

Abstract
The role of mineralocorticoid receptors (MRs) in human T-cell migration is not yet understood. We have recently shown that the MR antagonist spironolactone selectively increases the numbers of circulating naïve and central memory T cells during early sleep, which is the time period in the 24 h cycle hallmarked by predominant MR activation. To investigate whether this effect is specific to spironolactone's blockade of MRs and to study the underlying molecular mechanisms, healthy humans were given the selective MR-agonist fludrocortisone or placebo and numbers of eight T-cell subsets and their CD62L and CXCR4 expression were analyzed. Fludrocortisone selectively reduced counts of naïve CD4+, central memory CD4+, and naïve CD8+ T cells and increased CXCR4 expression on the naïve subsets. In complementing in vitro studies, fludrocortisone enhanced CXCR4 and CD62L expression, which was counteracted by spironolactone. Incubation of naïve T cells with spironolactone alone reduced CD62L and CCR7 expression. Our results indicate a regulatory influence of MR signaling on human T-cell migration and suggest a role for endogenous aldosterone in the redistribution of T-cell subsets to lymph nodes, involving CD62L, CCR7, and CXCR4. Facilitation of T-cell homing following sleep-dependent aldosterone release might thus essentially contribute to sleep's well-known role in supporting adaptive immunity.
Keywords: CD62L, CXCR4, Mineralocorticoid receptor, Naïve T cells, Sleep
Introduction
The central nervous system impacts T-cell migration, with many of the effects being mediated via hypothalamic-pituitary-adrenocortical (HPA) activity 1. Previous studies have focused on the role of glucocorticoids and glucocorticoid receptor (GR) activation for T-cell migration in the context of stress and circadian rhythm 2–6. In contrast, knowledge about the role of mineralocorticoids (such as aldosterone) and mineralocorticoid receptor (MR) signaling in the physiological regulation of T-cell trafficking is surprisingly sparse. Early nocturnal sleep is a period hallmarked by predominant MR activation due to nadir levels of cortisol (which preferentially activates MRs rather than GRs at low levels), and the concurrent increase in the sleep-dependent release of aldosterone 7–10. This hormonal constellation during early sleep has been linked to an enhanced T-cell extravasation 11 characterizing this period 12. Specifically the MR, which is expressed in T lymphocytes 13 has been proposed as a candidate for mediating effects of sleep on T-cell migration, as acute administration of the MR antagonist spironolactone to healthy humans attenuates the drop in numbers of circulating naïve and central memory T cells during early sleep 11. Intriguingly, the effects of spironolactone closely mimic the effects of one night without sleep on T-cell numbers 12. This led us to conclude that the reducing effect of sleep on blood T-cell counts is mediated via sleep-dependent release of endogenous aldosterone activating MRs. However, as spironolactone also has MR-independent effects on cytokine release 14, which might likewise affect T-cell migration, conclusive evidence is needed to show that the observed changes in T-cell numbers are indeed dependent on MR signaling. Moreover, the factors mediating these changes at a molecular level are not known.
In the present study, we investigated the impact of fludrocortisone, a synthetic mineralocorticoid similar to aldosterone, on the numbers of various circulating T-cell subsets in healthy humans during normal sleep. Based on the previously reported effects of spironolactone 11, we hypothesized that fludrocortisone would selectively decrease numbers of circulating naïve and central memory T cells, while leaving effector memory and effector T cells unaffected. As naïve and central memory T cells continuously migrate between blood, secondary lymphoid organs, and bone marrow 15, we also examined the impact of fludrocortisone on the subsets’ expression of CD62L (l-selectin) and CCR7, two essential molecules for directing naïve and central memory T cells to lymph nodes 16, and expression of the chemokine receptor CXCR4, which is a major regulator of bone marrow migration 17. To control for changes in T-cell subset composition of the analyzed blood samples following fludrocortisone administration in vivo, effects of the MR agonist (and its specific blockade by spironolactone) on the expression of these molecules were examined in additional in vitro experiments.
Results
Fludrocortisone intake reduces naïve T-cell counts in blood and increases their CXCR4 expression
Fludrocortisone administration induced a decline in circulating numbers of naïve CD4+ and CD8+ as well as central memory CD4+ T cells compared with placebo (p ≤ 0.006 for respective main effects of treatment). Post hoc pairwise comparisons revealed that the effect of fludrocortisone was first evident 3 and 4.5 h following administration of the drug, for the naïve subsets and for central memory CD4+ T cells, respectively, and persisted until the last point of measurement for the naïve subsets (i.e., 8.25 h post-drug administration) (p ≤ 0.029; Fig.1). The remaining subsets were not affected by fludrocortisone (Fig.1).
Figure 1.
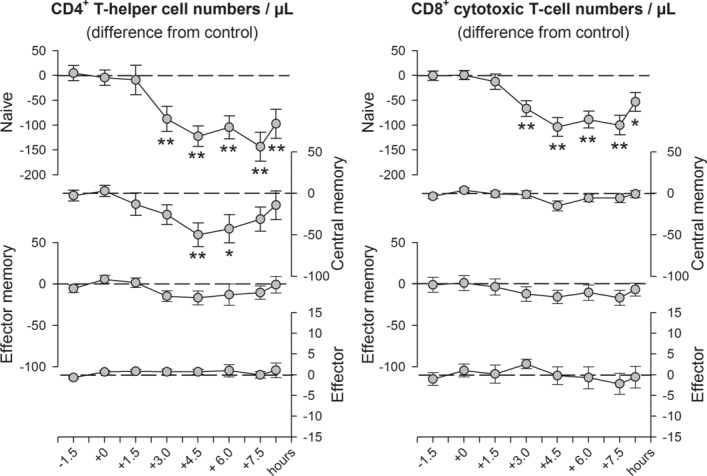
Changes in the numbers of circulating CD4+ and CD8+ T cells after fludrocortisone administration. Naive (CD45RA+CD62L+), central memory (CD45RA−CD62L+), effector memory (CD45RA−CD62L−) and effector (CD45RA+CD62L−) CD4+ (left) and CD8+ (right) T-cell counts were determined in whole blood by flow cytometry before (−1.5 and 0 h) and between 1.5 and 8.25 h after oral administration of fludrocortisone (0.2 mg) or placebo. Values for the fludrocortisone condition are indicated as difference from the placebo condition to eliminate the well-known strong circadian variation in T-cell numbers. Data are expressed as mean ± SEM of 13 healthy male subjects. All values are adjusted to baseline measures (i.e., the first two samples before drug administration) based on covariance analyses. *p < 0.05, **p < 0.01, for pairwise comparisons between the effects of fludrocortisone and placebo at single time points (paired t-tests). See text for ANOVA results.
In comparison to placebo, fludrocortisone markedly increased the expression of CXCR4 on naïve CD4+ and CD8+ T cells (p ≤ 0.004 for respective main effects of treatment), while the other subpopulations remained unaffected. The increase in CXCR4 expression was evident between 3 and 4.5 h following drug administration and again during the last two blood draws (p ≤ 0.036; Fig.2). Fludrocortisone did not affect expression of CD62L in vivo (Supporting Information Fig. 1).
Figure 2.
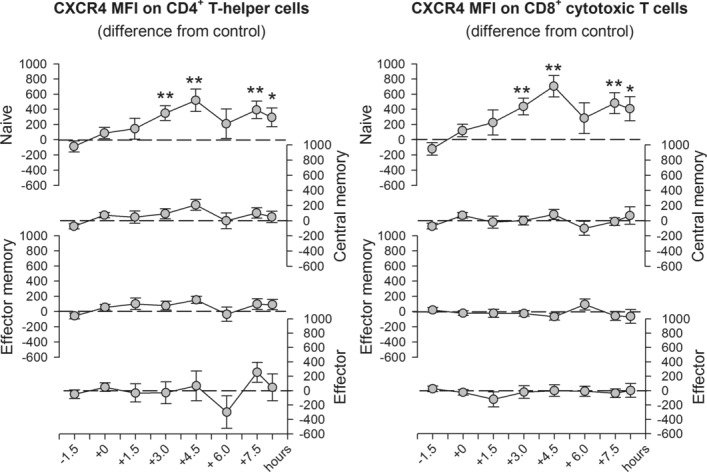
Changes in CXCR4 expression on circulating CD4+ and CD8+ T-cell subsets after fludrocortisone administration. CXCR4 expression was assessed by flow cytometry on naïve (CD45RA+CD62L+), central memory (CD45RA−CD62L+), effector memory (CD45RA−CD62L−) and effector (CD45RA+CD62L−) CD4+ (left) and CD8+ (right) T cells before (−1.5 and 0 h) and between 1.5 and 8.25 h after oral administration of fludrocortisone (0.2 mg) or placebo. Values for the fludrocortisone condition are indicated as difference from the placebo condition. All values are adjusted to baseline measures (i.e., the first two samples before drug administration) based on covariance analyses and are expressed as mean ± SEM of median fluorescence intensity (MFI) of 13 healthy male subjects. *p < 0.05, **p < 0.01, for pairwise comparisons between the effects of fludrocortisone and placebo at single time points (paired t-tests). See text for ANOVA results.
Hormones, blood pressure, and side effects following fludrocortisone administration
Compared with placebo, cortisol levels were reduced between 3.75 and 6.75 h following fludrocortisone administration (p = 0.021 for main effect of treatment; Fig.3 and Supporting Information Fig. 2) presumably reflecting a decreased adrenal sensitivity to adrenocorticotropic hormone (ACTH) upon MR activation 18. ACTH concentration itself and aldosterone levels were not significantly different between conditions (Supporting Information Fig. 2). In contrast to the findings by Charloux et al. 7,8, in our study aldosterone levels appeared to be higher after awakening than during sleep. However, this can be explained by our low blood sampling rate that did not allow the identification of single pulses in sleep-dependent aldosterone release, and by the orthostatic response occurring in the morning as our subjects got up and left the bed after awakening. Fludrocortisone produced no side effects and subjects were not able to correctly indicate whether they had received placebo or fludrocortisone. In line with previous studies, blood pressure was also unchanged after fludrocortisone administration 19–21.
Figure 3.
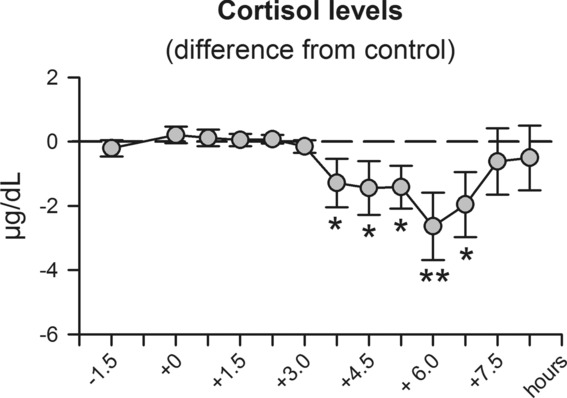
Changes in cortisol concentration after fludrocortisone administration. Serum cortisol concentration was measured before (−1.5 and 0 h) and between 1.5 and 8.25 h after oral administration of fludrocortisone (0.2 mg) or placebo. Values for the fludrocortisone condition are indicated as difference from the placebo condition to eliminate the well-known strong circadian variation in cortisol levels. Values shown as mean ± SEM of 13 healthy male subjects are adjusted to baseline measures (i.e., the first two samples before drug administration) based on covariance analyses. *p < 0.05, **p < 0.01, for pairwise comparisons between the effects of fludrocortisone and placebo at single time points (paired t-tests). See text for ANOVA results.
In vitro experiments reveal MR-mediated regulation of CD62L, CCR7, and CXCR4 expression
Compared with PBS control, incubation of blood with fludrocortisone for 2 h increased the expression of CD62L on naïve (p = 0.011), and central memory CD4+ T cells (p = 0.038) as well as on naïve CD8+ T cells, although this latter effect only approached significance (p = 0.052; see Fig.4). The impact of fludrocortisone on CD62L expression was blocked by the MR antagonist spironolactone, suggesting an MR-specific mechanism of the fludrocortisone-induced CD62L upregulation. Spironolactone alone, presumably by blocking activity of endogenous MR ligands, likewise decreased CD62L expression on these subsets, but only after a 4 h incubation period (p ≤ 0.021). The MR antagonist also reduced the expression of CCR7 on naïve CD4+ and CD8+ T cells after 2 h. Fludrocortisone upregulated expression of CCR7 only on central memory CD4+ T cells after 4 h and this effect was blocked by coincubation with spironolactone (Fig.4).
Figure 4.
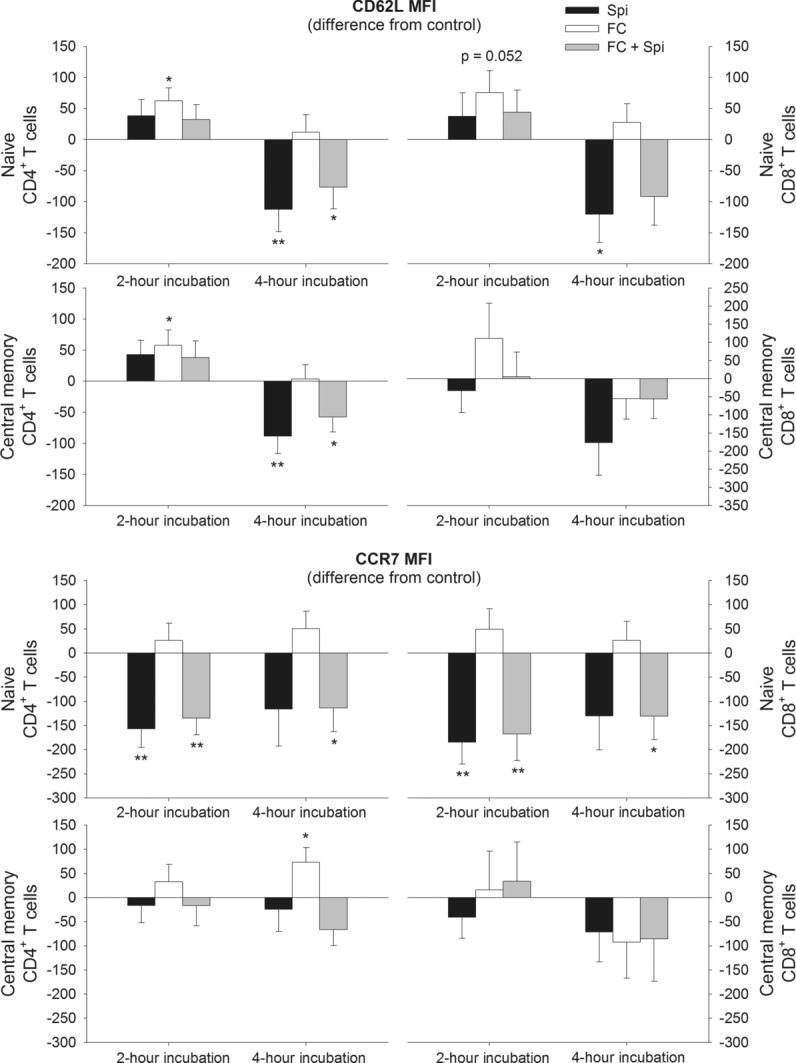
Changes in CD62L and CCR7 expression on naïve and central memory CD4+ and CD8+ T cells after incubation with spironolactone and fludrocortisone. Blood was sampled during sleep at 03:30 h and was then incubated for 2 or 4 h either with spironolactone (Spi), fludrocortisone (FC), fludrocortisone plus spironolactone (FC + Spi) or PBS as control. Expression of CD62L (top) and CCR7 (bottom) were assessed by flow cytometry on naïve (CD45RA+CD62L+) and central memory (CD45RA−CD62L+) CD4+ (left) and CD8+ (right) T cells and are shown as mean ± SEM of median fluorescence intensity (MFI) of 13 healthy male subjects. Data are expressed as difference from the PBS control. *p < 0.05, **p < 0.01, for pairwise comparisons between effects of the active agent(s) and PBS (paired t-tests).
Fludrocortisone increased CXCR4 expression on all subsets, except for effector CD4+ T cells, after the 2 h incubation interval. CXCR4 expression was further enhanced following the longer 4 h incubation interval (p < 0.001; Fig.5). The effect was blocked by spironolactone after 2 h in the naïve subsets but only attenuated in the remaining subsets or with longer incubation time. Spironolactone alone did not consistently affect CXCR4 expression on the naïve T-cell subsets (Fig.5). However, on effector memory CD4+ T cells as well as central memory, effector memory, and effector CD8+ T cells, spironolactone (such as fludrocortisone) paradoxically increased CXCR4 expression (p ≤ 0.044; Fig.5).
Figure 5.

Changes in CXCR4 expression on CD4+ and CD8+ T-cell subsets after incubation with spironolactone and fludrocortisone. Blood was sampled during sleep at 03:30 h and was then incubated for 2 or 4 h either with spironolactone (Spi), fludrocortisone (FC), fludrocortisone plus spironolactone (FC + Spi) or PBS as control. Expression of CXCR4 was assessed by flow cytometry on naïve (CD45RA+CD62L+), central memory (CD45RA−CD62L+), effector memory (CD45RA−CD62L−) and effector (CD45RA+CD62L−) CD4+ (left) and CD8+ (right) T cells and is shown as mean ± SEM of median fluorescence intensity (MFI) of 13 healthy male subjects. Data are expressed as difference from the PBS control. *p < 0.05, **p < 0.01, for pairwise comparisons between effects of the active agent(s) and PBS (paired t-tests).
Discussion
The role of MRs in the physiological regulation of T-cell migration has not yet been clearly defined. Previous animal studies showed that prolonged administration of aldosterone decreases CD4+ T-cell numbers 22 and T-cell percentages 23 in blood, whereas a single dose only counteracts stress-induced increases in lymphocyte numbers 3. Our study on the effects of fludrocortisone on numbers of various T-cell subsets and expression of various molecules involved in T-cell migration indicates a clear role for human MRs in the acute regulation of T-cell trafficking. We showed that a single administration of fludrocortisone to healthy men selectively reduces numbers of circulating naïve CD4+, central memory CD4+, and naïve CD8+ T cells. The results are in line with our recent study showing that the MR antagonist spironolactone increases blood cell counts of the same T-cell subsets during early sleep, while other T-cell subpopulations remain unaffected 11. The present findings provide conclusive evidence that the effects of spironolactone are specific to its MR blockade and not secondary to changes in cytokine release that have been found following spironolactone administration and are MR-independent 14. Moreover, although fludrocortisone displays some glucocorticoid potency, such activity is negligible at the low dose of the drug used in our experiments 24. Together, the highly complementary findings observed following fludrocortisone and spironolactone administration strongly suggest that the MR is the specific receptor involved in the effects of both drugs.
Our study also showed that peripheral MRs are not saturated during sleep and thus can be further activated by an exogenous ligand such as fludrocortisone, as has been similarly demonstrated for central nervous system MRs 18,25. This finding is important given that human leukocytes normally do not express the enzyme 11 β-hydroxysteroid dehydrogenase type 2, which protects MRs in the kidney from being constantly occupied by cortisol 26. Therefore, T cells seem to have other MR-protecting mechanisms, as has already been discussed for brain MRs 27. As a consequence of MR activation at the HPA axis level, cortisol concentration was also reduced in the present study, which could have influenced the observed effects on T cells. However, because cortisol diminishes T-cell numbers via GR activation 4,6, decreased cortisol levels are expected to counteract the effects of fludrocortisone to some extent.
In vitro, we identified increases in CD62L, CCR7, and CXCR4 expression as possible molecular mechanisms mediating the effect of MR signaling on T-cell redistribution. The failure of fludrocortisone to further upregulate CCR7 expression on naïve T cells was probably due to a ceiling effect, as expression of CCR7 on these subsets is already rather high. A role for MR signaling in CCR7 expression is still supported by the finding that spironolactone alone reduced CCR7 expression, presumably by blocking activity of endogenous MR ligands. On the other hand, fludrocortisone-induced increases in CD62L expression became evident in vitro but not in vivo. This discrepancy might be due to changes in the blood composition of T-cell subsets that occur in vivo following selective disappearance of cells with high CD62L expression from the circulation, which might have obscured the impact of fludrocortisone on this molecule.
Increases in the expression of CD62L and CCR7 suggest a redistribution of T cells to lymph nodes, as these are the main lymph node-homing receptors on T cells 16. This is in line with our findings that only T-cell subsets with lymph node-homing capacity (i.e., naïve and central memory T cells) were affected by fludrocortisone administration in this study, and by spironolactone in our former study 11. The reduction in T-cell numbers in blood does not provide definite proof for an extravasation of the cells, as they might have only attached to the endothelium, thus remaining undetected in the circulating blood. Nevertheless, an enhanced adhesion to the endothelium following increases in CD62L and CCR7 expression is the first step in the migratory cascade and would also favor a subsequent extravasation and homing to lymph nodes. The fact that glucocorticoids inhibit lymphocyte migration to lymph nodes 1,28 might explain why effects of an MR blockade were only seen during early sleep, when cortisol levels are low and there is only minor activation of GRs 11. Also in the present study, during high GR activation in the morning the effect of fludrocortisone on cell counts faded away, despite its high biological half-life of up to 36 h.
Taken together, predominant MR activation during periods of high aldosterone and low cortisol levels seems to provide the optimal endocrine milieu for facilitated homing of naïve T cells to the lymph nodes, i.e., the site where adaptive immune responses are initiated. This hormonal constellation indeed hallmarks early nocturnal sleep, which is known to support the formation of adaptive immune responses 29. Nocturnal sleep has been consistently found to benefit the formation of an antigen-specific immune response, using experimental vaccination in humans 30–34. An enhanced recruitment of naïve T cells to lymph nodes during sleep is a likely mechanism for this beneficial effect of sleep on adaptive immunity 29,31,35, a view that is further supported by data from animal studies 36,37. Aldosterone, which is released in a tightly sleep-dependent fashion and seems to be more relevant as endogenous agonist for lymphocytic MRs than cortisol 7,8,38,39, conceivably mediates this effect of sleep, as blocking MRs during sleep affects T-cell numbers in the same way as depriving subjects from nocturnal sleep 11,12,40. Our present results showing MR-mediated regulation of CD62L and CCR7 expression and reduced numbers of circulating naïve T cells following fludrocortisone administration corroborate and extend this concept (see also Fig.6).
Figure 6.
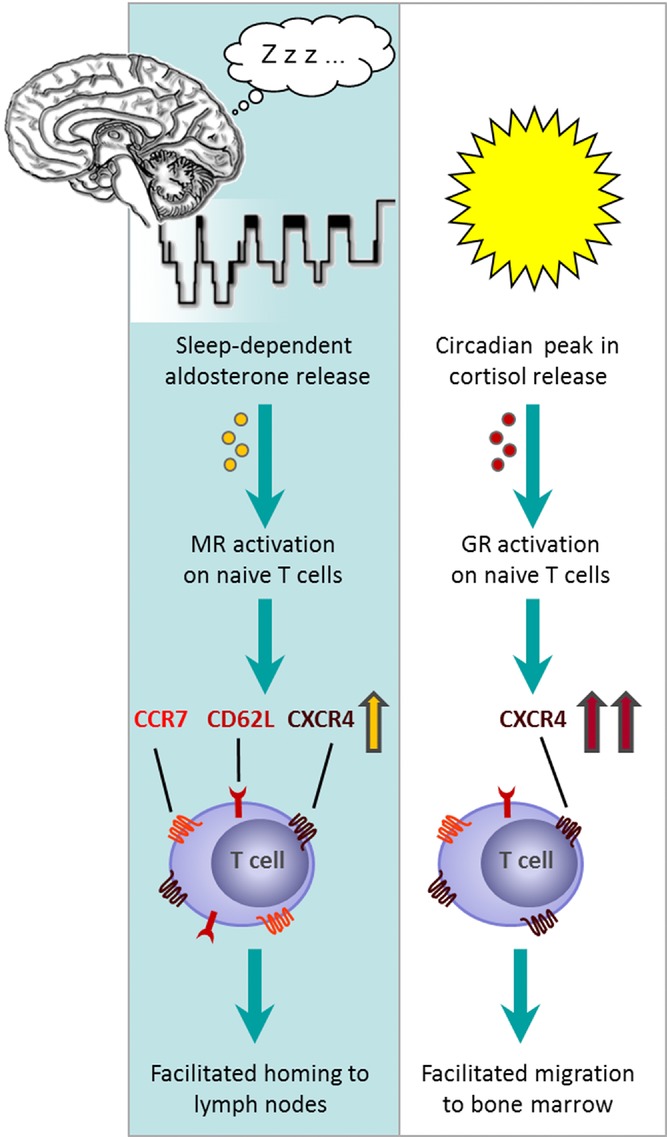
Concept: Nocturnal sleep facilitates T-cell homing to lymph nodes via aldosterone release, whereas cortisol facilitates migration to bone marrow during daytime wakefulness. Sleep induces an increase in aldosterone levels, which activates mineralocorticoid receptors (MRs) on naïve T cells (left). This leads to an increase in the expression of CCR7 and CD62L, the most important lymph node-homing receptors on T cells, as well as of CXCR4. As a consequence, migration to lymph nodes is facilitated. This concept could explain the beneficial effect of sleep on mounting an adaptive immune response. During daytime, the circadian peak in cortisol release leads to an activation of glucocorticoid receptors (GRs) on T cells, which diminishes lymph node homing (right). The further increase in CXCR4 expression leads to a preferential recruitment of these cells to the bone marrow, which produces high levels of the CXCR4 ligand CXCL12 during daytime wakefulness.
On naïve T-cell subsets, fludrocortisone also increased the expression of CXCR4 in vivo and in vitro. CXCR4 expression shows a circadian rhythm, with highest levels in the morning which are dependent on the morning cortisol rise 4,6. The impact of fludrocortisone was unexpected, as we and others have shown that the increasing effect of cortisol on CXCR4 expression is mediated via activation of GRs and most likely induces a redistribution of CXCR4+ T cells to the bone marrow in the morning hours 4,6,41,42. However, CXCR4 signaling is also involved in the migration of naïve and central memory T cells to lymph nodes 43,44. In combination with the enhanced expression of the lymph node-homing receptors CD62L and CCR7, it is therefore plausible to assume that CXCR4 upregulation following MR activation at a time of low cortisol levels preferentially redistributes these cells to the lymph nodes rather than to the bone marrow. This conclusion well fits with findings in rodents that, in the bone marrow, levels of CXCL12, the ligand of CXCR4 are lowest during the rest period 45. Thus attraction of T cells to this compartment is expected to be reduced during sleep. In contrast, during the active phase, when CXCL12 levels in the bone marrow are at maximum and high cortisol levels in blood interfere with lymph node homing, CXCR4 signaling will favor recruitment of T cells to the bone marrow 1,4,28,46. Therefore, the consequences of enhanced CXCR4 expression might crucially depend on the time of day (see also Fig.6). However, future animal and in vitro studies are warranted to clarify whether the changes we observed in the expression of different migration molecules following MR signaling indeed are functional in that they cause the extravasation and recruitment of T cells to lymph nodes or other body compartments during sleep.
In contrast to CD62L, spironolactone alone failed to consistently affect levels of CXCR4 on naïve subsets, which agrees with previous results showing no effects of spironolactone on CXCR4 expression in vivo 11. Nevertheless spironolactone effectively blocked the increase in CXCR4 expression on naïve subsets after 2 h of in vitro culture with fludrocortisone, which points to an essential involvement of MRs specifically for this subset. For the other T-cell subsets, also during the shorter incubation time, spironolactone failed to completely block the CXCR4-enhancing effects of fludrocortisone, which suggests additional MR-independent actions. Paradoxically, on these subsets, spironolactone alone increased CXCR4 expression, as did the MR agonist fludrocortisone. Such increasing effects of fludrocortisone and spironolactone on CXCR4 expression of nonnaïve subsets were not observed in vivo in the present and a former study 11, respectively, raising the suspicion that they reflect nonphysiological conditions of in vitro culture. Likewise, such MR-agonistic actions of spironolactone were revealed in other in vitro systems employing human cell lines 47 and isolated rat hearts 48.
MR effects are of major clinical relevance as hyperaldosteronism is causally linked to cardiovascular diseases. One underlying pathomechanism is an inflammatory response following prolonged MR activation. Interestingly, T cells were identified as key regulators in this process, which eventually leads to hypertension, cardiac fibrosis, heart failure, and endothelial dysfunction 49,50. In this context, a recent study in rodents showed that CXCR4-antagonism blocks the accumulation of T cells in heart and kidney that was induced by chronic mineralocorticoid excess 51. Although those authors suggested an MR-mediated increase in local CXCL12 levels as possible cause of the observed effects, results of the present experiments provide an additional explanation by showing a direct influence of MR activation on T-cell CXCR4 expression. Finally, also our findings on CD62L seem to be relevant in the clinical setting, as patients referred to coronary angiography show positive associations between plasma aldosterone and soluble CD62L levels 52. Besides these detrimental actions of hyperaldosteronism on inflammation and cardiovascular function the physiological role of MR signaling in T-cell migration could be used clinically to support beneficial immune responses. If fludrocortisone indeed facilitates recruitment of naïve T cells to lymph nodes, this substance might serve as an adjuvant during vaccination. Such application is also suggested by findings that aldosterone enhances the priming of naïve T cells via effects on dendritic cells 53.
In summary, our study provides the first evidence that MR signaling is essentially involved in the physiological regulation of T-cell migration in humans. To the best of our knowledge, this is the first study implying MRs in the regulation of CD62L and CCR7 expression and showing a direct impact of mineralocorticoids on CXCR4 expression. Our results further support the hypothesis that the beneficial effect of sleep on adaptive immunity is at least partially mediated by sleep-dependent increases in MR signaling, with a consequent facilitation of T-cell migration to lymph nodes.
Materials and methods
Subjects
Thirteen healthy men participated in the study (mean age ± SEM: 22.62 ± 0.86 years). All subjects had a regular sleep/wake pattern, did not take any medication at the time of the experiments, and were nonsmokers. Acute and chronic illness was excluded by medical history, physical examination, and routine laboratory investigation. The men were synchronized by daily activities and nocturnal rest. All subjects spent one adaptation night in the laboratory in order to become accustomed to the experimental setting. The study was approved by the Ethics Committee of the University of Lübeck and all participants gave written informed consent.
Experimental design
The study was performed according to a double-blind, randomized cross-over design. Each participant spent two experimental nights in the sleep laboratory (placebo versus fludrocortisone). The two nights were separated by at least 2 weeks to assure clearance of the drug. The order of conditions was balanced across subjects. On experimental nights, subjects arrived at the laboratory at 19:30 h for preparing blood sampling and polysomnographic recordings. Between 21:40 and 22:30 h subjects performed on psychological tasks, results of which (as part of an extended subject sample) are reported elsewhere 25. Sleep was allowed for a 7 h period starting around 23:00 h (lights off; ± 30 min). Subjects received either 0.2 mg of fludrocortisone (Merck Serono GmbH, Germany; peak plasma concentration after 1.7 h; biological half-life 18–36 h) or placebo orally right before lights were turned off.
Blood was sampled first 90 min and immediately before drug administration, and then every 45 min until 90 min after awakening for determination of cortisol, ACTH, and aldosterone levels. T-cell parameters were determined in blood sampled 90 min before drug administration and then every 1.5 h until 45 min as well as 90 min after awakening. Blood was sampled via an intravenous forearm catheter, which was connected to a long thin tube and enabled blood collection from an adjacent room without disturbing the subject's sleep. To prevent clotting, approximately 700 mL of saline solution were infused during the experimental period. Potential side effects of fludrocortisone were evaluated in the morning by questionnaires. Standard polysomnographic recordings were obtained to assure normal nocturnal sleep. Blood pressure was assessed before subjects went to sleep and again in the morning.
T-cell subpopulations
Absolute counts of CD3+ total T lymphocytes, CD4+ T-helper lymphocytes, and CD8+ cytotoxic T lymphocytes as well as their naïve (CD45RA+CD62L+), central memory (CD45RA−CD62L+), effector memory (CD45RA−CD62L−), and (terminally differentiated) effector (CD45RA+CD62L−) subsets were determined by a ‘‘lyse no-wash’’ flow cytometry procedure. Briefly, 50 μL of an undiluted blood sample was immunostained with anti-CD3/Horizon V500, anti-CD8/PerCP, anti-CD4/Horizon V450, anti-CD62L/FITC, anti-CD45RA/PE, and anti-CD184 (CXCR4)/allophycocyanin, in Trucount tubes (all from BD Biosciences, San Jose, USA). After 15 min of incubation at room temperature, 0.9 mL of fluorescence activated cell sorting (FACS) lysing solution (BD Biosciences) was added followed by incubation for 15 min. Finally, samples were mixed gently and at least 10 000 CD3+ cells were acquired on a BD LSRII Flow Cytometer using DIVA Software (BD Biosciences). The absolute number of cells per microliter blood was calculated using the following formula: Cells/μL = (acquired cell events in the respective gate) × (number of beads per tube)/((acquired bead events) × (sample volume (μL))). To study changes in relative quantity of CD184 (CXCR4) and CD62L (l-selectin) on the cell subsets, the median fluorescence intensity (MFI) of the labeled anti-CD184- and anti-CD62L-antibodies was analyzed.
Hormone assays and sleep analyses
Samples for measuring hormone concentrations were kept frozen at −80°C until assay. Serum cortisol, plasma ACTH, and serum aldosterone were measured using commercial assays (cortisol, ACTH: Immulite, DPC Biermann, Bad Nauheim, Germany; aldosterone: IBL International GmbH, Hamburg, Germany). Sensitivity was as follows: cortisol 0.2 μg/dL, ACTH 9 pg/mL, aldosterone 10 pg/mL. Intraassay and interassay coefficients of variation for all assays were < 10.4%. Sleep stages were determined off-line from polysomnographic recordings following standard criteria 54, and confirmed that sleep architecture was normal under laboratory conditions and was not significantly affected by fludrocortisones administration (see also 25).
In vitro experiments
Whole blood from additional 13 healthy subjects (mean age ± SEM: 23.46 ± 0.71 years) was sampled during early sleep (at 03:30 h). To test the influence of fludrocortisone and spironolactone on the expression of CXCR4, CD62L and CCR7, blood samples were incubated with PBS at 37°C in the absence or presence of 6.7 nM fludrocortisone (representing expected peak blood concentration after oral intake of 0.2 mg fludrocortisone 55), 5 μM spironolactone (MR antagonist) or both drugs in combination for 2 and 4 h. Samples were then labeled with anti-CD3/Horizon V500, anti-CD4/Horizon V450, anti-CD62L/FITC, anti-CD45RA/Alexa Fluor 700, anti-CD184 (CXCR4)/allophycocyanin, and anti-CD197 (CCR7)-PE (all from BD Biosciences), and anti-CD8/Qdot 605 (Invitrogen), and subsequently processed via a ‘‘lyse no-wash’’ flow cytometry procedure as described for the in vivo experiments.
Statistical analyses
Data are presented as mean ± SEM. Analyses generally relied on ANOVA including repeated measures factors for “Treatment” (fludrocortisone versus placebo) and “Time” (reflecting the different time points of the observation period). Differences between conditions in baseline measures (i.e., the first two blood samples) were used as covariates to correct for day-to-day variations in flow cytometer performance and baseline differences. Degrees of freedom were corrected using the Greenhouse-Geisser procedure. Paired t-tests were applied to analyze post hoc differences at single time points once ANOVA indicated significant effects, to assess results of in vitro experiments, and to analyze differences in sleep parameters and blood pressure. A p-value < 0.05 was considered significant.
Acknowledgments
We are grateful to Sabine Groch, Ulrike Steffen and Kerstin Lausen for technical assistance. This work was supported by a grant from the Deutsche Forschungsgemeinde (DFG), SFB 654 ‘‘Plasticity and Sleep’’ and by a grant from the University of Lübeck (H02-2012). L.B., B.L., J.B., and T.L. designed the research, L.B., B.L., and T.L. performed experiments, L.B., J.B., and T.L. analyzed data and wrote the manuscript.
Glossary
- ACTH
adrenocorticotropic hormone
- GR
glucocorticoid receptor
- HPA axis
hypothalamic-pituitary-adrenocortical axis
- MFI
median fluorescence intensity
- MR
mineralocorticoid receptor
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Additional supporting information may be found in the online version of this article at the publisher's web-site
References
- Ottaway CA, Husband AJ. Central nervous system influences on lymphocyte migration. Brain Behav. Immun. 1992;6:97–116. doi: 10.1016/0889-1591(92)90011-c. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, Miller AH, Stein M, McEwen BS, Spencer RL. Diurnal and acute stress-induced changes in distribution of peripheral blood leukocyte subpopulations. Brain Behav. Immun. 1994;8:66–79. doi: 10.1006/brbi.1994.1006. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, Miller AH, McEwen BS, Spencer RL. Stress-induced changes in blood leukocyte distribution. Role of adrenal steroid hormones. J. Immunol. 1996;157:1638–1644. [PubMed] [Google Scholar]
- Dimitrov S, Benedict C, Heutling D, Westermann J, Born J, Lange T. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood. 2009;113:5134–5143. doi: 10.1182/blood-2008-11-190769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonova ST, Zimmer J, Turner JD, Muller CP. Diurnal redistribution of human lymphocytes and their temporal associations with salivary cortisol. Chronobiol. Int. 2013;30:669–681. doi: 10.3109/07420528.2013.775654. [DOI] [PubMed] [Google Scholar]
- Besedovsky L, Born J, Lange T. Endogenous glucocorticoid receptor signaling drives rhythmic changes in human T-cell subset numbers and the expression of the chemokine receptor CXCR4. FASEB J. 2014;28:67–75. doi: 10.1096/fj.13-237958. [DOI] [PubMed] [Google Scholar]
- Charloux A, Gronfier C, Lonsdorfer-Wolf E, Piquard F, Brandenberger G. Aldosterone release during the sleep-wake cycle in humans. Am. J. Physiol. 1999;276:E43–E49. doi: 10.1152/ajpendo.1999.276.1.E43. [DOI] [PubMed] [Google Scholar]
- Charloux A, Gronfier C, Chapotot F, Ehrhart J, Piquard F, Brandenberger G. Sleep deprivation blunts the night time increase in aldosterone release in humans. J. Sleep Res. 2001;10:27–33. doi: 10.1046/j.1365-2869.2001.00235.x. [DOI] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Kalman BA, Spencer RL. Rapid corticosteroid-dependent regulation of mineralocorticoid receptor protein expression in rat brain. Endocrinology. 2002;143:4184–4195. doi: 10.1210/en.2002-220375. [DOI] [PubMed] [Google Scholar]
- Besedovsky L, Born J, Lange T. Blockade of mineralocorticoid receptors enhances naive T-helper cell counts during early sleep in humans. Brain Behav. Immun. 2012;26:1116–1121. doi: 10.1016/j.bbi.2012.07.016. [DOI] [PubMed] [Google Scholar]
- Born J, Lange T, Hansen K, Mölle M, Fehm HL. Effects of sleep and circadian rhythm on human circulating immune cells. J. Immunol. 1997;158:4454–4464. [PubMed] [Google Scholar]
- Armanini D, Endres S, Kuhnle U, Weber PC. Parallel determination of mineralocorticoid and glucocorticoid receptors in T- and B-lymphocytes of human spleen. Acta. Endocrinol.(Copenh) 1988;118:479–482. doi: 10.1530/acta.0.1180479. [DOI] [PubMed] [Google Scholar]
- Sonder SU, Mikkelsen M, Rieneck K, Hedegaard CJ, Bendtzen K. Effects of spironolactone on human blood mononuclear cells: mineralocorticoid receptor independent effects on gene expression and late apoptosis induction. Br. J. Pharmacol. 2006;148:46–53. doi: 10.1038/sj.bjp.0706700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rosa F. T-lymphocyte interaction with stromal, bone and hematopoietic cells in the bone marrow. Immunol. Cell Biol. 2009;87:20–29. doi: 10.1038/icb.2008.84. [DOI] [PubMed] [Google Scholar]
- von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- Mazo IB, Honczarenko M, Leung H, Cavanagh LL, Bonasio R, Weninger W, Engelke K, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity. 2005;22:259–270. doi: 10.1016/j.immuni.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Buckley TM, Mullen BC, Schatzberg AF. The acute effects of a mineralocorticoid receptor (MR) agonist on nocturnal hypothalamic-adrenal-pituitary (HPA) axis activity in healthy controls. Psychoneuroendocrinology. 2007;32:859–864. doi: 10.1016/j.psyneuen.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Morise T, Miyamori I, Hifumi S, Okamoto S, Ikeda M, Takeda Y, Koshida H, et al. Effect of 9 alpha-fluorocortisol on the excretion of urinary digoxin-like substance in normotensive men. Endocrinol. Jpn. 1986;33:279–283. doi: 10.1507/endocrj1954.33.279. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Pheley A, Schroeppel J, Schenck C, Marshall P, Kind A, Haugland JM, et al. A preliminary placebo-controlled crossover trial of fludrocortisone for chronic fatigue syndrome. Arch. Intern. Med. 1998;158:908–914. doi: 10.1001/archinte.158.8.908. [DOI] [PubMed] [Google Scholar]
- Westerdahl C, Bergenfelz A, Larsson J, Nerbrand C, Valdemarsson S, Wihl A, Isaksson A. Re-evaluation of the fludrocortisone test: duration, NaCl supplementation and cut-off limits for aldosterone. Scand. J. Clin. Lab. Invest. 2009;69:234–241. doi: 10.1080/00365510802483690. [DOI] [PubMed] [Google Scholar]
- Miller AH, Spencer RL, Hassett J, Kim C, Rhee R, Ciurea D, Dhabhar F, et al. Effects of selective type I and II adrenal steroid agonists on immune cell distribution. Endocrinology. 1994;135:1934–1944. doi: 10.1210/endo.135.5.7956914. [DOI] [PubMed] [Google Scholar]
- Ahokas RA, Warrington KJ, Gerling IC, Sun Y, Wodi LA, Herring PA, Lu L, et al. Aldosteronism and peripheral blood mononuclear cell activation: a neuroendocrine-immune interface. Circ. Res. 2003;93:e124–e135. doi: 10.1161/01.RES.0000102404.81461.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renold AE, Haydar NA, Reddy WJ, Goldfien A, St. Marc JR, Laidlaw JC. Biological effects of fluorinated derivatives of hydrocortisone and progesterone in man. Ann. N. Y. Acad. Sci. 1955;61:582–590. doi: 10.1111/j.1749-6632.1955.tb42510.x. [DOI] [PubMed] [Google Scholar]
- Groch S, Wilhelm I, Lange T, Born J. Differential contribution of mineralocorticoid and glucocorticoid receptors to memory formation during sleep. Psychoneuroendocrinology. 2013;38:2962–2972. doi: 10.1016/j.psyneuen.2013.08.006. [DOI] [PubMed] [Google Scholar]
- Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol. Cell Endocrinol. 2009;301:123–131. doi: 10.1016/j.mce.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez EP. Mineralocorticoid receptors in the brain and cardiovascular regulation: minority rule? Trends. Endocrinol. Metab. 2011;22:179–187. doi: 10.1016/j.tem.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackstein R, Borenstein M. The effects of corticosteroids on lymphocyte recirculation in humans: analysis of the mechanism of impaired lymphocyte migration to lymph node following methylprednisolone administration. J. Investig. Med. 1995;43:68–77. [PubMed] [Google Scholar]
- Besedovsky L, Lange T, Born J. Sleep and immune function. Pflugers. Arch. 2012;463:121–137. doi: 10.1007/s00424-011-1044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange T, Perras B, Fehm HL, Born J. Sleep enhances the human antibody response to hepatitis A vaccination. Psychosom. Med. 2003;65:831–835. doi: 10.1097/01.psy.0000091382.61178.f1. [DOI] [PubMed] [Google Scholar]
- Lange T, Dimitrov S, Bollinger T, Diekelmann S, Born J. Sleep after vaccination boosts immunological memory. J. Immunol. 2011;187:283–290. doi: 10.4049/jimmunol.1100015. [DOI] [PubMed] [Google Scholar]
- Spiegel K, Sheridan JF, Van Cauter E. Effect of sleep deprivation on response to immunization. JAMA. 2002;288:1471–1472. doi: 10.1001/jama.288.12.1471-a. [DOI] [PubMed] [Google Scholar]
- Prather AA, Hall M, Fury JM, Ross DC, Muldoon MF, Cohen S, Marsland AL. Sleep and antibody response to hepatitis B vaccination. Sleep. 2012;35:1063–1069. doi: 10.5665/sleep.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, Brytting M, Markstrom A, Broman JE, Schioth HB. Acute sleep deprivation has no lasting effects on the human antibody titer response following a novel influenza A H1N1 virus vaccination. BMC. Immunol. 2012;13:1. doi: 10.1186/1471-2172-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Dickstein JB, Hay JB, Lue FA, Moldofsky H. The relationship of lymphocytes in blood and in lymph to sleep/wake states in sheep. Sleep. 2000;23:185–190. [PubMed] [Google Scholar]
- Zager A, Andersen ML, Ruiz FS, Antunes IB, Tufik S. Effects of acute and chronic sleep loss on immune modulation of rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R504–R509. doi: 10.1152/ajpregu.00105.2007. [DOI] [PubMed] [Google Scholar]
- Armanini D, Strasser T, Weber PC. Characterization of aldosterone binding sites in circulating human mononuclear leukocytes. Am. J. Physiol. 1985;248:E388–E390. doi: 10.1152/ajpendo.1985.248.3.E388. [DOI] [PubMed] [Google Scholar]
- Wehling M, Kuhls S, Armanini D. Volume regulation of human lymphocytes by aldosterone in isotonic media. Am. J. Physiol. 1989;257:E170–E174. doi: 10.1152/ajpendo.1989.257.2.E170. [DOI] [PubMed] [Google Scholar]
- Lange T, Dimitrov S, Born J. Effects of sleep and circadian rhythm on the human immune system. Ann. N. Y. Acad. Sci. 2010;1193:48–59. doi: 10.1111/j.1749-6632.2009.05300.x. [DOI] [PubMed] [Google Scholar]
- Fauci AS. Mechanisms of corticosteroid action on lymphocyte subpopulations. I. Redistribution of circulating T and b lymphocytes to the bone marrow. Immunology. 1975;28:669–680. [PMC free article] [PubMed] [Google Scholar]
- Okutsu M, Ishii K, Niu KJ, Nagatomi R. Cortisol-induced CXCR4 augmentation mobilizes T lymphocytes after acute physical stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R591–R599. doi: 10.1152/ajpregu.00438.2004. [DOI] [PubMed] [Google Scholar]
- Bai Z, Hayasaka H, Kobayashi M, Li W, Guo Z, Jang MH, Kondo A, et al. CXC chemokine ligand 12 promotes CCR7-dependent naive T cell trafficking to lymph nodes and Peyer's patches. J. Immunol. 2009;182:1287–1295. doi: 10.4049/jimmunol.182.3.1287. [DOI] [PubMed] [Google Scholar]
- Scimone ML, Felbinger TW, Mazo IB, Stein JV, von Andrian UH, Weninger W. CXCL12 mediates CCR7-independent homing of central memory cells, but not naive T cells, in peripheral lymph nodes. J. Exp. Med. 2004;199:1113–1120. doi: 10.1084/jem.20031645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat. Rev. Immunol. 2013;13:190–198. doi: 10.1038/nri3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad C, Lombes M, Aggerbeck M, Rafestin-Oblin ME, Barouki R. Cell-specific, promoter-dependent mineralocorticoid agonist activity of spironolactone. Mol. Pharmacol. 1997;51:285–292. doi: 10.1124/mol.51.2.285. [DOI] [PubMed] [Google Scholar]
- Barbato JC, Mulrow PJ, Shapiro JI, Franco-Saenz R. Rapid effects of aldosterone and spironolactone in the isolated working rat heart. Hypertension. 2002;40:130–135. doi: 10.1161/01.hyp.0000025879.29822.24. [DOI] [PubMed] [Google Scholar]
- Shen JZ, Young MJ. Corticosteroids, heart failure, and hypertension: a role for immune cells? Endocrinology. 2012;153:5692–5700. doi: 10.1210/en.2012-1780. [DOI] [PubMed] [Google Scholar]
- Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59:324–330. doi: 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- Chu PY, Zatta A, Kiriazis H, Chin-Dusting J, Du XJ, Marshall T, Kaye DM. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circ. Heart Fail. 2011;4:651–658. doi: 10.1161/CIRCHEARTFAILURE.110.960831. [DOI] [PubMed] [Google Scholar]
- Tomaschitz A, Pilz S, Ritz E, Grammer T, Amrein K, Merger S, Meinitzer A, et al. Relationship between plasma aldosterone concentration and soluble cellular adhesion molecules in patients referred to coronary angiography. Exp. Clin. Endocrinol. Diabetes. 2011;119:649–655. doi: 10.1055/s-0031-1287791. [DOI] [PubMed] [Google Scholar]
- Herrada AA, Contreras FJ, Marini NP, Amador CA, Gonzalez PA, Cortes CM, Riedel CA, et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J. Immunol. 2010;184:191–202. doi: 10.4049/jimmunol.0802886. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Kales A. A Manual of Standardized Terminology, Techniques and Scoring System for Sleep of Human Subjects. Washington DC: United States Government Printing Office; 1968. [Google Scholar]
- Mitsky VP, Workman RJ, Nicholson WE, Vernikos J, Robertson RM, Robertson D. A sensitive radioimmunoassay for fludrocortisone in human plasma. Steroids. 1994;59:555–558. doi: 10.1016/0039-128x(94)90074-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.