

Abstract
Sporadic amyotrophic lateral sclerosis (ALS) is a fatal disease with unknown etiology, characterized by a progressive loss of motor neurons leading to paralysis and death typically within 3–5 years of onset. Recently, there has been remarkable progress in understanding inherited forms of ALS in which well defined mutations are known to cause the disease. Rodent models in which the superoxide dismutase-1 (SOD1) mutation is overexpressed recapitulate hallmark signs of ALS in patients. Early anatomical changes in mouse models of fALS are seen in the neuromuscular junctions (NMJs) and lower motor neurons, and selective reduction of toxic mutant SOD1 in the spinal cord and muscle of these models has beneficial effects. Therefore, much of ALS research has focused on spinal motor neuron and NMJ aspects of the disease. Here we show that, in the SOD1G93A rat model of ALS, spinal motor neuron loss occurs presymptomatically and before degeneration of ventral root axons and denervation of NMJs. Although overt cell death of corticospinal motor neurons does not occur until disease endpoint, we wanted to establish whether the upper motor neuron might still play a critical role in disease progression. Surprisingly, the knockdown of mutant SOD1 in only the motor cortex of presymptomatic SOD1G93A rats through targeted delivery of AAV9–SOD1–shRNA resulted in a significant delay of disease onset, expansion of lifespan, enhanced survival of spinal motor neurons, and maintenance of NMJs. This datum suggests an early dysfunction and thus an important role of the upper motor neuron in this animal model of ALS and perhaps patients with the disease.
Keywords: ALS, amyotrophic lateral sclerosis, motor neuron disease, neurodegenerative disorder, RNAi, SOD1
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by progressive loss of upper and lower motor neurons, typically leading to muscle atrophy, paralysis, and death within 3–5 years. Disease etiology remains unclear, with multiple causes of cell death under investigation (Ling et al., 2013). There is no effective treatment, and several promising drugs have failed recently in clinical trials (Goyal and Mozaffar, 2014). Most ALS cases present as sporadic, and, of the remaining familial cases, ∼20% are linked to mutations in the superoxide dismutase-1 (SOD1) gene. In addition, there is great interest in the recently discovered C9orf72 mutation (DeJesus-Hernandez et al., 2011; Renton et al., 2011). However, little is known about how this mutation causes disease, and there is no established animal model. Instead, transgenic rodents overexpressing mutant human SOD1 (SOD1G93A) recapitulate the hallmarks of ALS. This knock-in model and the fact that the absence of SOD1 does not cause motor neuron disease provide evidence for an acquired toxicity of mutant SOD1 (Gurney et al., 1994; Turner and Talbot, 2008).
Reducing mutant SOD1 can delay disease onset and extend survival in transgenic SOD1 mice. Nonretrogradely transported lentivirus-encoded siRNA to SOD1, injected into the muscle of transgenic mice, does not affect disease onset or progression (Miller et al., 2006). In contrast, retrogradely transported RNAi, injected into the muscle (Miller et al., 2005; Ralph et al., 2005) or directly into the spinal cord (Raoul et al., 2005; Foust et al., 2013) to knock down mutant SOD1 in spinal motor neurons, provides improved functional outcome, delayed disease onset, and extended survival. However, these experiments did not assess retrograde knockdown of SOD1 in the motor cortex nor survival of corticospinal motor neurons, leaving open the possibility that mutant SOD1 was significantly knocked down in the brain at an early age to contribute to the observed beneficial effects.
ALS research has primarily focused on the lower motor neurons and connectivity with the neuromuscular junction (NMJ), with little focus on upper motor neuron pathology. The SOD1G93A mouse model shows that NMJs are denervated early in disease progression (Fischer et al., 2004). However, transcranial magnetic stimulation of familial and SOD1-carrying ALS patients has established that cortical hyperexcitability occurs early in disease and even at presymptomatic stages and is linked to neurodegeneration (Vucic and Kiernan, 2006; Vucic et al., 2008, 2011). Furthermore, the mouse ALS model has shown that corticospinal motor neurons display early, presymptomatic signs of degeneration (Ozdinler et al., 2011). However, this aspect of disease onset and progression remains unclear. Given this suggestion of upper motor neuron involvement with ALS, we performed a detailed study of the progression of disease pathology in both upper and lower motor neurons using another larger rodent model of ALS: the SOD1G93A rat (Howland et al., 2002). Additionally, we used short hairpin RNA (shRNA) to knock down mutant SOD1 expression in only the motor cortex to establish how this region including the upper motor neurons may contribute to disease. This resulted in delayed disease onset, extended survival, and amelioration of both spinal motor neuron death and denervation of NMJs. Our findings highlight the importance of the motor cortex in ALS.
Materials and Methods
Animals.
Wild-type (WT) and SOD1G93A transgenic rats (Sprague Dawley background) were housed under National Institute of Health guidelines, and all experiments were conducted in accordance with the Cedars-Sinai Institutional Animal Care and Use Committee (IACUC Protocol 4260) and the Guide for the Care and Use of Laboratory Animals. This colony provides later onset and endpoint than the original model published by Howland et al. (2002). Reminiscent of human pathology, disease onset in hindlimbs and/or forelimbs is unpredictable in this model with overt paresis progressing to complete paralysis. Significant atrophy of trunk and neck muscles is also observed in some animals. No significant differences were observed in WT rats over time, so data were pooled. Male and female SOD1G93A rats showed no significant differences, so data were also pooled.
High-copy SOD1G93A mice were obtained from The Jackson Laboratory and bred within the laboratory of B.K.K. All procedures performed were in accordance with the National Institutes of Health guidelines and approved by the Research Institute at Nationwide Children's Hospital (IACUC Protocol AR11-00100). Animals were genotyped before the treatment to obtain SOD1G93A-expressing mice and their WT littermates.
Assessment of motor behavior.
The Basso, Beattie, and Bresnahan (BBB) locomotor rating scale (Basso et al., 1995) is used commonly to assess an animal's ability to walk around its environment and has been used to quantify the degree of limb paralysis in SOD1G93A rats. The 21-point BBB scoring is an open-field locomotor test of limb function, with a 21 score indicating coordinated limb movement, consistent toe clearance, and parallel paw placement, and a 0 score indicating no observable limb movement. BBB locomotor ratings provide an indication of when paralysis starts in any limb and the degree of progression continuing until the animal's endpoint.
SOD1G93A female and male rats (n ≥ 6) were randomized into groups for postnatal day 90 (P90), P120, early symptomatic (defined as when rats have lost 10–15% of their peak body weight), and endpoint (defined as when rats can no longer right themselves within 25 s after being placed on their side). Starting at ∼100 d and continuing until disease endpoint, an observer blinded for genotype and treatment assessed animals once or twice weekly for BBB scores and body weights to determine the average age of onset and survival (endpoint group only). If left and right limb scores were different, both scores were recorded but the average for each (hind/fore) limb were taken for the rat's overall score. Disease onset was classified as when an animal displayed a BBB score of ≤15.
Tissue collection.
Tissue for anatomical characterization was collected from animals euthanized with a ketamine/xylazine mixture and transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde (PFA). Brain, spinal cord, dorsal/ventral roots (lumbar L3, L4, and L5), and muscle tissue were postfixed in PFA overnight and stored in 30% sucrose. Brains and spinal cords were sectioned at 35 μm using a microtome and collected as free-floating sections for immunohistochemistry. Muscle tissue was sectioned, using a cryostat, directly onto precoated slides at 25 μm for immunohistochemistry.
Analysis of corticospinal motor neurons.
To identify corticospinal motor neurons, sections were stained for COUP-TF-interacting protein 2 (CTIP2; 1:250; Abcam) that stains the nuclei of motor neurons in layer V and fluorescent Nissl (Neurotrace; Life Technologies) to stain cell bodies. Before stereological analysis, fluorogold injections in the lumbar spinal cord were performed on a subset of animals to define hindlimb representation regions within the motor cortex. Based on fluorogold labeling in the cortex, stereological analysis of CTIP2-expressing (CTIP2+) cells was performed on three 35 μm sections at 420 μm apart, starting at 1 mm posterior to bregma. For each section, a standard size rectangular contour (dimensions, 300 μm height × 1400 μm width) was drawn encompassing layer V of the cortex (as determined by initial fluorogold experiments). The optical fractionator method was used with a counting frame of 100 × 100 μm and grid size of 200 × 200 μm (yielding 14–18 counting sites per section) with Gunderson coefficient of error <0.08. CTIP2+ cells were counted and cell bodies were traced to measure cell size using Neurotrace (fluorescent Nissl) staining and the nucleator method (MBF Bioscience software).
Analysis of spinal motor neurons, astrocytes, and microglia.
Spinal cord sections were immunostained using primary antibodies against choline acetyltransferase (ChAT; goat, 1:250; Millipore) for spinal motor neurons, glial fibrillary acidic protein (GFAP; mouse, 1:1000; Millipore) for astrocytes and ionized calcium binding adaptor molecule 1 (Iba1; rabbit, 1:1000; Wako) for microglia, followed by appropriate secondary antibodies. One 20× image stack per section (eight sections total, 420 μm apart) was captured encompassing the lateral ventral horn of the lumbar (L4–L5) spinal cord. ChAT+ cells were counted to determine total spinal motor neuron numbers, and cell body size was measured using NIH Image J software.
Analysis of L5 ventral root axons.
L5 ventral root axons were collected and postfixed in 3% glutaraldehyde for at least 2 d before staining for osmium tetroxide (2%). Samples were washed, dehydrated, embedded, sectioned at 1 μm, and stained for toluidine blue. Axons were quantified stereologically at 63× (using a counting frame of 75 × 75 μm and grid size of 150 × 150 μm) and classified as healthy (as defined by intact myelin sheath and clear lumen) or unhealthy based on the appearance of degeneration.
Analysis of NMJs.
NMJs were stained using α-bungarotoxin (1:100; Life Technologies) to label acetylcholine receptors located at the subsynaptic membrane and an antibody against synaptic vesicle protein 2 (SV2; mouse, 1:25; Developmental Studies Hybridoma Bank) to label synaptic vesicle proteins. NMJs were classified as fully innervated, semi-innervated, or denervated based on the extent of overlap of α-bungarotoxin and SV2 staining.
Pseudorabies virus.
For transneuronal tracing of the pathway providing input to gastrocnemius motor neurons, we used a transgenic recombinant of an attenuated pseudorabies virus strain (PRV-614). PRV-614 is derived from PRV-Bartha, which is an attenuated form of the parental strain PRV-Becker. PRV-614 carries the gene coding for red fluorescent protein (RFP) at the gG locus, which is constitutively expressed under control of the cytomegalovirus immediate early promoter (Smith et al., 2000). Previous studies have demonstrated that this virus strain is transported trans-synaptically in a retrograde manner (Card et al., 1993, 1998). Viral recombinants, kindly provided by Dr. Lynn Enquist (Princeton University, Princeton, NJ; Virus Center Grant P40RR018604), were harvested from pig kidney cell cultures at a titer of 108 pfu/ml, and a single 30 μl injection was made into the right gastrocnemius of WT and SOD1G93A rats at ∼90 and 120 d of age.
Injections of AAV9–GFP–SOD1–shRNA.
Presymptomatic rats at P70 were injected with adeno-associated virus serotype 9 containing the green fluorescent reporter protein (AAV9–GFP) detailed by Foust et al. (2009) or with AAV9–GFP–SOD1–shRNA (AAV9–SOD1–shRNA; SOD1–shRNA: –CATGGATTCCATGTTCATGA–) detailed by Foust et al. (2013). Virus particles at 5 × 1012 were injected in 2 μl of viral solution per site using a glass micropipette attached to a 10 μl Hamilton syringe, controlled by a Hamilton microinjection pump. For the initial knockdown experiment, bilateral injections were made at lateral × medial stereotaxic coordinates from bregma as follows: (1) 2 × 1 mm; (2) 2 × 0 mm; (3) 2 × −1 mm; (4) 2 × −2 mm; (5) 3 × 1 mm; (6) 3 × 0 mm; (7) 3 × −1 mm; and (8) 3 × −2 mm. These 16 injections were administered at a depth of 1.4 mm, relative to the brain surface. The second knockdown experiment consisted of 20 total injections through additional bilateral coordinates: (9) 2 × 2 mm; and (10) 3 × 2 mm.
To detect possible anterograde transport of AAV9 from brain to spinal cord, spinal cord sections were immunostained using an antibody against GFP (rabbit polyclonal or goat polyclonal, 1:500; Abcam). ChAT+ motor neurons, GFAP+ astrocytes, and Iba1+ microglia cell populations (in eight sections as described above) were quantified for GFP expression to assess the extent of viral transduction. Knockdown of misfolded SOD1 protein was assessed in brain and spinal cord sections using a D3H5 antibody (1:250; kindly supplied by Dr. Jean-Pierre Julien, Quebec, Quebec, Canada).
Laser-capture microdissection.
Lumbar spinal cord and brain frozen sections (12 μm) were collected onto polyethylene naphthalate membrane slides (Zeiss) and stained with 1% cresyl violet (Sigma) in methanol. Sections were air dried and stored at −80°C. After thawing, motor neurons were collected within 30 min from staining using the laser-capture microdissector (PALM Robo3; Zeiss) with the following settings: cut energy, 48; laser power controller (LPC) energy, 20; cut focus, 80/81; LPC focus, 1; position speed, 100; cut speed, 50. Approximately 300 lumbar motor neurons were collected per animal from the ventral horn, and ∼500 motor neurons were collected from layer V in the motor cortex.
qRT-PCR.
RNA was isolated from pooled laser-captured cells using the RNaqueous Micro kit (Ambion) according to manufacturer instructions. The RT2 HT First Strand kit was used for RT of RNA into cDNA (SABiosciences). A total of 12.5 ng of RNA were used in each qPCR reaction using SYBR Green (Invitrogen) to establish the relative quantity of human SOD1 and GFP transcripts in animals who had received the AAV9–SOD1–shRNA compared with control animals. Each sample was run in triplicate, and relative concentration was calculated using the ddCt values normalized to endogenous actin transcript.
Viral genome particles quantification.
DNA was isolated from ∼500 pooled laser-captured corticospinal motor neurons from rats injected with AAV9–SOD1–shRNA and controls using the QIAamp DNA Mini kit. Absolute quantification of viral genome (vg) particles per micrograms of sample DNA was calculated interpolating the cycle threshold Ct values obtained with a standard curve ranging from 25 to 2 × 106 copies of viral plasmid.
Inclusion criteria for behavioral and histological analysis.
Injections in the first knockdown experiment targeted the entire hindlimb motor region and not the entire forelimb motor region. It was not surprising that analysis of forelimb motor function revealed no differences in onset or behavioral function. However, there was evidence for delayed hindlimb onset and improved hindlimb behavioral function in AAV9–SOD1–shRNA-injected rats versus AAV9–GFP controls. However, this was not significant, likely because of the inclusion of data from rats exhibiting initial forelimb onset (data not shown). Because of the lack of target injections to the entire forelimb motor cortex region, we excluded rats (in both AAV9–GFP and AAV9–SOD1–shRNA groups) from behavioral analyses that showed exclusive forelimb onset. Therefore, SOD1G93A rats included in analysis for hindlimb BBB, onset, and survival either exhibited onset in hindlimb only (n = 5, exclusively the AAV9–GFP control group) or simultaneous forelimb/hindlimb onset (AAV9–GFP controls, n = 5; and AAV9–SOD1–shRNA, n = 7) for a total n = 10 AAV9–GFP controls and n = 7 AAV9–SOD1–shRNA. For the second knockdown experiment targeting both forelimb and hindlimb motor regions, in each group, only rats that had reached peak body weight and had begun to consistently lose weight were included in the behavioral and histological analyses (n = 4 AAV9–SOD1–shRNA, n = 4 non-injected controls).
AAV9-scrambled vector construction.
A scrambled shRNA (–GACTTGACCGACTTATGTAT–) construct was generated using Life Technologies design tool. The shRNA construct was first cloned into pRNA–H1 neo vector under human H1 promoter and tested in vitro. Scrambled–shRNA–SOD1 along with the H1 promoter was then PCR amplified and cloned at Kpn1 sites in AAV9–CBA–GFP vector carrying GFP under a chicken β-actin promoter.
Transfection.
Human embryonic kidney 293 (HEK293) cells were transfected using the calcium phosphate method with AAV9–scrambled–SOD1–shRNA plasmid to determine the effect on human SOD1 protein levels. Cell lysates were harvested 72 h after transfection and analyzed for the levels of human SOD1 protein by Western blot analysis (rabbit anti-hSOD1 at 1:750, Cell Signaling Technology; mouse anti-GAPDH at 1:1000, Millipore; anti-rabbit horse radish peroxidase (HRP) at 1:25000 or anti-mouse HRP at 1:10000, Jackson ImmunoResearch). AAV9–CBA–GFP and AAV9–SOD1–shRNA were used as negative and positive controls for SOD1 downregulation, respectively, and GAPDH was used for protein normalization.
Mouse injections.
For neonatal mouse injections, P1 SOD1G93A pups (n = 5) were used. A total volume of 50 μl containing 5 × 1011 (3.6 × 1014 vg/kg) DNase-resistant viral particles of AAV9–scrambled–SOD1–shRNA (Virapur) was injected through the temporal vein as described previously (Foust et al., 2009). After a correct injection was verified by blanching of the vein, pups were returned to their cage. For adult tail vein injections, SOD1G93A mice (n = 4) at P21 were placed in a restraint that positioned the tail in a lighted, heated groove. The tail was swabbed with alcohol and then injected intravenously with 200 μl viral solution containing 2 × 1012 DNase-resistant viral particles of AAV9–scrambled–SOD1–shRNA, for an average dose of 1.7 × 1014 vg/kg.
Mouse behavior and survival analysis.
Treated and non-injected control SOD1G93A mice were monitored throughout the disease course compared with non-injected control mice (n = 7). Mice were subjected to weekly assessment of hindlimb grip strength using a grip-strength meter (Columbus Instruments). Each weekly session consisted of three tests per animal. Onset and survival analyses were performed using Kaplan–Meier analyses. Endpoint was defined as the death point when an animal could no longer regain their upright position within 30 s after being placed on their back. Disease onset was determined from retrospective analysis of the data and defined as the age at which the animal reached its peak weight.
Statistical analysis.
Statistical analyses were performed using Prism software (GraphPad Software). Student's t tests, and one-way and two-way ANOVA using Bonferroni's post hoc analyses were performed to determine standard error of the mean (SEM) with a 95% confidence level. Kaplan–Meier survival analyses were analyzed by the log-rank test, and comparisons of median disease durations and survival times were analyzed by the Wilcoxon's signed-rank test.
Results
Spinal motor neurons degenerate presymptomatically
Although the disease onset and death occurring in SOD1G93A rodent models of ALS have been well characterized, there exist species and even colony differences. Hence, before initiating our detailed study of the progression of disease pathology in lower and upper motor neurons, we needed to characterize disease onset and survival in our colony of SOD1G93A rats used to model ALS. We showed that disease onset ranged from 140 to 201 d, with a median of 154 d (Fig. 1A), and that survival ranged from 147 to 218 d, with a median survival age of 172 d (Fig. 1B). Because no rats exhibited signs of paralysis at P120, both P90 and P120 were classified as “presymptomatic” time points for our subsequent analyses.
Figure 1.
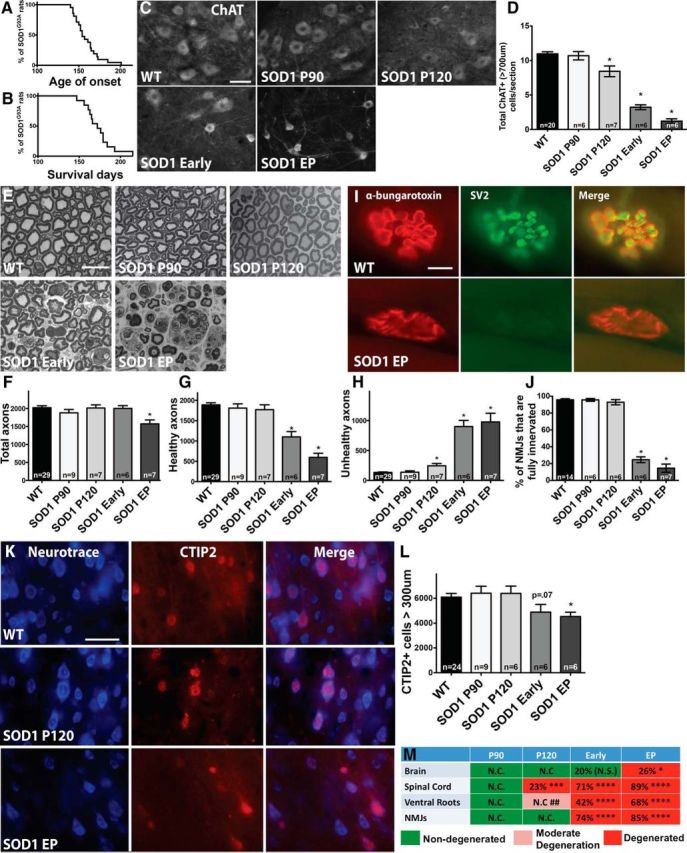
Spinal motor neurons are lost presymptomatically, before corticospinal motor neuron loss and before significant axon and muscle degeneration in the SOD1G93A rat. P90 and P120 were designated “presymptomatic” time points based on Kaplan–Meier analyses showing that disease onset for rats in this study ranged from 140 to 201 d, with a median of 154 d (n = 21) (A) and survival ranged from 147 to 218 d, with a median of 172 d (n = 13) (B). C, Immunostaining for ChAT+ spinal motor neurons in the lumbar spinal cord at P90, P120, early symptomatic, and endpoint (EP) showed qualitative changes between WT rats and SOD1G93A rats over time. D, Quantification revealed a significant reduction of large (>700 μm) ChAT+ spinal motor neurons starting at presymptomatic time point P120. E, Osmium tetroxide and toludine blue staining provided qualitative analysis of (L5) ventral root axons in WT versus SOD1G93A rats over time. Stereological quantification revealed that the number of total axons was not significantly reduced until rats reach endpoint (F), whereas the number of healthy axons is first significantly reduced when rats appear symptomatic (G) and a slight yet significant increase in degenerating (“unhealthy”) axons is seen starting at P120 (H). I, Immunostaining for α-bungarotoxin and SV2 in the muscle of WT versus SOD1G93A rats. J, Quantification revealed that NMJs do not become significantly denervated until the early symptomatic stages, after spinal motor neuron loss. K, Immunostaining of motor cortex layer V sections using CTIP2 that specifically stains nuclei of motor neurons, and Neurotrace, a fluorescent Nissl stain, in WT and SOD1G93A rats. L, Quantification revealed that the number of large corticospinal motor neurons was declined at early symptomatic stages and was significantly reduced at disease endpoint. Scale bars: C, K, 50 μm; E, I, 10 μm. *p < 0.05; error bars indicate SEM. M, Summary of the time course of degeneration in SOD1G93A rats showing the percentage of corticospinal motor neuron loss in the brain, spinal motor neuron loss, loss of healthy ventral root axons, and degenerated NMJs: *p < 0.05, **p < 0.001, ***p < 0.0001, ## indicates moderate degeneration designated attributable to a slight yet significant increase in numbers of unhealthy axons. N.C., No change; N.S., not significant.
We next examined the time course of degeneration of the widely studied spinal (“lower”) motor neurons within the lumbar spinal cord region of SOD1G93A rats. Quantifying the total number of ChAT+ cells in the ventral horn showed that spinal motor neurons in SOD1G93A rats were significantly reduced starting presymptomatically at P120 with an 11% reduction relative to WT rats and that there was an additional loss to 35% by endpoint (data not shown). Quantifying only the large (>700 μm) subpopulation of total spinal motor neurons in SOD1G93A rats showed an even more dramatic loss with a significant 23% decrease starting at P120 relative to WT rats and an 89% depletion by endpoint (Fig. 1C,D). We also assessed the cell size of spinal motor neurons that were slightly decreased presymptomatically at P90 and P120, reaching significance with 32% decrease at early symptomatic and 52% decrease at endpoint (data not shown).
Ventral root axons show degeneration and NMJs are denervated only after spinal motor neuron loss in symptomatic rats
We next assessed degeneration in L5 ventral root axons over time in SOD1G93A rats. Sections of ventral root axons stained with osmium tetroxide and toluidine blue (Fig. 1E) were quantified for total numbers of healthy and unhealthy axons. We found that there was no significant loss in the total number of axons (healthy plus unhealthy) relative to WT until disease endpoint (Fig. 1F). Numbers of healthy axons (as defined by intact myelin sheath and clear lumen) did not decrease until the early symptomatic stage (Fig. 1G). However, we did observe a significant increase in the number of apparently degenerating, “unhealthy” axons (damaged myelin sheath, dark lumen) beginning at P120 (Fig. 1H).
We also examined NMJs in the gastrocnemius over time by staining for α-bungarotoxin and SV2. NMJs were classified as fully innervated, semi-innervated, or denervated based on the extent of overlap of these two markers (Fig. 1I). At presymptomatic time points, NMJs in SOD1G93A rats were still fully innervated relative to WT rats (Fig. 1J). At P120, when a significant loss of spinal motor neurons was observed, 93% of NMJs were still fully innervated in SOD1G93A rats, which was similar to the 96% seen in WT rats. However, by the time SOD1G93A rats had reached the early symptomatic stage, there was a significant 74% decrease in the percentage of fully innervated NMJs, and this continued to drop off as SOD1G93A rats reached endpoint. This careful temporal analysis provided the unexpected finding that ventral root axons were still intact and appeared generally healthy and that NMJs remained fully innervated at the presymptomatic time point when spinal motor neurons were already lost. We next turned to understanding the health and survival of corticospinal motor neurons in SOD1G93A rat.
Corticospinal motor neurons are not lost until disease endpoint
Corticospinal motor neurons reside in layer V of the motor cortex and express the CTIP2 protein (Arlotta et al., 2005). To determine the time course of corticospinal (“upper”) motor neuron degeneration, we quantified the number of large CTIP2+ cells present in this defined region over time. A fluorescent Nissl stain was used to visualize cell size, followed by stereological analysis to calculate the number of large (>300 μm) CTIP2+ cells (Fig. 1K). Results showed that the number of larger corticospinal motor neurons in the SOD1G93A rat brain was similar during presymptomatic time periods (P90 and P120) relative to WT rats (Fig. 1L). Total numbers of large corticospinal motor neurons did not decline until rats were in the early symptomatic stage, and this only became significant when rats had reached clinical endpoint (26% reduction in CTIP2+ cells relative to WT at endpoint; Fig. 1L). Additionally, overall numbers of CTIP2+ cells in layer V, regardless of cell size, were not significantly different relative to WT or over time in SOD1G93A rats (data not shown). Collectively, results from the spinal cord, ventral roots, NMJs, and now the brain suggest that the death of motor neurons starts at P120 in the spinal cord and radiates out to the muscle and brain over time (Fig. 1M).
The brain–muscle circuitry is disrupted in presymptomatic rats at P120
Although corticospinal motor neurons are apparently not dying presymptomatically, it is possible that they are present but are dysfunctional. Pseudorabies virus injected into the gastrocnemius muscle retrogradely labels spinal and corticospinal motor neurons (Kerman et al., 2003). Based on this, we injected pseudorabies virus at presymptomatic time points before muscle denervation occurs to determine whether the pathway from brain to muscle is disrupted (Fig. 2A). There was no difference in the number of retrogradely labeled corticospinal motor neurons at P90. Importantly, there was a significant decrease in the number of labeled corticospinal motor neurons in SOD1G93A rats at the presymptomatic P120 time point when NMJs were fully innervated in SOD1G93A rats relative to WT controls (Fig. 2B,C).
Figure 2.

The brain–muscle circuitry is disrupted in presymptomatic rats at P120. A, The brain-to-muscle pathway was retrogradely labeled by injecting RFP-tagged pseudorabies virus into the right gastrocnemius. B, Image of RFP-labeled corticospinal motor neurons at P120 in WT and SOD1G93A rats. C, Quantification of RFP-labeled motor neurons in the left motor cortex revealed a significant reduction in SOD1G93A rats at P120, but not at P90, relative to WT controls. D, Image of RFP-labeled spinal motor neurons at P120 in a WT rat. E, Quantification of RFP-labeled motor neurons in the right lumbar spinal cord revealed a significant reduction in SOD1G93A rats at P120, but not at P90, relative to WT controls. Scale bars, 75 μm. *p < 0.05; error bars indicate SEM.
Quantifying the number of spinal motor neurons labeled with pseudorabies virus showed that, similar to corticospinal motor neurons quantifications, there was no difference in the number of spinal motor neurons at P90, but there was a significant reduction in labeled spinal motor neurons in SOD1G93A rats relative to WT controls at P120 (Fig. 2D,E). Although the detailed health and exact numbers of upper and lower motor neurons cannot be fully assessed using these pseudorabies virus experiments, collectively, loss of both spinal and corticospinal motor neurons but not NMJs at the P120 presymptomatic time point supports a disruption in the brain-to-muscle pathway that is likely to originate at sites proximal to the NMJ. However, because the reduction in corticospinal and spinal motor neurons occurred at the same time, additional investigation is required to identify where in the pathway disruption first occurs (e.g., within corticospinal motor neurons innervating the spinal cord or spinal motor neurons innervating the muscle).
Injection of AAV9–SOD1–shRNA in the SOD1G93A rat brain induces local suppression of mutant, misfolded SOD1 protein
Although our initial anatomical studies did not show that the brain is first to degenerate in the SOD1G93A rat model, we hypothesized that, whereas corticospinal motor neurons are not dying, they could be dysfunctional and hence that the brain could be playing a role in initiating disease onset in a manner other than overt cortical cell death. To test the hypothesis that the brain is involved in the initiation of ALS disease pathology, we used shRNA to knock down mutant SOD1 expression in the motor cortex of SOD1G93A rats at 70 d of age. The shRNA to mutant SOD1 was fused to the GFP reporter, and both genes were packaged in AAV9, known to infect neurons (Foust et al., 2009). We first injected AAV9–SOD1–shRNA unilaterally into the motor cortex. Histological analysis showed robust GFP expression in the ipsilateral motor cortex, with no GFP expression in the motor cortex contralateral to the injection site (Fig. 3A,B, green, respectively). Importantly, immunohistochemistry using the D3H5 antibody that recognizes misfolded SOD1 protein showed successful local knockdown of mutant SOD1 ipsilateral, but not contralateral, to the injection site (Fig. 3A,B, red, respectively).
Figure 3.

Injection of AAV9–SOD1–shRNA in the SOD1G93A rat brain induces local suppression of mutant, misfolded SOD1 protein. A, A single injection of AAV9–SOD1–shRNA into the motor cortex of SOD1G93A rats induces local GFP expression (shown by histology) and suppression of mutant SOD1 (shown by immunohistochemistry using the D3H5 antibody in red that recognizes misfolded SOD1 protein) in these cells. B, Because AAV9–SOD1–shRNA does not cross into the contralateral side of the brain, there is no GFP expression and mutant SOD1 protein is still produced. C, Motor cortex functional map shows the location of AAV9 injection sites. Eight injections per hemisphere (total of 16 injections) were administered to target the posterior/hindlimb cortex. FL, Forelimb; HL, Hindlimb; W, whisker; N, neck; T, tail representation within the motor cortex. Immunostaining for GFP (green), CTIP2 (red), and D3H5 (blue) in coronal brain sections shows that a large number of CTIP2+ corticospinal motor neurons were virally transduced and that mutant SOD1 (D3H5) was still highly expressed in cells infected with AAV9–GFP control virus (D) but was knocked down in cells infected with AAV9–SOD1–shRNA (E). Low-power coronal brain images revealed that AAV9–GFP control-injected SOD1G93A rats display robust GFP expression and a remaining abundant expression of misfolded, mutant SOD1 protein in the motor cortex (F), whereas AAV9–SOD1–shRNA-injected SOD1G93A rats show robust GFP expression with a corresponding significant 40% reduction of misfolded SOD1 protein (G). H, Immunostaining using antibodies against GFP and D3H5 in the spinal cord revealed that very few (3.6%) total spinal motor neurons expressed GFP (and therefore AAV9–SOD1–shRNA), and the majority of these GFP+ cells did not show an abundant level of mutant SOD1 knockdown, resulting in only ∼1.2% of all spinal motor neurons with reduced levels of mutant protein (right, pink bar) (I). J, qRT-PCR analysis of laser-captured motor neurons from the motor cortex of SOD1G93A rats injected with AAV9–SOD1–shRNA revealed significant amounts of GFP vector expression and a corresponding significant reduction of mutant SOD1 expression compared with non-injected SOD1G93A rats (K). L, Similar qRT-PCR analysis of laser-captured motor neurons from the spinal cord revealed no loss of mutant SOD1 expression in these spinal motor neurons, suggesting a lack of vector expression. Immunostaining in the spinal cord after delivery of AAV9 to the motor cortex revealed that <1% of GFAP+ astrocytes (M) and Iba1+ microglia (N) were anterogradely transduced with AAV9 and therefore GFP+. Scale bars: A, B, M, N, 100 μm; D, E, H, 50 μm; F, G, 750 μm. ***p < 0.001; error bars indicate SEM.
We next administered eight injections per hemisphere (16 total injections) into the motor cortex to suppress mutant SOD1 in a much larger region (Fig. 3C). After the 16 cortical injections, we assessed the motor cortex for GFP expression using histology and for misfolded SOD1 protein using the D3H5 antibody. We found widespread GFP expression after administration of both AAV9–GFP and AAV9–SOD1–shRNA that colocalized with a large number of CTIP2+ corticospinal motor neurons (Fig. 3D,E). However, a corresponding knockdown of misfolded SOD1 protein was only observed with administration of AAV9–SOD1–shRNA (Fig. 3D–G).
We also assessed the spinal cord for GFP expression to confirm that knockdown of mutant SOD1 was isolated to the brain. Histological analysis of GFP fluorescence without using a GFP antibody revealed no observable GFP expression in the spinal cord. Using an antibody against GFP for signal amplification showed rare GFP expression in spinal motor neurons (Fig. 3H) and astrocytes, as well as the presence of GFP in the dorsal columns (data not shown). Quantification of these immunostained sections showed that GFP expression occurred in only 3.6% of total spinal motor neurons, whereas the remaining 96.4% of spinal motor neurons had no visible GFP expression. Assessment of the 3.6% GFP+ cells for double labeling with the D3H5 antibody showed that, in terms of total spinal motor neurons, 2.4% were still positive for D3H5, suggesting no knockdown of SOD1 protein, and just 1.2% were negative for D3H5, suggesting knockdown of SOD1 protein (Fig. 3I). To confirm that there was minimal GFP and shRNA expression in spinal motor neurons, we used laser-capture microdissection to isolate individual neurons from the cortex and spinal cord of rats injected with AAV9–SOD1–shRNA and non-injected controls. Analysis by qRT-PCR showed that pooled corticospinal motor neurons from AAV9–SOD1–shRNA-injected rats had significantly increased levels of GFP (Fig. 3J) and, correspondingly, significantly reduced levels of mutant SOD1 (Fig. 3K) compared with non-injected controls. In stark contrast, pooled spinal motor neurons had undetectable GFP expression (data not shown) and no significant reduction of mutant SOD1 relative to non-injected controls (Fig. 3L). Additionally, quantitative RNA analysis of whole spinal cord sections did not show significant GFP expression or reduction of mutant SOD1 (data not shown). Finally, quantification of cells double stained with GFP and GFAP for astrocytes (Fig. 3M) or Iba1 for microglia (Fig. 3N) confirmed that <1% of each of these non-neuronal cells in the spinal cord had been transduced (0.65% and 0.57%, respectively). Collectively, these data show that very little AAV9–SOD1–shRNA traveled anterogradely from the cortex and that, in most cases, the level was not sufficient to reduce mutant SOD1 protein levels in the spinal cord.
Suppression of mutant SOD1 in the posterior motor cortex leads to improved hindlimb function (but not forelimb function) with an associated delay in disease onset and extension of survival in the SOD1G93A rat
Disease onset and progression in this SOD1G93A rat model is heterogeneous in regards to forelimb and hindlimb onset. As predicted, SOD1G93A rats injected (16 sites; Fig. 3C) with AAV9–GFP control virus at P70 showed a balance of both forelimb and hindlimb onset (Fig. 4A). However, when we used AAV9–SOD1–shRNA injections to suppress mutant SOD1 in the posterior motor cortex, which has been shown to be associated with hindlimb motor function (Fonoff et al., 2009; Fig. 3C), we found that none of the injected SOD1G93A rats (n = 14) developed exclusive hindlimb paralysis (Fig. 4A, arrow). Because we specifically targeted the posterior motor cortex, we limited our BBB analyses to those rats that had exclusive hindlimb or simultaneous hindlimb/forelimb onset. We found that hindlimb motor function was significantly improved in AAV9–SOD1–shRNA-injected rats relative to AAV9–GFP-injected controls (Fig. 4B). Furthermore, local motor cortex suppression of mutant SOD1 expression by AAV9–SOD1–shRNA in SOD1G93A rats delayed disease onset by 25 d relative to AAV9–GFP control-injected SOD1G93A rats (median onset, 168 d for AAV9–SOD1–shRNA vs 143 d for AAV9–GFP; p < 0.05; Fig. 4C). Finally, local motor cortex suppression of mutant SOD1 expression extended the SOD1G93A rat survival time by ∼20 d (median survival, 183 d for AAV9–SOD1–shRNA vs 164 d for AAV9–GFP; p < 0.05; Fig. 4D). Disease onset and survival after AAV9–GFP control injections (Fig. 4) did not differ from previously published SOD1G93A controls (Suzuki et al., 2007), showing that GFP alone was having no negative effects. Additionally, it has been shown that systemic injection of AAV9–SOD1–shRNA in mice leads to delayed disease onset and extended survival (Foust et al., 2013), suggesting that the beneficial effects observed among the current experiments are likely specific to the knockdown caused by AAV9–SOD1–shRNA. To confirm this, we tested AAV9–scrambled–shRNA first by transfection into a human cell line and subsequently by intravenous injection in SOD1G93A mice at P1 and P21. Western blot analysis and quantification showed that AAV9–scrambled–shRNA did not significantly decrease human SOD1 protein levels in HEK293 cells (Fig. 5A,B). Importantly, disease onset (Fig. 5C,D) and survival (Fig. 5E,F) remained unchanged after systemic delivery of AAV9–scrambled–shRNA relative to non-injected SOD1G93A controls.
Figure 4.

Local suppression of mutant, misfolded SOD1 protein in the posterior motor cortex of SOD1G93A rat brain leads to improved hindlimb but not forelimb function, delayed disease onset, and an extension of survival. A, SOD1G93A rats administered AAV9–GFP control virus (n = 14) showed an expected variation in disease onset location in solely forelimb (n = 4; FL ONLY), solely hindlimb (n = 5; HL ONLY), or both (n = 5; BOTH FL,HL same time). In contrast, no SOD1G93A rats injected with AAV9–SOD1–shRNA developed exclusive hindlimb onset (arrow, A). This was likely attributable to the targeting field that knocked down mutant SOD1 primarily in the posterior/hindlimb region, while providing incomplete targeting in the anterior/forelimb motor cortex. In light of the targeting strategy, rats in both groups that developed exclusive forelimb onset were excluded from analysis. B, BBB hindlimb analysis showed that SOD1G93A rats administered AAV9–SOD1–shRNA had significant behavioral improvements relative to AAV9–GFP control-injected SOD1G93A rats. In addition, knockdown of mutant SOD1 expression in the motor cortex of SOD1G93A rats resulted in delayed hindlimb onset by ∼24 d (C) and extended survival time by ∼20 d (D).
Figure 5.
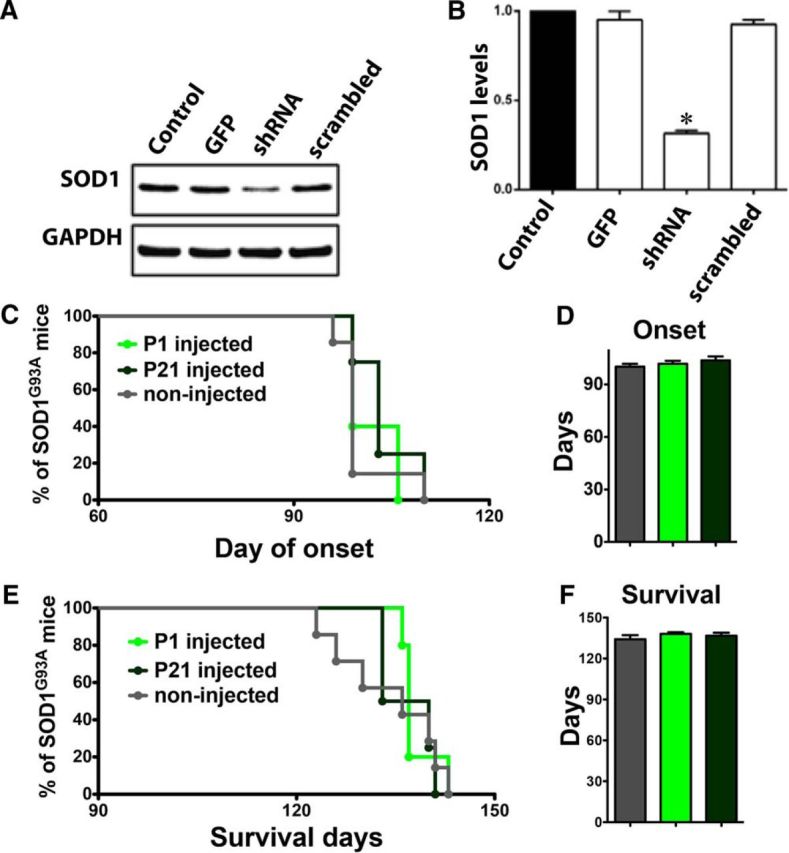
AAV9–scrambled–shRNA has no effect on human SOD1 protein levels and does not improve motor function or survival. A, AAV9–scrambled–shRNA or AAV9–SOD1–shRNA were transfected into HEK293 cells, and 72 h later, cell lysates were collected for analysis by Western blot. Unlike AAV9–SOD1–shRNA-treated cells, AAV9–scrambled–shRNA treatment did not reduce human SOD1 levels compared with control conditions. B, Quantification of Western blot analysis of human SOD1 knockdown showed that AAV9–SOD1–shRNA-treated cells, but not AAV9–scrambled–shRNA-treated cells, showed a significant reduction (*p < 0.05) compared with control conditions. C–F, SOD1G93A mice were treated with a single intravenous injection of AAV9–scrambled–shRNA at P1 (n = 5; light green) or P21 (n = 4; dark green) and were monitored throughout the disease course compared with non-injected control mice (n = 7; gray). AAV9–scrambled–shRNA treatment did not significantly delay the disease onset (C, D) or increase survival (E, F).
Mutant SOD1 suppression in the anterior and posterior cortices of SOD1G93A rat leads to improvements in both forelimb and hindlimb function
We next extended the mutant SOD1 knockdown experiments to include the anterior motor cortex by adding two rostral motor cortex injection sites (four total bilaterally). This new total of 20 injection sites should now target not only the hindlimb region of the motor cortex but also the forelimb region (Fig. 6A). Targeting the hindlimb representation region of the motor cortex with shRNA to mutant SOD1 once again provided significantly better behavioral scores in the hindlimb region relative to non-injected controls (Fig. 6B). Critically, the additional targeting of the forelimb representation region of the motor cortex also provided significantly improved behavioral scores in the forelimb region (Fig. 6C).
Figure 6.
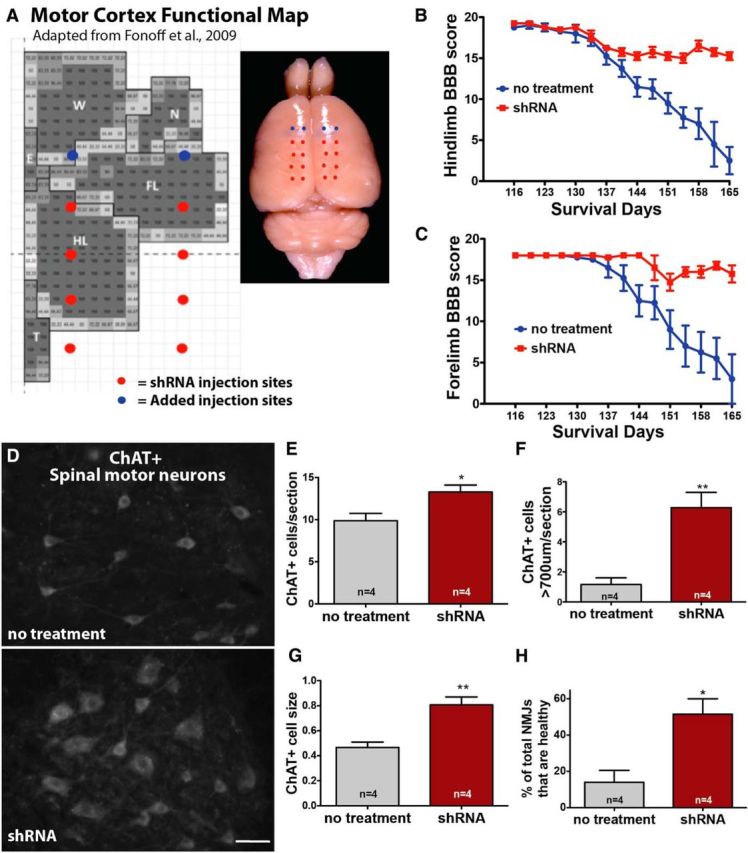
Local suppression of mutant SOD1 in both the anterior and posterior motor cortices of SOD1G93A rats leads to improved forelimb and hindlimb function, enhanced spinal motor neuron survival, and increased muscle innervation. A, Previous experiments were repeated, with two additional AAV9 injection sites per hemisphere to the anterior motor cortex to target more of the forelimb region (total = 20 injections). This time, BBB analysis showed that both hindlimb (B) and forelimb (C) motor function were significantly improved in AAV9–SOD1–shRNA-injected rats relative to control rats. D, Immunostaining showed that SOD1G93A rats treated with AAV9–SOD1–shRNA in the motor cortex had preserved ChAT+ motor neurons in the spinal cord compared with non-treated SOD1G93A rats. Quantification confirmed that the enhanced survival was significant for both total ChAT+ spinal motor neurons (E) and large (>700 μm) spinal motor neurons (F). G, In addition, the average cell body size (μm2) of the spinal motor neurons was significantly larger in AAV9–SOD1–shRNA-treated SOD1G93A rats relative to nontreated control SOD1G93A rats. H, Finally, knockdown of mutant SOD1 in the brain also lead to increased muscle innervation relative to non-treated controls. Scale bar, 50 μm. *p < 0.05, **p < 0.01; error bars indicate SEM. FL, Forelimb; HL, hindlimb; W, whisker; N, neck; T, tail representation within the motor cortex.
Suppression of SOD1 in the cortex of SOD1G93A rats leads to enhanced survival of spinal motor neurons and improved innervation of NMJs
Importantly, we wanted to determine whether the mutant SOD1 suppression in the anterior and posterior cortices of the SOD1G93A rat leads not only to improvements in forelimb and hindlimb function but also to enhanced spinal motor neuron survival and NMJ integrity. Because our previous shRNA knockdown experiment assessed animal lifespan, all animals were at the same endpoint behavioral state and therefore likely at the same endpoint anatomical state. In contrast, this cohort of AAV9–SOD1–shRNA-treated and untreated rats was euthanized earlier at ∼168 d, when behavior function was significantly different between groups and therefore when anatomical state may also be different.
Histological analyses were used to test whether knockdown of mutant SOD1 in the motor cortex of SOD1G93A rats delays disease onset by enhancing spinal motor neuron survival and NMJs. We found that rats injected with AAV9–SOD1–shRNA had a significant ∼35% increase in the number of total ChAT+ motor neurons in the lumbar spinal cord at ∼168 d and a 4.5-fold greater number of large (>700 μm) ChAT+ spinal motor neurons relative to non-injected controls (Fig. 6D–F). Additionally, the average cell size of ChAT+ spinal motor neurons was significantly greater, by 72%, in AAV9–SOD1–shRNA-injected rats versus controls (Fig. 6G). Finally, we found that SOD1G93A rats that received brain injections of AAV9–SOD1–shRNA had a significantly greater percentage of NMJs that were fully innervated relative to non-injected controls (52 vs 14%, AAV9–SOD1–shRNA vs non-injected, respectively; Fig. 6H). Together, these results provide strong evidence that the brain is playing an important role in initiating disease onset with a dying-forward cascade.
Discussion
The motor cortex primarily initiates and modulates voluntary movement, because the residing corticospinal motor neurons are responsible for collecting, integrating, translating, and transferring information to the spinal cord. If these cells do not function properly, system breakdown occurs. Because the motor neuron circuitry clearly degenerates over time in ALS patients and animal models, it is surprising that the motor cortex containing the initiating component (corticospinal motor neurons) has received limited attention among the ALS research community.
Here, we have shown for the first time that suppression of mutant SOD1 at the “top” of the motor neuron pathway (e.g., the motor cortex) results in a significant delay in ALS disease onset and extension in survival in transgenic SOD1G93A rats. This shRNA-based knockdown in the brain results in concomitant enhanced survival of spinal motor neurons and improved innervation of NMJs, supporting a proposed model of degeneration whereby ALS originates in the brain and progresses in a dying-forward manner (summary in Fig. 7). Anterograde transport of AAV9 and consequent knockdown of mutant SOD1 in the spinal cord or muscle is unlikely to contribute to the beneficial functional effects observed because only 1.2% of all spinal motor neurons showed signs of AAV9 infection with associated reduced levels of mutant SOD1, and even <1% of non-neuronal cells showed infection. The findings from these studies provide critical support for a cortical basis in the development of ALS, providing scientists and drug screeners with a novel focus for future ALS research and treatments.
Figure 7.
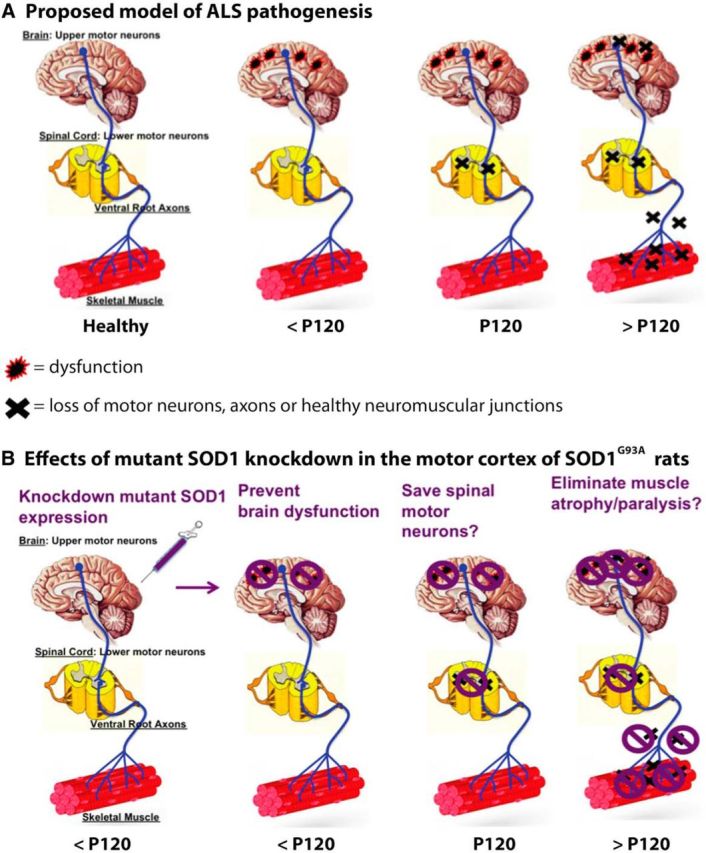
Summary of the proposed model of ALS disease pathology and effects of mutant SOD1 knockdown in the brain. A, Proposed model of ALS disease progression whereby dysfunction (red/black symbols) but not cell death (black × symbols) of corticospinal motor neurons occurs at early, presymptomatic time points leading to presymptomatic loss of spinal motor neurons, which then results in the degeneration of ventral root axons and denervation of NMJs and concomitant appearance of disease symptoms. B, Consequences of knocking down mutant SOD1 in the brain based on model above (A), whereby the reduction of mutant SOD1 eliminates the dysfunction (purple symbol) of corticospinal motor neurons, which delays disease progression by preventing cell loss in the spinal cord and consequently preventing axon degeneration and NMJ denervation.
A wide range of factors are likely to contribute to ALS pathogenesis, including mitochondrial dysfunction, protein misfolding/aggregation, oxidative damage, neuronal excitotoxicity, non-cell-autonomous effects/neuroinflammation, axonal transport defects, neurotrophin depletion, effects from extracellular mutant SOD1, and aberrant RNA processing (Eisen et al., 1992; Chou and Norris, 1993; Williamson, 1999; Kieran et al., 2005; De Winter et al., 2006; Parkhouse et al., 2008; Schmidt et al., 2009; Bilsland et al., 2010; Brockington et al., 2010; Li et al., 2010; Dadon-Nachum et al., 2011). However, the extent to which each factor is involved and the overall pathology of the disease are still not understood. A common feature of ALS is glutamate toxicity in the spinal cord, suggesting that upper motor neuron hyperexcitability may induce anterior horn degeneration via an anterograde glutamatergic mechanism (Eisen et al., 1992; Trotti et al., 1999; Rothstein et al., 2005). Hyperexcitability has also been shown recently to be a feature of induced pluripotent stem cell-derived motor neurons from ALS patients harboring SOD1 mutations (Wainger et al., 2014). Because cortical hyperexcitability and signs of corticospinal motor neuron degeneration have been shown to be early events in ALS patients and mice (Vucic et al., 2008; Ozdinler et al., 2011; Menon et al., 2014), future studies focusing on cortical dysfunction will likely yield insight into the importance of the brain in disease pathology.
Electrical recording techniques and low-resolution imaging are used to assess global changes in ALS patient cortical function but cannot successfully determine changes at the cellular level, such as motor neuron death at early time points. Using detailed histological analysis of SOD1G93A rats over time, we showed that the first signs of cell death occurred in the spinal cord, with this loss of spinal motor neurons preceding significant degeneration of distal ventral root axons and denervation of NMJs. The decrease in spinal motor neuron cell bodies before an equal loss of NMJs suggested that there is a window of time in this rat model of ALS when the cell body is cleared and the NMJs appear intact. Although this was an unexpected finding, it mirrors transection models of axon degeneration that show a latency period after acute axonal injury when distal axons may remain morphologically intact and functionally excitable for brief periods of time (Lubińska, 1982; Tsao et al., 1994). Indeed, changing the physiological conditions can alter the timeframe during which injured axons are in a vulnerable yet functional state (George et al., 1995; Tsao et al., 1999; Conforti et al., 2000). In the current report, a survival or trophic factor within the distal axons may be maintained at a sufficient level to preserve the distal axons even when spinal motor neurons are lost (for review, see Wang et al., 2012).
Our anatomical studies showed no evidence that corticospinal motor neurons are lost early on at presymptomatic time points. This is in contrast to the presymptomatic death of corticospinal motor neurons observed in the SOD1G93A mouse, with associated structural differences in apical dendrites of these layer V pyramidal cells (Ozdinler et al., 2011; Jara et al., 2012). The lack of overt cell death within the motor cortex at early time points in the transgenic rat compared with the transgenic mouse can likely be attributed to differences in animal models. Indeed, many promising preclinical trials using mouse models have not successfully translated to humans (Gordon et al., 2007; Berry and Cudkowicz, 2011; Philips and Robberecht, 2011). Relative to the transgenic mouse model of ALS, the rat model is larger and the timing and location of disease onset is less predictable. Interestingly, patients also show variable location of onset and progression rates. Although the rat model is valuable for additional studies investigating ALS pathogenesis and therapeutic approaches, ALS mouse models are more amenable to transgenic strategies that may elucidate fundamental mechanisms of the disease. For instance, the UCHL1–eGFP mouse was generated recently in which corticospinal motor neurons selectively expressed GFP under the control of the ubiquitin C-terminal hydrolase-L1 (UCHL1) promoter (Yasvoina et al., 2013). Crossbreeding UCHL1–GFP and SOD1G93A mice generated hSOD1G93A–U–GFP mice that recapitulated the reduced numbers of corticospinal motor neurons with disease progression reported in the SOD1G93A mice (Ozdinler et al., 2011).
It is well established that ALS involves non-cell-autonomous toxicity and that unknown toxic factors secreted by mutant SOD1 astrocytes and microglia are capable of killing healthy motor neurons (Clement et al., 2003; Di Giorgio et al., 2008; Haidet-Phillips et al., 2011; Sargsyan et al., 2011; Frakes et al., 2014; Re et al., 2014). shRNA knockdown of SOD1 in astrocytes eliminates this toxicity, confirming that this is mediated through glial SOD1 synthesis (Haidet-Phillips et al., 2011; Frakes et al., 2014). It has been proposed that a prion-like mechanism is involved in the spread of ALS and that perhaps toxicity is transferred through an unidentified misfolded SOD1 initiator that may trigger prion-like aggregation of normal SOD1 in neighboring cells (for review, see Polymenidou and Cleveland, 2011; Grad and Cashman, 2014). We found that shRNA-based knockdown of mutant SOD1 in the posterior motor cortex provided functional improvements in the hindlimb and increased lifespan. Interestingly, unlike localized injections targeting only the hindlimb region, adding shRNA injection sites to knockdown SOD1 in an increased area that included the forelimb motor cortex resulted in additional functional improvements within the forelimb region. Although SOD1 knockdown in specific regions of the motor cortex did not completely prevent disease onset and progression, it is quite possible that targeting the entire motor cortex to eliminate a greater area of toxicity and the potential for toxic spread may lead to an even better outcome. The presence of GFP expression and the absence of mutant SOD1 expression within layers I–VI both suggest that AAV9–SOD1–shRNA transduced all cells in this region of the motor cortex, which includes corticospinal motor neurons, astrocytes, and interneurons. Although our data strongly suggest that the brain plays an important role in ALS pathology, the exact mechanisms and involvement of various cell types require additional investigation, such as experiments using promoter-specific expression or knockdown of mutant SOD1. It is likely that non-cell-autonomous toxicity occurs in the brain as in the spinal cord. It has taken years of research by numerous laboratory groups to show a central role of the spinal cord in ALS, and we hope that these novel data will initiate a new era of experiments that focus on understanding the mechanisms underlying the critical contribution of the brain to ALS.
AAV9–SOD1–shRNA might be a valid candidate to translate AAV9-mediated suppression of SOD1 synthesis to the human setting for clinical trials in ALS. Indeed, direct cerebrospinal fluid infusion at the lumbar level in nonhuman primates has been deemed safe and has been shown to produce substantial SOD1 reduction by targeting both motor neurons and non-neuronal cells (Foust et al., 2013). However, caution is still required given differences not only between rodent models and the human condition but also given differences between the motor neuron pools most affected with distinct human mutations (Cudkowicz et al., 1998; Lopate et al., 2010). In addition, it remains controversial whether SOD1 toxicity is involved in both sporadic and familial cases of ALS (Haidet-Phillips et al., 2011; Re et al., 2014). Clearly, SOD1 toxicity involvement in both sporadic and familial cases would substantially broaden the application of this therapeutic approach for ALS. Although the exact mechanism of ALS is still unclear and is likely dependent on multiple factors, we show here that a toxic/dysfunctional brain environment can trigger downstream disruption of the motor neuron pathway. Thus, our study further highlights the importance of the upper motor neurons in ALS.
Conclusions
There is a large body of evidence to suggest that the onset of clinical symptoms in ALS patients and animal models is preceded by a long presymptomatic period (Eisen et al., 2014). It is very likely that dysfunction at a cellular level (without cell death) exists and goes undetected for years before clinical diagnosis. It is only after reaching and exceeding a threshold that extensive motor neuron circuitry failure occurs and clinical expression is observed. The current lack of successful therapeutic options for ALS patients may be attributed, in part, to the timing of treatment initiation, because attempts at slowing disease progression typically occur after diagnosis, which is after an irreversible cascade of motor neuron circuitry failure has set in. Therefore, it is essential to identify the key players in early dysfunction in order to identify and treat those at high risk of developing ALS. Because it is becoming clear that the motor cortex plays a significant role in ALS pathology, it will be essential to focus future studies on identifying the mechanisms and key molecular/cellular components within the brain that may initiate system breakdown and the onset of ALS.
Footnotes
This work was supported by the Board of Governors Regenerative Medicine Institute (C.N.S.), NIH Grant RO1 NS644912 (B.K.K.), and the ALS Association (C.N.S. and B.K.K.). We thank Dr. Soshana Svendsen for critical review and manuscript editing, Dr. Patrick Card, Dr. Lynn Enquist, and the Center for Neuroanatomy with Neurotropic Viruses (National Institutes of Health Grant P40 OD010996) for providing pseudorabies virus, and Dr. Jean-Pierre Julien for providing D3H5 antibody.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Berry JD, Cudkowicz ME. New considerations in the design of clinical trials for amyotrophic lateral sclerosis. Clin Investig (Lond) 2011;1:1375–1389. doi: 10.4155/cli.11.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A. 2010;107:20523–20528. doi: 10.1073/pnas.1006869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockington A, Heath PR, Holden H, Kasher P, Bender FL, Claes F, Lambrechts D, Sendtner M, Carmeliet P, Shaw PJ. Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFdelta/delta mouse model of amyotrophic lateral sclerosis. BMC Genomics. 2010;11:203. doi: 10.1186/1471-2164-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Levitt P, Enquist LW. Different patterns of neuronal infection after intracerebral injection of two strains of pseudorabies virus. J Virol. 1998;72:4434–4441. doi: 10.1128/jvi.72.5.4434-4441.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Rinaman L, Lynn RB, Lee BH, Meade RP, Miselis RR, Enquist LW. Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of viral replication, transport, and pathogenesis. J Neurosci. 1993;13:2515–2539. doi: 10.1523/JNEUROSCI.13-06-02515.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SM, Norris FH. Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve. 1993;16:864–869. doi: 10.1002/mus.880160810. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci U S A. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudkowicz ME, McKenna-Yasek D, Chen C, Hedley-Whyte ET, Brown RH., Jr Limited corticospinal tract involvement in amyotrophic lateral sclerosis subjects with the A4V mutation in the copper/zinc superoxide dismutase gene. Ann Neurol. 1998;43:703–710. doi: 10.1002/ana.410430604. [DOI] [PubMed] [Google Scholar]
- Dadon-Nachum M, Melamed E, Offen D. The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- De Winter F, Vo T, Stam FJ, Wisman LA, Bär PR, Niclou SP, van Muiswinkel FL, Verhaagen J. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci. 2006;32:102–117. doi: 10.1016/j.mcn.2006.03.002. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Eisen A, Kiernan M, Mitsumoto H, Swash M. Amyotrophic lateral sclerosis: a long preclinical period? J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2013-307135. Advance online publication. Retrieved October 14, 2014. [DOI] [PubMed] [Google Scholar]
- Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve. 1992;15:219–224. doi: 10.1002/mus.880150215. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Fonoff ET, Pereira JF, Jr, Camargo LV, Dale CS, Pagano RL, Ballester G, Teixeira MJ. Functional mapping of the motor cortex of the rat using transdural electrical stimulation. Behav Brain Res. 2009;202:138–141. doi: 10.1016/j.bbr.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H, Meyer K, Schmelzer L, Braun L, Cleveland DW, Kaspar BK. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther. 2013;21:2148–2159. doi: 10.1038/mt.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H, Neville HE, Boylan K, Mozaffar T, Belsh JM, Ravits J, Bedlack RS, Graves MC, McCluskey LF, Barohn RJ, Tandan R. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–1053. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- Goyal NA, Mozaffar T. Experimental trials in amyotrophic lateral sclerosis: a review of recently completed, ongoing and planned trials using existing and novel drugs. Expert Opin Investig Drugs. 2014;23:1541–1551. doi: 10.1517/13543784.2014.933807. [DOI] [PubMed] [Google Scholar]
- Grad LI, Cashman NR. Prion-like activity of Cu/Zn superoxide dismutase: implications for amyotrophic lateral sclerosis. Prion. 2014;8:33–41. doi: 10.4161/pri.27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara JH, Villa SR, Khan NA, Bohn MC, Ozdinler PH. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis. 2012;47:174–183. doi: 10.1016/j.nbd.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman IA, Enquist LW, Watson SJ, Yates BJ. Brainstem substrates of sympatho-motor circuitry identified using trans-synaptic tracing with pseudorabies virus recombinants. J Neurosci. 2003;23:4657–4666. doi: 10.1523/JNEUROSCI.23-11-04657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, Chun SJ, Roy T, Winer L, Yates JR, Capaldi RA, Cleveland DW, Miller TM. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107:21146–21151. doi: 10.1073/pnas.1014862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopate G, Baloh RH, Al-Lozi MT, Miller TM, Fernandes Filho JA, Ni O, Leston A, Florence J, Schierbecker J, Allred P. Familial ALS with extreme phenotypic variability due to the I113T SOD1 mutation. Amyotroph Lateral Scler. 2010;11:232–236. doi: 10.3109/17482960902898069. [DOI] [PubMed] [Google Scholar]
- Lubińska L. Patterns of Wallerian degeneration of myelinated fibres in short and long peripheral stumps and in isolated segments of rat phrenic nerve. Interpretation of the role of axoplasmic flow of the trophic factor. Brain Res. 1982;233:227–240. doi: 10.1016/0006-8993(82)91199-4. [DOI] [PubMed] [Google Scholar]
- Menon P, Kiernan MC, Vucic S. Cortical dysfunction underlies the development of the split-hand in amyotrophic lateral sclerosis. PLoS One. 2014;9:e87124. doi: 10.1371/journal.pone.0087124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Kaspar BK, Kops GJ, Yamanaka K, Christian LJ, Gage FH, Cleveland DW. Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann Neurol. 2005;57:773–776. doi: 10.1002/ana.20453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Kim SH, Yamanaka K, Hester M, Umapathi P, Arnson H, Rizo L, Mendell JR, Gage FH, Cleveland DW, Kaspar BK. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:19546–19551. doi: 10.1073/pnas.0609411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdinler PH, Benn S, Yamamoto TH, Güzel M, Brown RH, Jr, Macklis JD. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G(9)(3)A transgenic ALS mice. J Neurosci. 2011;31:4166–4177. doi: 10.1523/JNEUROSCI.4184-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhouse WS, Cunningham L, McFee I, Miller JM, Whitney D, Pelech SL, Krieger C. Neuromuscular dysfunction in the mutant superoxide dismutase mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:24–34. doi: 10.1080/17482960701725646. [DOI] [PubMed] [Google Scholar]
- Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147:498–508. doi: 10.1016/j.cell.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- Raoul C, Abbas-Terki T, Bensadoun JC, Guillot S, Haase G, Szulc J, Henderson CE, Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sargsyan SA, Blackburn DJ, Barber SC, Grosskreutz J, De Vos KJ, Monk PN, Shaw PJ. A comparison of in vitro properties of resting SOD1 transgenic microglia reveals evidence of reduced neuroprotective function. BMC Neurosci. 2011;12:91. doi: 10.1186/1471-2202-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ER, Pasterkamp RJ, van den Berg LH. Axon guidance proteins: novel therapeutic targets for ALS? Prog Neurobiol. 2009;88:286–301. doi: 10.1016/j.pneurobio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Smith BN, Banfield BW, Smeraski CA, Wilcox CL, Dudek FE, Enquist LW, Pickard GE. Pseudorabies virus expressing enhanced green fluorescent protein: A tool for in vitro electrophysiological analysis of transsynaptically labeled neurons in identified central nervous system circuits. Proc Natl Acad Sci U S A. 2000;97:9264–9269. doi: 10.1073/pnas.97.16.9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Tork C, Shelley B, McHugh J, Wallace K, Klein SM, Lindstrom MJ, Svendsen CN. Sexual dimorphism in disease onset and progression of a rat model of ALS. Amyotroph Lateral Scler. 2007;8:20–25. doi: 10.1080/17482960600982447. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- Tsao JW, Brown MC, Carden MJ, McLean WG, Perry VH. Loss of the compound action potential: an electrophysiological, biochemical and morphological study of early events in axonal degeneration in the C57BL/Ola mouse. Eur J Neurosci. 1994;6:516–524. doi: 10.1111/j.1460-9568.1994.tb00295.x. [DOI] [PubMed] [Google Scholar]
- Tsao JW, George EB, Griffin JW. Temperature modulation reveals three distinct stages of Wallerian degeneration. J Neurosci. 1999;19:4718–4726. doi: 10.1523/JNEUROSCI.19-12-04718.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain. 2006;129:2436–2446. doi: 10.1093/brain/awl172. [DOI] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540–1550. doi: 10.1093/brain/awn071. [DOI] [PubMed] [Google Scholar]
- Vucic S, Cheah BC, Yiannikas C, Kiernan MC. Cortical excitability distinguishes ALS from mimic disorders. Clin Neurophysiol. 2011;122:1860–1866. doi: 10.1016/j.clinph.2010.12.062. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TAC. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. 1999 doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Yasvoina MV, Genç B, Jara JH, Sheets PL, Quinlan KA, Milosevic A, Shepherd GM, Heckman CJ, Özdinler PH. eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J Neurosci. 2013;33:7890–7904. doi: 10.1523/JNEUROSCI.2787-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]