightly different conditions than the voltage-clamp recordings and were generally much shorter (<15 min vs >30 min), which may have influenced the activity of these channels.
Our AP waveform analysis also assumes that the dendritic and/or axonal projections are similar in both the lean and DIO mice: if the projections in the DIO mice are smaller/less extensive, this could decrease the capacitive load and shorten the AP. It is unlikely that such an effect would occur in the fasted mice (which had similar AP properties as the DIO mice), and the projections in DIO mice would have to be substantially shorter, since we likely only have adequate current clamp over the soma and proximal dendrites. Considering that we observe substantial synaptic input onto the NPY neurons from DIO mice (data not shown), we consider a dramatic remodeling of the dendritic tree to be unlikely.
ARH NPY neurons receive substantial excitatory and inhibitory input (Shanley et al., 2001; Pinto et al., 2004; Villanueva and Myers, 2008; Bouret et al., 2012), which certainly plays a significant role in shaping the activity of these neurons. There have been elegant optogenetic (Aponte et al., 2010; Atasoy et al., 2012) and pharmacogenetic (Krashes et al., 2011) approaches developed to investigate both the importance of ARH NPY neurons themselves in feeding behaviors as well as to explore the downstream targets of these neurons. There is also intensive exploration of the afferent inputs to this neuronal population (Pinto et al., 2004; Vong et al., 2011; Yang et al., 2011; Liu et al., 2012). However, the identity and origin of this input is still unknown, making it extremely difficult to target for therapeutic or experimental purposes. On the other hand, Kv2.1 represents a clearly defined molecular target for the potential development of therapeutic approaches for obesity that requires no specific knowledge about the synaptic inputs to ARH NPY neurons. Our results also raise the intriguing possibility that other contributors to intrinsic neuronal excitability (e.g., Nav channels, Cav channels, HCN channels, etc.) may also represent possible targets for modulation by peripheral signals such as leptin and/or the animal's energy state, opening a significant new line of inquiry into the intrinsic function and modulation of these critical neurons that complements the current investigation of the synaptic regulation of these circuits.
Footnotes
We thank Dr. Brad Lowell for the hrGFP-NPY mice; Drs. Cathy Proenza, Roger Bannister, and Dale Oskar for critical reading of this manuscript; and Alana Antoine and Dr. Fu-Ming Zhou for technical assistance.
The authors declare no competing financial interests.
References
- Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nature. 2010;14:351–356. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488:172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baver SB, O'Connell KM. The C-terminus of neuronal Kv2.1 channels is required for channel localization and targeting but not for NMDA-receptor-mediated regulation of channel function. Neuroscience. 2012;217:56–66. doi: 10.1016/j.neuroscience.2012.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Bates SH, Chen S, Myers MG, Jr, Simerly RB. Distinct roles for specific leptin receptor signals in the development of hypothalamic feeding circuits. J Neurosci. 2012;32:1244–1252. doi: 10.1523/JNEUROSCI.2277-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda O, Trimmer JS. Analysis and functional implications of phosphorylation of neuronal voltage-gated potassium channels. Neurosci Lett. 2010;486:60–67. doi: 10.1016/j.neulet.2010.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/S0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, Belsham DD, Sarruf DA, Schwartz MW, Bennett AM, Shanabrough M, Mobbs CV, Yang X, Gao XB, Horvath TL. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med. 2011;17:1121–1127. doi: 10.1038/nm.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Haak LL, Phillips-Tansey E, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv2.1. J Physiol. 2000;522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/S0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- El-Kholy W, Macdonald PE, Lin JH, Wang J, Fox JM, Light PE, Wang Q, Tsushima RG, Wheeler MB. The phosphatidylinositol 3-kinase inhibitor LY294002 potently blocks Kv currents via a direct mechanism. FASEB J. 2003;17:720–722. doi: 10.1096/fj.02-0802fje. [DOI] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Escoubas P, Diochot S, Célérier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- Heida NM, Leifheit-Nestler M, Schroeter MR, Muller JP, Cheng IF, Henkel S, Limbourg A, Limbourg FP, Alves F, Quigley JP, Ruggeri ZM, Hasenfuss G, Konstantinides S, Schafer K. Leptin enhances the potency of circulating angiogenic cells via Src kinase and integrin v 5: implications for angiogenesis in human obesity. Arterioscler Thromb Vasc Biol. 2010;30:200–206. doi: 10.1161/ATVBAHA.109.192807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldsinger A, Grabauskas G, Song I, Owyang C. Synergistic interaction between leptin and cholecystokinin in the rat nodose ganglia is mediated by PI3K and STAT3 signaling pathways: implications for leptin as a regulator of short term satiety. J Biol Chem. 2011;286:11707–11715. doi: 10.1074/jbc.M110.198945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanstyne TO, Kihira Y, Misono K, Deitchler A, Yanagawa Y, Misonou H. Immunolocalization of the voltage-gated potassium channel Kv2.2 in GABAergic neurons in the basal forebrain of rats and mice. J Comp Neurol. 2010;518:4298–4310. doi: 10.1002/cne.22457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Pineda R, Chow A, Amarillo Y, Moreno H, Saganich M, Vega-Saenz de Miera EC, Hernández-Cruz A, Rudy B. Kv3.1-Kv3.2 channels underlie a high-voltage-activating component of the delayed rectifier K+ current in projecting neurons from the globus pallidus. J Neurophysiol. 1999;82:1512–1528. doi: 10.1152/jn.1999.82.3.1512. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Hwang PM, Cunningham AM, Peng YW, Snyder SH. CDRK and DRK1 K+ channels have contrasting localizations in sensory systems. NSC. 1993;55:613–620. doi: 10.1016/0306-4522(93)90427-h. [DOI] [PubMed] [Google Scholar]
- Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammälä CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6:229–235. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Li Z, Rui L. Leptin stimulates both JAK2-dependent and JAK2-independent signaling pathways. J Biol Chem. 2008;283:28066–28073. doi: 10.1074/jbc.M805545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihira Y, Hermanstyne TO, Misonou H. Formation of heteromeric Kv2 channels in mammalian brain neurons. J Biol Chem. 2010;285:15048–15055. doi: 10.1074/jbc.M109.074260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011;121:1424–1428. doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F. Pharmacology of neuronal background potassium channels. Neuropharmacology. 2003;44:1–7. doi: 10.1016/S0028-3908(02)00339-8. [DOI] [PubMed] [Google Scholar]
- Liu T, Kong D, Shah BP, Ye C, Koda S, Saunders A, Ding JB, Yang Z, Sabatini BL, Lowell BB. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron. 2012;73:511–522. doi: 10.1016/j.neuron.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A, Sutherland C, Ashford ML. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 2004;5:54. doi: 10.1186/1471-2202-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005;25:11184–11193. doi: 10.1523/JNEUROSCI.3370-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Menegola M, Mohapatra DP, Guy LK, Park KS, Trimmer JS. Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J Neurosci. 2006;26:13505–13514. doi: 10.1523/JNEUROSCI.3970-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Trimmer JS. The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. J Neurosci. 2006;26:685–695. doi: 10.1523/JNEUROSCI.4620-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Siino DF, Trimmer JS. Interdomain cytoplasmic interactions govern the intracellular trafficking, gating, and modulation of the Kv2.1 channel. J Neurosci. 2008;28:4982–4994. doi: 10.1523/JNEUROSCI.0186-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Münzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- O'Connell KM, Tamkun MM. Targeting of voltage-gated potassium channel isoforms to distinct cell surface microdomains. J Cell Sci. 2005;118:2155–2166. doi: 10.1242/jcs.02348. [DOI] [PubMed] [Google Scholar]
- O'Connell KM, Rolig AS, Whitesell JD, Tamkun MM. Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J Neurosci. 2006;26:9609–9618. doi: 10.1523/JNEUROSCI.1825-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Loftus R, Tamkun MM. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc Natl Acad Sci U S A. 2010;107:12351–12356. doi: 10.1073/pnas.1003028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Roepke TA, Qiu J, Smith AW, Rønnekleiv OK, Kelly MJ. Fasting and 17-estradiol differentially modulate the M-current in neuropeptide Y neurons. J Neurosci. 2011;31:11825–11835. doi: 10.1523/JNEUROSCI.1395-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/S0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. (1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau YS, Huang PT, Liou HH, Liaw YC, Shiau YY, Lou KL. Structural basis of binding and inhibition of novel tarantula toxins in mammalian voltage-dependent potassium channels. Chem Res Toxicol. 2003;16:1217–1225. doi: 10.1021/tx0341097. [DOI] [PubMed] [Google Scholar]
- Song MY, Hong C, Bae SH, So I, Park KS. Dynamic modulation of the Kv2.1 channel by Src-dependent tyrosine phosphorylation. J Proteome Res. 2012;11:1018–1026. doi: 10.1021/pr200770v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Foehring R. A mechanism for homeostatic plasticity. Nat Neurosci. 2004;7:691–692. doi: 10.1038/nn0704-691. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Cone RD. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology. 2005;146:1043–1047. doi: 10.1210/en.2004-1397. [DOI] [PubMed] [Google Scholar]
- Tamargo J, Caballero R, Gómez R, Valenzuela C, Delpón E. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9–33. doi: 10.1016/j.cardiores.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Tiran Z, Peretz A, Attali B, Elson A. Phosphorylation-dependent regulation of Kv2.1 channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J Biol Chem. 2003;278:17509–17514. doi: 10.1074/jbc.M212766200. [DOI] [PubMed] [Google Scholar]
- Tiran Z, Peretz A, Sines T, Shinder V, Sap J, Attali B, Elson A. Tyrosine phosphatases epsilon and alpha perform specific and overlapping functions in regulation of voltage-gated potassium channels in Schwann cells. Mol Biol Cell. 2006;17:4330–4342. doi: 10.1091/mbc.E06-02-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, Broberger C. Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. J Neurosci. 2009;29:4622–4639. doi: 10.1523/JNEUROSCI.3249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Top M, Lee K, Whyment AD, Blanks AM, Spanswick D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat Neurosci. 2004;7:493–494. doi: 10.1038/nn1226. [DOI] [PubMed] [Google Scholar]
- VanDongen AM, Frech GC, Drewe JA, Joho RH, Brown AM. Alteration and restoration of K+ channel function by deletions at the N- and C-termini. Neuron. 1990;5:433–443. doi: 10.1016/0896-6273(90)90082-Q. [DOI] [PubMed] [Google Scholar]
- Villanueva EC, Myers MG. Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 2008;32:S8–S12. doi: 10.1038/ijo.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Sohn JW, Donato J, Jr, Lee CE, Zhao JJ, Elmquist JK, Elias CF. The acute effects of leptin require PI3K signaling in the hypothalamic ventral premammillary nucleus. J Neurosci. 2011;31:13147–13156. doi: 10.1523/JNEUROSCI.2602-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MJ, Wang F, Wang JH, Wu WN, Hu ZL, Cheng J, Yu DF, Long LH, Fu H, Xie N, Chen JG. PI3K integrates the effects of insulin and leptin on large-conductance Ca2+-activated K+ channels in neuropeptide Y neurons of the hypothalamic arcuate nucleus. Am J Physiol Endocrinol Metab. 2010;298:E193–E201. doi: 10.1152/ajpendo.00155.2009. [DOI] [PubMed] [Google Scholar]
- Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 2011;146:992–1003. doi: 10.1016/j.cell.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]